用于免疫病况的治疗的MK2途径抑制剂的口服组合物
文献发布时间:2023-06-19 18:27:32
相关申请的交叉引用
本申请要求于2020年03月27日提交的美国临时专利申请序列号63,000,746;于2020年04月24日提交的序列号63/015,241;于2020年05月01日提交的序列号63,018/954;于2020年05月08日提交的序列号63/022,301;于2020年05月08日提交的序列号63/022,298;于2020年05月13日提交的序列号63/024,160;于2020年07月20日提交的序列号63/053,903;于2020年09月10日提交的序列号63/076,689;于2020年12月16日提交的序列号63/126,173;于2020年12月21日提交的序列号63/128,523;于2021年01月11日提交的序列号63/136,080;于2021年01月13日提交的序列号63/136,967;于202年01月18日提交的序列号63/138,672;于2021年01月21日提交的序列号63/140,116;以及于2021年02月13日提交的序列号63/149,230(其中的每个通过引用被整体并入本文)的权益。
概述
本公开针对治疗炎性病况的方法,方法包括向患有炎性病况的人类受试者施用5mg/天至300mg/天的口服剂量的具有下列结构的化合物I:
或其衍生物以治疗所述炎性病况。
本公开进一步针对包括化合物I或化合物I的衍生物的口服药物组合物,其中口服组合物包括5mg至300mg的化合物I以及药学上可接受的载体。
如贯穿本公开所使用的,“化合物I”的叙述涵盖如下公开的以从4:1((P)-I:(M)-I)至999:1的任意摩尔比的阻转异构体化合物(P)-I和(M)-I,并且还包含其中化合物(P)-I基本上不含化合物(M)-I的实施方案。化合物(P)-I和(M)-I可以是如本文所描述的任何形式(例如,游离碱、结晶形式等)。
附图说明
图1是示出在禁食条件下单次剂量后ATI-450的平均(SD)血药浓度-时间曲线的图表,半对数标度。
图2是示出在禁食条件下BID(每天两次)给药7天后ATI-450的平均(SD)血药浓度-时间曲线的图表,半对数标度。
图3示出从用安慰剂或50mg/BID ATI-450给药的受试者采集的血液样品中体外LPS刺激的细胞因子/趋化因子产生的ATI-450调控。
图4A-4E是示出从用10mg、30mg、50mg、80mg和120mg的ATI-450或安慰剂给药的受试者采集的第7天的血液样品中体外LPS刺激的pHSP27(图4A,上部)、TNF-α(图4B,上部)、IL-1β(图4C,上部)、IL-8(图4D,上部)、IL-6(图4E,上部)的产生图表。图4A-4E还示出对比第1天给药前的值(设定为100%)与第7天给药后4小时的值(大约C
图5是示出从用安慰剂或80mg或120mg的ATI-450给药的受试者采集的血液样品中,体外刺激的IL-10与TNF-α和IL-1β的差分调控的图表。
图6描绘了比较LPS和IL-1β刺激的TNF-α(最左边)、IL-6(中间)和IL-8(最右边)产生的ATI-450调控的三个图表。
图7A-7C示出80mg或120mg BID施用后ATI-450的药代动力学(PK)数据。图7A的图表示出BID给药第1天(上部)和第7天(中间和下部)后ATI-450的平均血药浓度-时间曲线,半对数标度。图7B是示出以120mg BID给药的ATI-450的平均(±标准偏差(std.dev.))血药浓度-时间曲线的图表。图7C是示出120mg BID给药后第1天(左)和第7天(右)单独的受试者中的ATI-450的血药浓度-时间曲线的图表。
图8是示出在治疗的第1天、第7天、第14天、第28天、第42天、第56天和第84天以及第112天治疗结束后安慰剂组和ATI-450(50mg/BID)治疗组中所有患者的自基线的DAS28-CRP的中值变化的图表。每条线上的数字代表每个时间点的患者人数。
图9是示出在治疗过程中(直至第84天)具有DAS-28<2.6和<3.2的患者百分比的图表。
图10是示出在治疗的第1天、第7天、第14天、第28天、第42天、第56天和第84天以及第112天治疗结束后安慰剂组和ATI-450(50mg/BID)治疗组中所有患者的高敏性C反应蛋白(hsCRP)(mg/mL)自基线的中值百分比变化的图表。每条线上的数字代表每个时间点的患者人数。
图11是示出在治疗的第1天、第7天、第14天、第28天、第42天、第56天和第84天以及第112天治疗结束后安慰剂组和ATI-450(50mg/BID)治疗组中所有患者的肿胀关节数自基线的中值百分比变化的图表。每条线上的数字代表每个时间点的患者人数。
图12是示出在治疗的第1天、第7天、第14天、第28天、第42天、第56天和第84天以及第112天治疗结束后安慰剂组和ATI-450(50mg/BID)治疗组中所有患者的关节压痛数自基线的中值百分比变化的图表。每条线上的数字代表每个时间点的患者人数。
图13A-13D是示出疾病活动度的患者可视模拟标度(VAS)(图13A)、关节炎疼痛的患者VAS(图13B)、健康评估问卷(HAQ)功能障碍指数(DI)(图13C)和疾病活动度的医师VAS评分(图13C)的中值百分比变化的图表。
图14示出安慰剂组和ATI-450(50mg/BID)治疗组在研究过程中(直至第112天)的ACR20/50/70响应。
图15是示出通过手腕MRI RAMRIS评估对ATI-450治疗响应的患者的百分比。RAMRIS提供针对CARLOS(软骨损失)、侵蚀、骨炎、滑膜炎的分项评分,并且图15的图表示出治疗和手部的这些终点中的百分比响应性。如果观察到任何一只手至少1分的改善,则认为患者是有响应的。
图16是示出安慰剂患者和ATI-450(50mg BID)患者中CARLOS、侵蚀、骨炎和滑膜炎终点在第84天的基线变化的图表。数据代表手腕MRI RAMRIS评估的双手的平均值。菱形代表平均值。
图17是示出安慰剂患者和ATI-450(50mg BID)患者中每只手的CARLOS、侵蚀、骨炎和滑膜炎终点在第84天的基线变化的图表。数据代表手腕MRI RAMRIS评估的每只手的平均值。菱形代表平均值。
图18A-18B是示出来自安慰剂组和ATI-450(50mg/BID)治疗组中的患者的血液样品中第1天和第84天体外LPS-刺激的细胞因子(包含TNF-α和IL-1β(图18A)以及IL-6和IL-8(图18B))的图表。
图19A-19B示出在治疗的第1天、第28天、第56天和第84天,ATI-450治疗(50mg/BID)对促炎性细胞因子TNF-α(图19A)和抗炎性细胞因子IL-1RA(图19B)的内源性血浆水平的影响。
图20A-20D示出在治疗的第1天、第28天、第56天和第84天,ATI-450治疗(50mg/BID)对TNF-α(图20A)、IL-8(图20B)、IL-6(图20C)和MIP1β(图20D)的内源性血浆水平的影响。
图21是示出在小鼠胶原蛋白诱导关节炎模型中T调节(Treg)细胞中ATI-450介导的增加的图表。图表上的时间线描绘了胶原蛋白施用后小鼠中ATI-450和
图22A-22C是示出TLR4介导的细胞因子释放(图22A)、TLR3介导的细胞因子释放(图22B)和TLR7,8介导的细胞因子释放(图22C)的ATI-450抑制的图表。
图23是示出ATI-450剂量依赖性地抑制对于TNF-α产生的TLR2介导的诱导。
图24示出ATI-450抑制TH17细胞中的IL-17A产生。
图25示出ATI-450抑制IL-1β刺激的TNF-α、Il-6和IL-8产生。
图26A-26B示出ATI-450抑制人类外周血单核细胞中的IL-1α生物合成(图26A)和中性粒细胞中的IL-1α活性(图26B)两者。
图27示出化合物(P)-I的PXRD。
图28示出化合物(P)-I的TGA(上部轨迹)和DSC(下部轨迹)曲线。
详细说明
定义
在描述本发明的组合物和方法之前,应当理解的是,本发明不限于所描述的特定过程、制剂、组合物、或方法,因为这些可以变化。还应当理解的是,说明书中使用的术语仅出于描述特定型式或实施方案的目的,而非旨在限制本文实施方案的范围,本文实施方案的范围将仅由所附权利要求限定。除非另外定义,否则本文中使用的全部技术术语和科学术语具有与本领域普通技术人员通常理解的含义相同的含义。尽管与本文描述的那些方法和材料相似或等同的任何方法和材料都可以用于本文实施方案的实践或测试中,但现在描述优选的方法、装置、和材料。本文提及的全部出版物都通过引用被整体并入。本文中的任何内容均不应当解释为承认本文的实施方案无权借助在先发明而早于这样的公开。
除非上下文另外明确指出,否则如本文和所附权利要求中所使用的,单数形式“一(a)”、“一(an)”和“所述(the)”包含复数提及。因此,例如,对“MK2抑制剂”的提及是对本领域技术人员已知的一种或更多种MK2抑制剂及其等同物等的提及。
与“包含”、“含有”或“特征在于”同义的过渡术语“包括”是包含性的或开放式的,并且不排除附加的未列举的要素或方法步骤。相反,过渡短语“由……组成(consistingof)”排除未在权利要求中未指定的任何要素、步骤或成分。过渡短语“基本上由……组成(consisting essentially of)”将权利要求的范围限定至指定的材料或步骤“以及实质上不影响所要求的保护的发明的基本特性和新颖特性的材料或步骤”。本公开的组合物和方法可以包括公开的组分或步骤、基本上由公开的组分或步骤组成、或者由公开的组分或步骤组成。
如本文所使用的,术语“约”意为与其一起使用的数字的数值加或减10%。因此,约50%意为在45%-55%的范围内。
当与治疗剂结合使用时,“施用”意为将治疗剂施用至患者,由此治疗剂正向地影响其所靶向的组织。因此,如本文所使用的,当与MK2化合物结合使用时,术语“施用”可以包含(但不限于):将MK2抑制剂化合物提供到靶标组织内或靶标组织上;通过例如口服施用向患者全身性地提供MK2抑制剂化合物,由此使治疗剂到达靶标组织。
如本文所使用的,术语“其衍生物”指其盐、其药学上可接受的盐、其酯、其游离酸形式、其游离碱形式、其溶剂合物、其氘化衍生物、其水合物、其N-氧化物、其笼合物、其前体药物、其多晶型物、其立体异构体、其几何异构体、其互变异构体、其互变异构体的混合物、其对映异构体、其非对映异构体、其外消旋体、其立体异构体的混合物、其同位素(例如,氚、氘)、或其组合。
如本文所单独地或组合地使用的,术语“基本上不含”指在分析方法(如核磁共振(NMR)、气相色谱/质谱(GC/MS)、高效液相色谱法(HPLC)或液相色谱/质谱(LC/MS))的检测限度内不含异构体。
如本文所使用的,术语“病况”旨在与术语“紊乱”、“病征”、和“疾病”大体上同义并且可互换地使用,因为全部都反映人体或动物体或其部分之一的异常状况,该异常状况损害正常功能,通常表现为有区别的病征和症状,并且使人或动物具有降低的寿命或生活品质。
术语“组合疗法”意为施用两种或更多种治疗剂以治疗本公开中描述的治疗病况或紊乱。这样的施用涵盖以基本上同时的方式共同施用这些治疗剂,如在具有固定比例的活性成分的单个胶囊中、或者在每种活性成分的多个单独的胶囊中。此外,这样的施用还涵盖以连续方式使用每种类型的治疗剂。在任一情况下,治疗方案将在治疗本文中描述的病况或紊乱中提供药物组合的有益作用。
“MK2抑制剂”在本文中用于指展现出不超过约100μM并且更典型地不超过约50μM的针对丝裂原激活蛋白激酶激活蛋白激酶2(“MK2”)活性的IC
如本文所使用的,术语“药学上可接受的盐”指由碱或酸制备的对于施用至患者(如哺乳动物)可接受的盐。术语“药学上可接受的盐”包括通常用于形成碱金属盐和形成游离酸或游离碱的加成盐的盐。盐的性质并不重要,只要其是药学上可接受的。这样的盐可以来源于药学上可接受的无机或有机碱以及药学上可接受的无机或有机酸。
可以由无机酸或有机酸制备本文实施方案的化合物的适合的药学上可接受的酸加成盐。可以通过常规手段由本文实施方案的相应化合物通过例如用适当的酸或碱处理化合物来制备所有这些盐。
药学上可接受的酸包含无机酸(例如,盐酸、氢溴酸、氢碘酸、硝酸、碳酸、硫酸、磷酸和焦磷酸);和有机酸(例如,甲酸、乙酸、三氟乙酸、丙酸、琥珀酸、乙醇酸、恩波酸(扑酸)、甲磺酸、乙磺酸、2-羟基乙磺酸、泛酸、苯磺酸、甲苯磺酸、磺胺酸、甲磺酸(mesylic acid)、环己基氨基磺酸、硬脂酸、藻酸(algenic acid)、β-羟丁酸、丙二酸、半乳酸(galacticacid)、半乳糖醛酸、柠檬酸、富马酸、葡萄糖酸、谷氨酸、乳酸(lactic acid)、马来酸、苹果酸、扁桃酸、粘酸、抗坏血酸、草酸、泛酸、琥珀酸、酒石酸、苯甲酸、乙酸、昔萘酸(xinafoicacid)(1-羟基-2-萘甲酸)、萘二磺酸(napadisilic acid)(1,5-萘二磺酸(1,5-naphthalenedisulfonic acid))等)两者。
适用于本文所描述的制剂的来源于药学上可接受的无机碱的盐包含铝、铵、钙、铜、铁、亚铁、锂、镁、锰(manganic)、锰(manganous)、钾、钠、锌等。来源于药学上可接受的有机碱的盐包含伯胺、仲胺和叔胺的盐,伯胺、仲胺和叔胺包含烷基胺、芳烷基胺、杂环基胺、环胺、天然存在的胺等,如精氨酸、甜菜碱、咖啡因、胆碱、氯普鲁卡因、二乙醇胺、N-甲葡糖胺、N,N’-二苄基乙二胺、二乙胺、2-二乙基氨基乙醇、2-二甲基氨基乙醇、乙醇胺、乙二胺、N-乙基吗啉、N-乙基哌啶、葡糖胺、葡萄糖胺、组氨酸、哈胺(hydrabamine)、异丙胺、赖氨酸、甲葡糖胺、吗啉、哌嗪、哌啶、多胺树脂、普鲁卡因、嘌呤、可可碱、三乙胺、三甲胺、三丙胺、氨丁三醇等。
根据本文实施方案的其他优选的盐是季铵化合物,其中阴离子(X-)的当量与N原子的正电荷有关。X-可以是各种无机酸的阴离子(例如,氯离子、溴离子、碘离子、硫酸根、硝酸根、磷酸根)、或者有机酸的阴离子(例如,乙酸根、马来酸根、延胡索酸根、柠檬酸根、草酸根、琥珀酸根、酒石酸根、苹果酸根、扁桃酸根、三氟乙酸根、甲磺酸根和对甲苯磺酸根)。X-优选为选自氯离子、溴离子、碘离子、硫酸根、硝酸根、乙酸根、马来酸根、草酸根、琥珀酸根或三氟乙酸根的阴离子。更优选地,X-为氯离子、溴离子、三氟乙酸盐或甲磺酸根。
本文实施方案的化合物可以以未溶剂化形式和溶剂化形式两者存在。术语溶剂合物在本文中用于描述包括本文实施方案的化合物与一定量的一种或更多种药学上可接受的溶剂分子的分子复合物。当所述溶剂是水时,采用术语水合物。溶剂合物形式的实例包含(但不限于)与水、丙酮、二氯甲烷、2-丙醇、乙醇、甲醇、二甲基亚砜(DMSO)、乙酸乙酯、乙酸、乙醇胺、或其混合物缔合的本文实施方案的化合物。特别预期的是,在本文实施方案中,一个溶剂分子可以与本文实施方案的化合物的一个分子缔合,如水合物。
在本文的一些实施方案中,一个溶剂分子可以与本文所述的化合物的一个分子缔合,如水合物。在一些实施方案中,超过一个溶剂分子可以与本文所述的化合物的一个分子缔合,如二水合物。此外,在本文的一些实施方案中,少于一个溶剂分子可以与本文所述的化合物的一分子缔合,如半水合物。此外,本文实施方案的溶剂合物被预期为保留化合物的非溶剂合物形式的生物有效性的本文所述的化合物的溶剂合物。
本文的实施方案还包含本文实施方案的经同位素标记的化合物,其中一个或更多个原子被具有相同原子序数但原子质量或质量数不同于自然界中通常发现的原子质量或质量数的原子代替。适合于包含在本文实施方案的化合物中的同位素的实例包含氢的同位素(如
通常可以使用适当的经同位素标记的试剂代替以其他方式采用的未经标记的试剂,通过本领域技术人员已知的常规技术或者通过与本文所描述的那些方法类似的方法来制备本文实施方案的经同位素标记的化合物。
优选的经同位素标记的化合物包含本文实施方案的化合物的氘化衍生物。如本文所使用的,术语氘化衍生物包含其中特定位置处的至少一个氢原子被氘代替的本文实施方案的化合物。氘(D或
氢氘交换(氘并入)是其中共价键合的氢原子被氘原子代替的化学反应。所述交换(并入)反应可以是全部的或部分的。
典型地,本文实施方案的化合物的氘化衍生物对于在化合物上被指定为潜在氘化位点的位点处存在的每个氘具有至少3500(52.5%氘并入)的同位素富集因子(同位素丰度与该同位素的天然丰度之间的比率,氘在分子中给定位置处代替氢的并入百分比)。
在一些实施方案中,同位素富集因子为至少5000(75%氘)。在一些实施方案中,同位素富集因子为至少6333.3(95%氘并入)。在一些实施方案中,同位素富集因子为至少6633.3(99.5%氘并入)。应当理解的是,在被指定为氘化位点的位点处存在的每个氘的同位素富集因子与其他氘化位点无关。
如本文所使用的且与“患者”可互换地使用的,术语“受试者”包含(但不限于)人类和非人类脊椎动物,如野生、家养和农场动物。在某些实施方案中,本文所描述的受试者是动物。在某些实施方案中,受试者是哺乳动物。在某些实施方案中,受试者是人类。在某些实施方案中,受试者是非人类动物。在某些实施方案中,受试者是非人类的哺乳动物。在某些实施方案中,受试者是驯养的动物,如狗、猫、牛、猪、马、绵羊或山羊。在某些实施方案中,受试者伴侣动物,如狗或猫。在某些实施方案中,受试者是家畜动物,如牛、猪、马、绵羊或山羊。在另外的实施方案中,受试者是研究动物,如啮齿动物、狗或非人类灵长类动物。在某些实施方案中,受试者是非人类转基因动物,如转基因小鼠或转基因猪。
短语“治疗有效的”旨在限定用于疾病或紊乱的治疗或者临床终点的影响的活性成分的量。
术语“治疗上可接受的”指适用于与患者的组织接触而没有过度毒性、刺激和过敏反应,与合理的利益/风险比相称,并且可有效用于其预期用途的那些化合物及其衍生物。
如本文所使用的,术语“治疗(treat)”、“治疗(treated)”、“治疗(treating)”或“治疗(treatment)”指治疗性治疗和预防疾病的措施或预防性措施两者,其中目的是预防或减缓(减轻)不期望的生理病况、紊乱或疾病,或者是获得有益的或期望的临床结果。为了本发明的目的,有益的或期望的临床结果包含(但不限于)症状的缓和;病况、紊乱或疾病的程度的减弱;病况、紊乱或疾病的状态的稳定(即,不恶化);病况、紊乱或疾病的发作的延迟或病况、紊乱或疾病的进展的减缓;病况、紊乱或疾病状态的减轻;和病况、紊乱或疾病的缓解(无论是部分或全部的,无论是诱导或维持),无论是可检测的还是不可检测的,或病况、紊乱或疾病的改进(enhancement)或改善。治疗包含引起临床上显著的响应,而无过度水平的副作用。与未接受治疗的预期存活相比,治疗还包含延长存活。治疗也可以在本质上是先发制人的,即,其可以包含疾病的预防。疾病的预防可以涉及完全免于疾病(例如,如在预防病原体感染的情况下),或者可以涉及疾病进展的预防。例如,疾病的预防可能不意味着在任何水平上与疾病有关的任何影响的完全消除,而是可能意味着将疾病的症状预防到临床上显著的或可检测的水平。疾病的预防也可以意味着预防疾病进展到疾病晚期,并且与未接受治疗的无疾病存活相比延长无疾病存活,并且与未接受治疗的无疾病存活相比延长无疾病存活。
本文实施方案针对抑制MK2活性的口服药物组合物以及涉及向有需要的受试者施用口服剂量的MK2抑制剂化合物的治疗方法。一些实施方案包含用于在有需要的受试者中治疗疾病的方法,方法包括口服施用本文所描述的MK2抑制剂化合物。
本文公开的口服组合物具有防止p38 MAP激酶介导的炎性信号传导的特异性MK2抑制剂,并且因此可以被用于其中p38 MAP激酶炎性信号传导发挥积极作用的疾病或病况的治疗或预防治疗中。因此,实施方案提供包括本文公开的MK2抑制剂和药学上可接受的载体的口服药物组合物以及用于使用化合物和组合物的方法。某些实施方案提供用于使用本文实施方案的化合物抑制p38 MAP激酶炎性信号传导的方法。其他实施方案提供用于在需要这样的治疗的患者中治疗p38 MAP激酶介导的紊乱的方法,方法包括向所述患者施用治疗有效量的根据本公开的MK2抑制剂化合物或包括MK2抑制剂化合物的组合物。还提供本文公开的特异性MK2抑制剂用于在制造用于通过p38 MAP激酶的抑制减轻的疾病或病况的治疗的药物的应用。
还提供其中本文所描述任意实施方案可与与这些实施方案中的任意一个或更多个组合的实施方案,条件是组合不是相互排斥的。
口服组合物
本文实施方案针对配制用于口服施用的药物组合物(“口服药物组合物”),药物组合物包括约5mg至约300mg的如下所示的化合物I
或其衍生物和药学上可接受的载体。
化合物I在本文中也通过化合物I的化学名称(即,3-氯-4-((3,5-二氟吡啶-2-基)甲氧基)-2'-(2-(2-羟基丙烷-2-基)嘧啶-4-基)-5',6-二甲基-2H-[1,4'-联吡啶]-2-酮)来指代化合物I。
可以根据美国专利号9,115,089(其通过引用被整体并入本文)中描述的方法制备化合物I。也可以从Aclaris Therapeutics,Inc(640Lee Road,Suite 200,Wayne,PA19087,USA)获得化合物I。
存在两种3-氯-4-((3,5-二氟吡啶-2-基)甲氧基)-2'-(2-(2-羟基丙烷-2-基)嘧啶-4-基)-5',6-二甲基-2H-[1,4'-联吡啶]-2-酮的阻转异构体,两种阻转异构体在下文被描绘为化合物(P)-I((P)-3-氯-4-((3,5-二氟吡啶-2-基)甲氧基)-2'-(2-(2-羟基丙烷-2-基)嘧啶-4-基)-5',6-二甲基-2H-[1,4'-联吡啶]-2-酮)和化合物(M)-I((M)-3-氯-4-((3,5-二氟吡啶-2-基)甲氧基)-2'-(2-(2-羟基丙烷-2-基)嘧啶-4-基)-5',6-二甲基-2H-[1,4'-联吡啶]-2-酮)。
术语“阻转异构现象”指由于取代基的空间应变导致的围绕单键的旋转受阻的异构类型。该现象产生显示轴向手性的立体异构体。可以经由使用二氧化碳和乙醇/甲醇的流动相的超临界流体色谱法来分离(解析)阻转异构体。在本文实施例中描述了3-氯-4-((3,5-二氟吡啶-2-基)甲氧基)-2'-(2-(2-羟基丙烷-2-基)嘧啶-4-基)-5',6-二甲基-2H-[1,4'-联吡啶]-2-酮的P和M阻转异构体的手性拆分。
如贯穿本公开所使用的,“化合物I”的叙述涵盖如上文描述的以从4:1((P)-I:(M)-I)至999:1的任意摩尔比的阻转异构体化合物(P)-I和(M)-I,并且还包含其中化合物(P)-I基本上不含化合物(M)-I的实施方案。化合物(P)-I和(M)-I可以是如本文所描述的任何形式(例如,游离碱、结晶形式等)。
在任意实施方案中,如本文公开的口服组合物的化合物I包括以约4:1((P)-I:(M)-I)至约999:1的摩尔比的化合物(P)-I和化合物(M)-I。在任意实施方案中,(P)-I与(M)-I的摩尔比为约4.3:1、约4.6:1、约4.9:1、约5.25:1、约5.7:1、约6.1:1、约6.7:1、约7.3:1、约8.1:1、约9:1、约10:1、约11.5:1、约13.3:1、约15.7:1、约19:1、约24:1、约32.3:1、约49:1、约91:1、约110.1:1、约124:1、约141.9:1、约165.7:1、约199:1、约249:1、约332.3:1、约399:1、约499:1和约999:1。在优选的实施方案中,(P)-I与(M)-I的摩尔比为约399:1。
换句话说,在任意的实施方案中,如本文公开的口服组合物的化合物I包括至少80mol%的化合物(P)-I。在任意的实施方案中,如本文公开的口服组合物包括至少81mol%的化合物(P)-I、至少82mol%的化合物(P)-I、至少83mol%的化合物(P)-I、至少84mol%的化合物(P)-I、至少85mol%的化合物(P)-I、至少86mol%的化合物(P)-I、至少87mol%的化合物(P)-I、至少88mol%的化合物(P)-I、至少89mol%的化合物(P)-I、至少90mol%的化合物(P)-I、至少91mol%的化合物(P)-I、至少92mol%的化合物(P)-I、至少93mol%的化合物(P)-I、至少94mol%的化合物(P)-I、至少95mol%的化合物(P)-I、至少96mol%的化合物(P)-I、至少97mol%的化合物(P)-I、至少98mol%的化合物(P)-I、至少99mol%的化合物(P)-I、至少99.1mol%的化合物(P)-I、至少99.2mol%的化合物(P)-I、至少99.3mol%的化合物(P)-I、至少99.4mol%的化合物(P)-I、至少99.5mol%的化合物(P)-I、至少99.6mol%的化合物(P)-I、至少99.7mol%的化合物(P)-I、至少99.8mol%的化合物(P)-I、至少99.9mol%的化合物(P)-I。在优选的实施方案中,如本文公开的口服组合物的化合物I包括基本上不含化合物(M)-I的化合物(P)-I。
在任意实施方案中,本文公开的口服药物组合物包括10mg的化合物I。在任意实施方案中,本文公开的口服药物组合物包括40mg的化合物I。在任意实施方案中,本文公开的口服药物组合物包括50mg的化合物I。在任意实施方案中,本文公开的口服药物组合物包括60mg的化合物I。在任意实施方案中,本文公开的口服药物组合物包括80mg的化合物I。在任意实施方案中,本文公开的口服药物组合物包括100mg的化合物I。在任意实施方案中,本文公开的口服药物组合物包括120mg的化合物I。在任意实施方案中,本文公开的口服药物组合物包括160mg的化合物I。在任意实施方案中,本文公开的口服药物组合物包括200mg的化合物I。在任意实施方案中,本文公开的口服药物组合物包括240mg的化合物I。
在任意实施方案中,本文所描述的口服药物组合物包括以约5mg至约200mg的量的化合物I。在任意实施方案中,化合物I以约5mg至约300mg、约7.5mg至约300mg、约10mg至约300mg、约12.5mg至约300mg、约15mg至约300mg、约17.5mg至约300mg、约20mg至约300mg、约22.5mg至约300mg、约25mg至约300mg、约27.5mg至约300mg、约30mg至约300mg、约32.5mg至约300mg、约35mg至约300mg、约37.5mg至约300mg、约40mg至约300mg、约42.5mg至约300mg、约45mg至约300mg、约47.5mg至约300mg、约50mg至约300mg、约50mg至约290mg、约50mg至约280mg、约50mg至约270mg、约50mg至约260mg、约50mg至约250mg、约50mg至约240mg、约50mg至约230mg、约50mg至约220mg、约50mg至约210mg、约50mg至约200mg约50mg至约190mg、约50mg至约180mg、约50mg至约170mg、约50mg至约160mg、约50mg至约150mg、约50mg至约140mg、约50mg至约130mg、约50mg至约120mg、约50mg至约110mg、约50mg至约100mg、约50mg至约90mg、约50mg至约80mg、约50mg至约70mg、约50mg至约60mg、约40mg至约50mg、约30mg至约60mg、约20mg至约70mg、约15mg至约80mg、约10mg至约90mg、约5mg至约100mg的量、或者之间的任意量存在于如本文所描述的药物组合物中。在优选的实施方案中,本文所描述的口服药物组合包括以约50mg至约240mg的量的化合物I。
在任意实施方案中,化合物I以约5mg、7.5mg、10mg、12.5mg、15mg、17.5mg、20mg、22.5mg、25mg、27.5mg、30mg、32.5mg、35mg、37.5mg、40mg、42.5mg、45mg、47.5mg、50mg、52.5mg、55mg、57.5mg、60mg、62.5mg、65mg、67.5mg、70mg、72.5mg、75mg、77.5mg、80mg、82.5mg、85mg、87.5mg、90mg、92.5mg、95mg、97.5mg、100mg、105mg、107.5mg、110mg、112.5mg、115mg、117.5mg、120mg、122.5mg、125mg、127.5mg、130mg、132.5mg、135mg、137.5mg、140mg、142.5mg、145mg、147.5mg、150mg、152.5mg、155mg、157.5mg、160mg、162.5mg、165mg、167.5mg、170mg、172.5mg、175mg、177.5mg、180mg、182.5mg、185mg、187.5mg、190mg、192.5mg、195mg、197.5mg、200mg、200mg、205mg、207.5mg、210mg、212.5mg、215mg、217.5mg、220mg、222.5mg、225mg、227.5mg、230mg、232.5mg、235mg、237.5mg、240mg、242.5mg、245mg、247.5mg、250mg、252.5mg、255mg、257.5mg、260mg、262.5mg、265mg、267.5mg、270mg、272.5mg、275mg、277.5mg、280mg、282.5mg、285mg、287.5mg、290mg、292.5mg、295mg、297.5mg、或者300mg的量存在于如本文所描述的药物组合物中。在优选的实施方案中,化合物I以50mg、80mg、100mg、120mg、160mg或240mg的量存在于本文所描述的药物组合物中。
在任意实施方案中,如本文公开的口服组合物的化合物I包括游离碱。在任意实施方案中,如本文公开的口服组合物的化合物I包括药学上可接受的盐。
在任意实施方案中,口服组合物的化合物I包括以游离碱形式的如本文公开的化合物(P)-I和化合物(M)-I。在任意实施方案中,口服组合物的化合物I包括以药学上可接受的盐形式的如本文公开的化合物(P)-I和化合物(M)-I。
在任意实施方案中,药学上可接受的盐是酸加成盐。适合的酸加成盐包含由有机酸和无机酸形成的酸加成盐。药学上可接受的酸包含无机酸(例如,盐酸、氢溴酸、氢碘酸、硝酸、碳酸、硫酸、磷酸和焦磷酸)和有机酸(例如,甲酸、乙酸、三氟乙酸、丙酸、琥珀酸、乙醇酸、恩波酸(扑酸)、甲磺酸、乙磺酸、2-羟基乙磺酸、泛酸、苯磺酸、甲苯磺酸、磺胺酸、甲磺酸(mesylic acid)、环己基氨基磺酸、硬脂酸、藻酸(algenic acid)、β-羟丁酸、丙二酸、半乳酸(galactic acid)、半乳糖醛酸、柠檬酸、富马酸、葡萄糖酸、谷氨酸、乳酸(lactic acid)、马来酸、苹果酸、扁桃酸、粘酸、抗坏血酸、草酸、泛酸、琥珀酸、酒石酸、苯甲酸、乙酸、昔萘酸(xinafoic acid)(1-羟基-2-萘甲酸)、萘二磺酸(napadisilic acid)(1,5-萘二磺酸(1,5-naphthalenedisulfonic acid))等)两者。
在任意实施方案中,药学上可接受的盐是碱加成盐。通过使羧基基团与适合的碱(如金属阳离子的氢氧化物、碳酸盐、或碳酸氢盐)或者与氨或有机伯胺、仲胺、或叔胺反应,可以在化合物最终分离和纯化期间制备碱加成盐。治疗上可接受的盐的阳离子包含锂、钠、钾、钙、镁、和铝,以及无毒季胺阳离子(如铵、四甲铵、四乙铵、甲胺、二甲胺、三甲胺、三乙胺、二乙胺、乙胺、三丁胺、吡啶、N,N-二甲基苯胺、N-甲基哌啶、N-甲基吗啉、二环己胺、普鲁卡因、二苄胺、N,N-二苄基苯乙胺、1-二苯羟甲胺和N,N’-二苄基乙二胺)。对于碱加成盐的形成有用的其他代表性有机胺包含乙二胺、乙醇胺、二乙醇胺、哌啶和哌嗪。
在任意实施方案中,口服组合物的化合物I是非溶剂形式或溶剂化形式。在本文的任意实施方案中,一个溶剂分子可以与本文所描述的化合物I的一个分子缔合,如水合物。在一些实施方案中,超过一个溶剂分子可以与如本文所描述的化合物I的一个分子缔合,如二水合物。此外,在本文的一些实施方案中,少于一个溶剂分子可以与本文所描述的化合物I的一个分子缔合,如半水合物。此外,本文实施方案的溶剂合物预期为保留化合物I的非溶剂化物形式的生物学有效性的如本文所描述的化合物I的溶剂合物。
在任意实施方案中,口服组合物的化合物I是氘化衍生物。如本文所使用的,术语氘化衍生物包含其中特定位置处的至少一个氢原子被氘代替的本文实施方案的化合物。氘(D或
在任意实施方案中,如本文公开的口服组合物的化合物I包括以结晶形式的化合物(P)-I(游离碱)。在任意实施方案中,化合物(P)-I的结晶形式是如本文公开的和表征的结晶形式A。
例如,口服组合物的化合物(P)-I的结晶形式A可以通过其PXRD图谱表征。因此,在任意实施方案中,化合物(P)-I的结晶形式A的特征在于具有以在约9.78±0.2的角度2θ表示的峰的PXRD图谱。在任意实施方案中,化合物(P)-I的结晶形式A的特征在于具有以在9.78±0.2和15.51±0.2的角度2θ表示的峰的PXRD图谱。在任意实施方案中,化合物(P)-I的结晶形式A的特征在于具有以在9.78±0.2、15.51±0.2、19.6±0.2和25.92±0.2的角度2θ表示的峰的PXRD图谱。在任意实施方案中,化合物(P)-I的结晶形式A的特征在于具有以在9.78±0.2、15.34±0.2、15.51±0.2、19.6±0.2、20.57±0.2、21.01±0.2、25.92±0.2、29.05±0.2和29.48±0.2的角度2θ表示的峰的PXRD图谱。在任意实施方案中,化合物(P)-I的结晶形式A的特征在于图27的PXRD图谱。
本文公开的口服组合物的化合物(P)-I的结晶形式A可以另外地或可替代地通过热重量分析(TGA)表征。(P)-I的结晶形式A的样品产生TGA曲线,TGA曲线揭示在分析的样品中,观察到可忽略不计的重量损失。对于游离碱结晶形式A,通过TGA在25℃和256℃之间观察到重量损失(0.7%),表明(P)-I的结晶形式A是基本上无水的。
(P)-I的结晶形式A可以另外地或可替代地通过差示扫描量热法(DSC)表征。因此,在任意实施方案中,化合物(P)-I的结晶形式A的特征在于DSC曲线,DSC曲线包括具有约188℃的起始温度的初始吸热熔融事件,随后在约196℃的放热再结晶事件以及在约254℃的最终急剧吸热熔融事件。
在任意实施方案中,本公开的口服组合物包括通过与药学上可接受的载体或赋形剂混合配制的如本文公开的化合物I。在某些实施方案中,药物组合物包含治疗有效量的化合物I和生理学上可接受的稀释剂或载体。在某些实施方案中,药物组合物还包含一种或更多种附加的治疗组分或佐剂。
在任意实施方案中,本文公开的口服组合物还可以包括药学上可接受的稀释剂、填充剂、崩解剂、粘结剂、润滑剂、表面活性剂、疏水性溶媒、水溶性溶媒、乳化剂、缓冲剂、湿润剂、保湿剂、增溶剂、防腐剂等。用于制备或施用的手段和方法是本领域已知的,并且技术人员可以参考各种药学参考文献作为指导。例如,可以查阅Banker,G.S.,&Rhodes,C.T.(2002).Modern pharmaceutics.New York:Marcel Dekker.;和Goodman,L.S.,Brunton,L.L.,Chabner,B.,&Knollmann,B.C.(2011).Goodman&Gilman's pharmacological basisof therapeutics.New York:McGraw-Hill。
可以通过将化合物I与本领域公知的药学上可接受的载体组合来容易地配制如本文公开的口服药物组合物。这样的载体使得本文实施方案的化合物能够被配制为纳米颗粒、纳米颗粒混悬剂、片剂、糖锭剂、丸剂、糖衣丸、胶囊剂、粉剂、液体剂、凝胶剂、糖浆、浆液、混悬剂等用于待治疗的受试者口服摄入。还可以使用用作漱口剂的液体载体制备口服组合物,其中液体载体中的化合物被口服施加以及漱口(swish)和吐出或吞咽。可以通过添加固体赋形剂、可选地研磨所得到的混合物以及加工颗粒混合物,在添加适合的助剂(如果期望的话)以获得片剂或糖衣丸芯来获得用于口服施用的药物制剂。适合的赋形剂包含(但不限于)填充剂(如糖,包含(但不限于)乳糖、蔗糖、甘露糖醇和山梨糖醇)、纤维素制剂(如(但不限于)玉米淀粉、小麦淀粉、稻淀粉、马铃薯淀粉、明胶、黄芪胶、甲基纤维素、羟丙基甲基纤维素(HPMC)、羧甲基纤维素钠(CMC)和聚乙烯吡咯烷酮(PVP))。如果期望的话,可以添加崩解剂(如(但不限于)交联PVP、琼脂或海藻酸或其盐(如海藻酸钠))。
糖衣丸芯可以被提供有适合的包衣。为此目的,可以使用浓缩的糖溶液,浓缩的糖溶液可以可选地含有阿拉伯树胶、滑石、PVP、卡巴普凝胶、聚乙二醇(PEG)、和/或二氧化钛、漆溶液、和适合的有机溶剂或溶剂混合物。可以将着色剂或色素添加到片剂或糖衣丸包衣中,以鉴别或表征活性化合物剂量的不同组合。
可以口服使用的药物制剂包含(但不限于)由明胶制成的推入配合式胶囊剂(push-fit capsule)、以及由明胶和增塑剂(如甘油或山梨糖醇)制成的密封式软胶囊剂。推入配合式胶囊剂可以含有与一种或更多种填充剂(例如,乳糖)、一种或更多种粘结剂(例如,淀粉)、和/或一种或更多种润滑剂(例如,滑石或硬脂酸镁)、以及可选地一种或更多种稳定剂混合的活性成分。在软胶囊剂中,活性化合物可以溶解于或悬浮于适合的液体(如脂肪油、液体石蜡、或液体PEG)中。此外,可以添加稳定剂。用于口服施用的所有组合物应当是以适用于这样的施用的剂量(例如,约5mg至约300mg)。
在一些实施方案中,口服组合物可以采用通过常规方式用下列制备的例如锭剂、片剂或胶囊剂的形式:药学上可接受的赋形剂(如粘合剂(例如,预胶化的玉米淀粉、PVP或HPMC));填充剂(例如,乳糖、微晶纤维素或磷酸氢钙);润滑剂(例如,硬脂酸镁、滑石或二氧化硅);崩解剂(例如,马铃薯淀粉或羧甲淀粉钠);或者润湿剂(例如,十二烷基硫酸钠)。可以通过本领域公知的方法,用例如糖包衣、膜包衣或肠溶包衣包覆片剂。此外,如本文公开的含有化合物I的药物组合物可以是以适用于口服使用的任意形式,包含例如糖锭剂、锭剂、水性或油性混悬剂、分散性粉剂或颗粒、乳剂、硬或软胶囊剂、或糖浆或酏剂。可以根据本领域已知的用于制造药物组合物的任意方法制备旨在用于口服使用的组合物,并且这样的组合物可以含有一种或更多种选自由下列组成的组的剂:甜味剂、矫味剂、着色剂和防腐剂,以便提供药学上精致且适口的制剂。
在一实施方案中,如本文公开的口服药物组合物是片剂。片剂可以含有与适用于片剂的制造的无毒的药学上可接受的赋形剂混合的化合物I。这些赋形剂可以是例如惰性稀释剂(如碳酸钙、碳酸钠、乳糖、磷酸钙或磷酸钠);成粒剂和崩解剂(例如,玉米淀粉或海藻酸);粘结剂(例如,淀粉、明胶或阿拉伯树胶);和润滑剂(例如硬脂酸镁、硬脂酸或滑石)。片剂可以是未包覆的或者其可以通过已知的技术被包覆,以延缓胃肠道中的分解和吸收,并且由此提供在较长的时间段内的持续的作用。在一些实施方案中,片剂被配制用于立即释放(immediate release)。在一些实施方案中,片剂被配制用于控制释放。例如,可以采用时间延迟材料(time delay material)(如单硬脂酸甘油酯或二硬脂酸甘油酯)。也可以通过美国专利号4,256,108;4,166,452;和4,265,874中描述的技术对其进行包覆,以形成用于控制释放的渗透治疗片剂。本文所描述的药物组合物还可以是水包油乳剂的形式。
在一些实施方案中,口服组合物包括化合物I和缓冲剂。在一些实施方案中,缓冲剂可以选自由柠檬酸单水合物、磷酸钠、水及其组合组成的组。在一些实施方案中,口服组合物包括化合物I和稳定剂。在一些实施方案中,稳定剂选自由聚维酮、苯甲酸钠、水、十二烷基硫酸钠及其组合组成的组。在一些实施方案中,口服组合物还包含缓冲剂、酸、苯甲酸钠、磷酸钠、柠檬酸或其组合物。在一些实施方案中,口服组合物包括化合物I以及稳定剂和缓冲剂。在一些实施方案中,口服组合物还包括润滑剂、pH调节剂、粘结剂、稀释剂、成粒剂、助流剂、崩解剂、填充剂、吸附剂、抗粘剂、着色剂、压缩辅助物(compression aid)、包衣材料、增甜剂、防腐剂、抗氧化剂或其组合。在一些实施方案中,化合物I是以治疗有效量的(例如,约5mg至约200mg)。在一些实施方案中,口服组合物是混悬剂、片剂、胶囊剂、纳米颗粒粉剂、纳米颗粒混悬剂、扁囊剂、小丸剂、丸剂、粉剂、颗粒或其组合。
在一些实施方案中,润滑剂可以选自由硬脂酸或其盐(例如,硬脂酸镁、硬脂酸钙)、十二烷基硫酸钠、PEG、矿物油、苯甲酸钠、硬脂酸棕榈酸甘油酯、山嵛酸甘油酯、硬脂富马酸钠及其组合组成的组。
在一些实施方案中,pH调节剂可以是酸(例如,盐酸、乙酸、柠檬酸、磷酸、硫酸或其组合)。
在一些实施方案中,粘结剂可以选自由天然或合成聚合物(例如,淀粉、糖、糖醇或纤维素衍生物)(如明胶、葡萄糖、乳糖、山梨糖醇、木糖醇、麦芽糖醇、甲基纤维素、微晶纤维素(MCC)、乙基纤维素、HPMC、羟丙基纤维素(HPC)、淀粉、PVP、PEG、海藻酸钠、CMC及其组合物)组成的组。
在一些实施方案中,压缩辅助物(compression aid)可以选自由硅化微晶纤维素、微晶纤维素、MCC-胶态二氧化硅的物理混合物及其组合物组成的组。
在一些实施方案中,崩解剂可以选自由淀粉、纤维素衍生物和海藻酸盐、PVP、交联羧甲基纤维素钠、羧甲淀粉钠及其组合组成的组。
在一些实施方案中,填充剂可以选自由乳糖、蔗糖、葡萄糖、甘露糖醇、山梨糖醇、碳酸钙、硬脂酸镁、植物纤维素、二碱式磷酸钙、二碱式磷酸钠、植物脂肪和植物油及其组合组成的组。
在一些实施方案中,稀释剂可以选自由糖化合物(例如,蔗糖、乳糖、糊精、葡萄糖、山梨糖醇等)、无机化合物(例如,硅酸盐、钙盐或镁盐)、氯化钠、氯化钾及其组合组成的组。
在一些实施方案中,防腐剂可以选自由抗氧化剂(例如,维生素A、维生素E、维生素C、棕榈酸视黄酯和硒)、氨基酸(例如,半胱氨酸或甲硫氨酸)、柠檬酸、柠檬酸钠、合成防腐剂(例如,尼泊金酯(如尼泊金甲酯或尼泊金丙酯))及其组合组成的组。
在一些实施方案中,助流剂可以选自由胶态无水二氧化硅和其他二氧化硅化合物(如气相二氧化硅)、碳酸镁、胶态二氧化硅
在一些实施方案中,包括化合物I的口服组合物是胶囊剂。在一些实施方案中,胶囊剂包括高脂乳剂制作的内包衣。在一些实施方案中,胶囊剂包括在胶囊剂的内侧上或外侧上的高脂包衣。在一些实施方案中,胶囊剂包括HPMC。在一些实施方案中,胶囊剂可以是HPMC胶囊剂。在一些实施方案中,胶囊剂可以是有肠溶包衣的。在一些实施方案中,胶囊剂可以是二氧化硅胶囊剂,如以商品名
在一些实施方案中,包括化合物I的口服剂型是片剂。在一些实施方案中,片剂含有以纳米颗粒形式的化合物I。在一些实施方案中,片剂可以被包覆。在一些实施方案中,片剂可以被包覆有肠溶包衣。在一些实施方案中,片剂可以被包覆有选自下列的包衣:糖包衣、膜包衣、有机膜包衣、水性膜包衣、锅包衣、浸蘸包衣、静电包衣、压制包衣、增塑剂干包衣、热干包衣、静电干包衣等。用于包衣的一些成分可以包含水性丙烯酸肠溶系统(如以商品名
在一些实施方案中,口服组合物包括以纳米颗粒混悬剂(纳米混悬剂)形式的化合物I。在一些实施方案中,纳米混悬剂包括化合物I、稳定剂和缓冲剂。在一些实施方案中,纳米混悬剂还可以包括pH调节剂。在一些实施方案中,pH调节剂可以选自由盐酸、乙酸、柠檬酸、磷酸、硫酸及其组合组成的组。在一些实施方案中,pH调节剂可以是盐酸。在一些实施方案中,盐酸可以是1.0N盐酸。在一些实施方案中,稳定剂可以选自由聚维酮、十二烷基硫酸钠、苯甲酸钠、WFI品质水(如以商品名HYCLONE
可以通过将活性颗粒悬于赋形剂中、使用研磨介质在磨机中将颗粒减小至期望的颗粒尺寸、并且然后将混悬液稀释至最终体积来制造纳米颗粒混悬剂。在一些实施方案中,使用的研磨介质可选自陶瓷、玛瑙、氮化硅、烧结刚玉、氧化锆、不锈钢、铬钢、Cr-Ni钢、碳化钨、玻璃(钇稳定的)、交联聚苯乙烯树脂、塑料聚酰胺、珍珠或其组合。在一些实施方案中,磨机可以是固定的搅拌容器或循环磨机。
可以通过将纳米悬浮液(上文)喷洒到蔗糖上形成喷雾造粒中间体,将喷雾造粒中间体与赋形剂进行造粒以形成最终制粒,并且压缩最终制粒以形成片剂来制造片剂。蔗糖可以是任何糖(包含,例如葡萄糖、果糖、麦芽糖、半乳糖、乳糖等)。在一些实施方案中,用于片剂制剂的赋形剂可以包含乳糖单水合物、PVP、硅化微晶纤维素(例如,以商品名
在一些实施方案中,如本文所描述的口服组合物是干粉剂。在一些实施方案中,干粉剂可以被封装或制作成混悬剂。在一些实施方案中,混悬剂可以是纳米颗粒混悬剂或经研磨的混悬剂。
在一些实施方案中,用于口服施用的液体制剂可以采用例如酏剂、溶液剂、糖浆或混悬剂的形式,或者其可以被呈现为干制品,用于在使用前与水或其他适合的溶媒构成。可以通过常规手段用药学上可接受的添加剂(如悬浮剂(例如,山梨糖醇糖浆、纤维素衍生物或氢化可食用脂肪);乳化剂(例如,卵磷脂或阿拉伯树胶);非水性溶媒(例如,杏仁油、油性酯、乙醇、
化合物的药物组合物还可以包括适合的固相或凝胶相载体或赋形剂。这样的载体或赋形剂的实例包含(但不限于)碳酸钙、磷酸钙、各种糖类、淀粉、纤维素衍生物、明胶和聚合物(例如,PEG)。
组合物可以进一步包含一种或更多种附加的药物药剂(如抗炎药物、抗动脉硬化药物、免疫抑制药物、免疫调控药物、细胞稳定药物(cytostatic drug)、血管生成抑制剂、激酶抑制剂、细胞因子阻断剂和细胞粘附分子的抑制剂)。
使用方法
本公开的另外的方面涉及用于有需要的受试者中炎性病况的治疗的方法。该方法包括向患有炎性病况的人类受试者施用5mg/天至300mg/天的口服剂量的化合物I(即,以如上文所描述的任意摩尔比和以如上文所描述的任意形式(例如,游离形式、结晶形式)的化合物(P)-I和(M)-I)。用于在本文所描述的方法中使用的化合物I的特定有用的口服剂量包括100mg/天至240mg/天的口服剂量的包括大于80mol%的化合物(P)-I的化合物I。
可以根据本文公开的方法治疗的适合的炎性病况包含涉及p38 MAP激酶介导的炎性信号传导的任意炎性病况。在一些实施方案中,炎性病况是慢性炎性病况。在一些实施方案中,炎性病况是急性炎性病况。在一些实施方案中,炎性病况是自身炎性病况。在一些实施方案中,炎性病况是自身免疫病况。在一些实施方案中,炎性病况是炎症小体病。
例如,在任意实施方案中,本文公开的方法和组合物适用于治疗慢性或急性炎性或自身免疫性胃肠道紊乱、炎性或自身免疫性皮肤紊乱、神经炎性紊乱、炎性心脏病、炎性肺病、炎性肌病、炎性骨紊乱或疾病、周期性发热综合征以及与任意上述疾病相关的疼痛或瘙痒。
在任意实施方案中,化合物I还被用于治疗瘢痕形成/纤维化疾病或紊乱以及各种类型的癌症和过度增殖性紊乱。
例如,在任意实施方案中,本文公开的方法和组合物适用于治疗炎性关节炎(如类风湿性关节炎(RA)、脊柱关节炎(如强直性脊柱炎)、银屑病关节炎、反应性关节炎和赖特综合征、幼年型类风湿性关节炎(JIA)、全身起病型幼年型类风湿性关节炎、特发性关节炎(JIA)(包含全身型(SJIA))和痛风);冷凝比林(cryopyrin)相关的自身炎性综合征(CAPS)(包含Muckle-Wells综合征(MWS))、新生儿起病多系统炎性疾病(NOMID)和家族性寒冷性自身炎性综合征(FCAS));慢性阻塞性肺病(COPD)(包含肺气肿、慢性支气管炎和哮喘(过敏性和非过敏性))、炎性皮肤病况(包含(但不限于)化脓性汗腺炎(HS),银屑病,如斑块状银屑病,坏疽性脓皮病,IL-17相关性皮肤病况,瘙痒);炎性肠病(IBD)引起的结肠炎(如克罗恩病或溃疡性结肠炎),和炎性肠病相关关节炎;心包炎,包含急性心包炎、复发性心包炎和慢性心包炎;肺部炎症或纤维化,包含特发性肺纤维化和间质性肺病;转移性乳腺癌和胰腺癌。
在任意实施方案中,本文公开的方法和组合物适用于治疗家族性地中海热(FMF);肿瘤坏死因子受体相关周期性综合征(TRAPS);成年起病型斯蒂尔病;坏疽性脓皮病;骨吸收紊乱(如与癌症(例如,乳腺癌)相关的紊乱);转移性黑色素瘤;卡斯尔门病;和慢性非典型中性粒细胞性皮肤病伴脂肪代谢障碍(CANDLE)。
在一些实施方案中,根据本文描述的方法治疗的病况是瘙痒,其可能与任意其他病症相关,例如与化脓性汗腺炎相关的瘙痒、与炎症相关的瘙痒、与类风湿性关节炎相关的瘙痒、与银屑病相关的瘙痒以及与TH17相关炎症相关的瘙痒。
在任意实施方案中,本文公开的方法和组合物适用于治疗莱姆病;细胞因子释放综合征(CRS);急性呼吸窘迫综合征(ARDS);慢性或急性支气管炎;大疱性表皮松解症(EB);大疱性类天疱疮;幼年型皮肌炎;炎性白癜风(包含边缘性);寻常型天疱疮;小肠结肠炎;多发性肌炎;肌炎,骨癌;肺癌;炎性骨紊乱(如慢性复发性多发性骨髓炎(CRMO)、滑膜炎、痤疮、脓疱病、骨质增生和骨炎(SAPHO)综合征、马吉德综合征、白介素-1受体拮抗剂缺乏症(DIRA)和家族性巨颌症;骨吸收(如与自身免疫性疾病有关);神经炎性疾病(如阿尔茨海默病(AD)、帕金森病(PD)、多发性硬化(MS)、急性播散性脑脊髓炎(ADEM)、急性视神经炎(AON)、横贯性脊髓炎和视神经脊髓炎(NMO);白塞病;内毒素性休克(例如,中毒性休克综合征(TSS)和其他全身性革兰氏阴性细菌感染);起止点炎;结节性多动脉炎(PAN);慢性疼痛;风湿性多肌痛;慢性同种异体移植排斥;斯耶格伦综合征;和施尼茨勒(Schnitzler)综合征(SchS)。
在任意实施方案中,根据本文所描述的方法治疗的病况是关节炎,如炎性关节炎、类风湿性关节炎(RA)、脊柱关节炎(如强直性脊柱炎)、银屑病关节炎、反应性关节炎和赖特综合征、幼年型类风湿性关节炎(JIA)、全身起病型幼年型类风湿性关节炎、特发性关节炎(JIA)(包括全身型(SJIA))和痛风)和炎性肠病相关关节炎。
在任意实施方案中,待治疗的病况是类风湿性关节炎、强直性脊柱炎或银屑病关节炎。
在任意实施方案中,治疗的病况是炎性皮肤病况,如化脓性汗腺炎、银屑病(如斑块状银屑病)、坏疽性脓皮病、IL-17相关性炎性皮肤病况和IL-1α相关性炎性皮肤病况。
在任意实施方案中,待治疗的病况是冷凝比林相关的周期性综合征(CAPS),包含MWS、NOMIDS和FCAS。
在任意实施方案中,待治疗的病况是肠易激病,包含结肠炎、克罗恩病和溃疡性结肠炎。
在任意实施方案中,待治疗的病况是细胞因子释放综合征(例如CAR-T细胞诱导的细胞因子释放综合征)和急性呼吸窘迫综合征。
在任意实施方案中,待治疗的病况是癌症,包含(但不限于)转移性乳腺癌、胰腺癌、结肠直肠癌和肺癌。
在任意实施方案中,待治疗的病况是慢性阻塞性肺病(COPD),包含肺气肿、慢性支气管炎和哮喘(过敏性和非过敏性)。
在任意实施方案中,待治疗的病况是肺纤维化,包含特发性肺纤维化和间质性肺病。
可以根据本文公开的方法和组合物治疗的其他病况包含炎性和/或自身免疫病况,如过敏性和非过敏性哮喘、胰腺炎、自身免疫性脑脊髓炎、自身免疫性肌炎、巨细胞动脉炎、巩膜表层炎、肾小球肾炎、桥本氏甲状腺炎、角膜炎、狼疮性肾炎、心肌炎、肠炎、中性粒细胞性汗腺炎、非酒精性脂肪性肝炎、牙周炎、多发性软骨炎、原发性硬化性胆管炎、巩膜炎(schleritis)、窦炎、小血管炎、大血管炎、高安氏动脉炎(Takayasu’s arteritis)、特异性皮炎、自身免疫性和炎症性肝炎、自身免疫性萎缩性胃炎、自身免疫性睾丸炎、细支气管炎、闭塞性细支气管炎、心脏炎、声带炎(chortitis)、食道炎、丙型肝炎、视神经炎,葡萄膜炎、头癣(tinea capitis)、寻常痤疮以及下垂体炎、狼疮、重症肌无力、恶性贫血、I型糖尿病、阿狄森氏病、恰加斯病、周围神经病变、缺氧或局部缺血引起的炎症、自身免疫性肾病、瘢痕性类灭疱疮、古德帕斯彻病(Goodpasture’s Disease)、葛瑞夫兹氏病(Graves’disease)、组织细胞中性皮肤病(histiocytoid neutrophilic dermatosis)、嗜酸细胞增多综合征、干扰素基因刺激蛋白相关的婴儿期起病型血管病变(SAVI)、查格-施特劳斯综合征、吉兰-巴雷综合症、免疫介导的肾小球肾炎、线性IgA疾病、结节病、交感性眼炎和3型超敏反应病。
可以根据本文公开的方法和组合物治疗的其他病况包含其中MK2抑制在治疗上有益的任何病况,如癌症(例如,头/颈癌、膀胱癌、肠癌、非小细胞肺癌、星形细胞瘤、小细胞肺癌、结肠癌、结肠直肠癌、食管癌、纤维化癌症、肝癌、白血病、肾癌、喉癌、多发性骨髓瘤、梅克尔细胞癌、口腔或喉癌、神经癌、非黑色素细胞皮肤癌、卵巢癌、前列腺癌、肾脏癌、精原细胞瘤、鳞状细胞癌、胃癌、施万细胞瘤、畸胎癌、睾丸癌、骨肉瘤、横纹肌肉瘤、韦格纳氏肉芽肿病、角化棘皮瘤、卡波西氏肉瘤、成胶质细胞瘤、神经胶质瘤、T细胞淋巴瘤、咽喉癌、甲状腺癌、甲状腺滤泡状癌和子宫癌)、纤维化病况(例如,心房纤维化、心脏纤维化、囊状纤维化、心内膜心肌纤维化症、肝纤维化、原发性骨髓纤维化(IMF)、纵膈纤维化、骨髓纤维化、肾源性系统纤维化、肾脏纤维化、腹膜后纤维化、纤维腺瘤、纤维肌痛、纤维肉瘤、纤维硬化、平滑肌瘤(fibroids)、(fibroma)纤维性脱发(fibrosing alopecia)、肾小球硬化症、幼年型硬皮病、膜性肾小球病、硬皮症(schleroderma)和心脏移植物血管病变)和胃肠道纤维化)、视网膜病变、肝硬化和角膜病变。
如上文所描述的,适用于根据本文所描述的方法施用的口服组合物包括治疗有效量(例如,约50mg/天至约300mg/天)的化合物I(即,以4:1至999:1的化合物(P)-I:化合物(M)-I的摩尔比)或者包括治疗有效量(例如,约50mg/天至约300mg/天)的化合物I(即,以4:1至999:1的化合物(P)-I:化合物(M)-I的摩尔比)的口服药物组合物。
任何特定患者的具体剂量水平将取决于多种因素,所述因素包含所采用的具体化合物的活性、年龄、体重、总体健康状况、性别、饮食、施用时间、排泄率、药物组合、正在治疗的确切紊乱、以及正在治疗的适应症或病况的严重程度。
在一些实施方案中,化合物I(即,以4:1至999:1的化合物(P)-I:化合物(M)-I的摩尔比)或者包括化合物I(即,以4:1至999:1的化合物(P)-I:化合物(M)-I的摩尔比)的口服药物组合物的施用对于使成为疾病特征的症状至少部分缓解是有效的。在一些实施方案中,本文实施方案的本文公开的口服组合物中的一种或更多种的施用对于使成为疾病特征的症状至少完全缓解是有效的。
在任意实施方案中,包括如本文所描述的化合物I的组合物的施用对于引起p38MAP激酶-介导的促炎性信号传导(而非抗炎性信号传导)的抑制是有效的。在一些实施方案中,本文公开的本文实施方案的口服组合物中的一种或更多种的施用对于引起MK2炎性信号传导的抑制是有效的。p38 MAP激酶和MK2介导的炎性信号传导的抑制可以通过一种或更多种炎性细胞因子(包含(但不限于)TNF-α、IL-1β、IL-6、IL-8、IFNγ、IL-17、IL-18、IL-1α和MIP1β)的体内血清水平被测量或评估。因此,在任意实施方案中,相比于在用包括化合物I的组合物治疗受试者之前对应细胞因子的体内血清水平,以本文所描述的剂量(例如,100mg/天、160mg/天或240mg/天)施用包括化合物I的组合物(特别是包括至少80mol%、至少90mol%、至少95mol%或至少99mol%的化合物(P)-I的组合物)降低受试者中选自TNF-α、IL-1β、IL-6、IL-8、IFNγ、IL-17、IL-18、IL-1α和MIP1β的一种或更多种细胞因子的体内血清水平。
在一实施方案中,根据本文所描述的方法和包括化合物I的组合物的待治疗的炎性病况是关节炎,特别是中度至重度类风湿性关节炎。在一些实施方案中,根据如本文所描述的方法和包括化合物I的组合物的患有中度至重度类风湿性关节炎的受试者的治疗(单独或者与甲氨蝶呤组合)对于抑制关节损伤的发展、改善滑膜炎或者两者是有效的(如通过磁共振成像(MRI)评估的)。
在一些实施方案中,根据如本文所描述的方法和包括化合物I的组合物的患有中度至重度类风湿性关节炎的受试者的治疗(单独或者与甲氨蝶呤组合)对于降低受试者中高敏性C反应蛋白(hsCRP)水平是有效的。在一些实施方案中,hsCRP相对于基线水平(即,治疗前的hsCRP水平)降低至少10%、至少20%、至少30%、至少40%或大于40%。优选地,用化合物I的治疗使hsCRP水平相对于基线降低大于40%。
在一些实施方案中,根据如本文所描述的方法和包括化合物I的组合物的患有中度至重度类风湿性关节炎的受试者的治疗(单独或者与甲氨蝶呤组合)对于降低受试者的28个关节的疾病活动度评分(DAS28)是有效的。DAS28由下列变量的综合评分组成:关节压痛数、肿胀关节数、CRP和患者的疾病活动度评分的整体评估。DAS28(CRP)疾病活动度测量的解释是以0至9.4为标准,其中:<2.6被认为是缓解,≥2.6至<3.2被认为是低度/最低,≥3.2至≤5.1被认为是中度,并且>5.1被认为是高度/重度(Anderson et al.,“Rheumatoidarthritis disease activity measures:American College of Rheumatologyrecommendations for use in clinical practice,”Arthritis Care Res(Hoboken)64(5):640-7(2012),其通过引用被整体并入本文)。因此,在一些实施方案中,根据如本文所描述的方法和包括化合物I的组合物的中度至重度类风湿性关节炎的治疗对于在治疗过程中将DAS28(CRP)评分降低至≤3.2(作为缓解的低疾病活动度的指标)是有效的。在一些实施方案中,根据如本文所描述的方法和包括化合物I的组合物的重度类风湿性关节炎的治疗对于在治疗过程中将DAS28(CRP)评分降低至<2.6(作为缓解的指标)是有效的。
在一些实施方案中,根据如本文所描述的方法和包括化合物I的组合物的患有中度至重度类风湿性关节炎的受试者的治疗(单独或者与甲氨蝶呤组合)对于实现肿胀关节数和关节压痛数的至少20%的改善和下列5个测量值中的至少3个的至少20%的改善是有效的:(i)患者的疾病活动度的整体评估(VAS),(ii)患者的关节炎疼痛的评估(VAS),(iii)患者的身体功能/健康评估问卷-功能障碍指数(HAQ-DI)的评估,(iv)医师的疾病活动度的整体评估(VAS),(v)如通过hsCRP测量的急性期反应物。在一些实施方案中,使用如本文所描述的包括化合物I的组合物的治疗(单独或者与甲氨蝶呤组合)对于实现肿胀关节数和关节压痛数的至少50%的改善和上述5个测量值中的至少3个的至少50%的改善是有效的。在一些实施方案中,使用如本文所描述的包括化合物I的组合物的治疗(单独或者与甲氨蝶呤组合)对于实现肿胀关节数和关节压痛数的至少70%的改善和上述5个测量值中的至少3个的至少70%的改善是有效的。
在一些实施方案中,根据如本文所描述的方法和包括化合物I的组合物的患有中度至重度类风湿性关节炎的受试者的治疗(单独或者与甲氨蝶呤组合)对于显著降低一种或更多种炎性细胞因子的给药前水平是有效的。在一些实施方案中,使用包括化合物I的组合物的患有重度类风湿性关节炎的受试者的治疗(单独或者与甲氨蝶呤组合)对于显著降低TNF-α、IL-6、IL-8、IL-1β和MIP1β中的一种或更多种的给药前水平是有效的。在任意实施方案中,在施用如本文实施例中公开的包括化合物I的组合物后,上述细胞因子水平降低至少10%、至少15%、至少20%、至少30%、至少40%、至少50%、至少60%、至少70%或大于75%。
在一些实施方案中,根据如本文所描述的方法和包括化合物I的组合物的患有中度至重度活动性银屑病关节炎的受试者的治疗(单独或者与甲氨蝶呤组合)对于实现相比于关节压痛数(TJC)、肿胀关节数(SJC)的基线的至少20%的降低(改善)和5个剩余ACR核心集测量值中的至少3个的至少20%的改善是有效的:(i)患者的疼痛评估,(ii)患者的疾病活动度的整体评估(PtGA);(iii)医师的疾病活动度的整体评估(PhGA),(iv)健康评估问卷-功能障碍指数(HAQ-DI),和(iv)高敏性C反应蛋白(hsCRP)。
在一些实施方案中,根据如本文所描述的方法和包括化合物I的组合物的患有中度至重度活动性银屑病关节炎的受试者的治疗(单独或者与甲氨蝶呤组合)对于实现0或1的银屑病静态研究者整体评估(sIGA)和自基线(治疗前)sIGA水平至少2点的改善是有效的。
在一些实施方案中,根据如本文所描述的方法和包括化合物I的组合物的患有中度至重度活动性银屑病关节炎的受试者的治疗(单独或者与甲氨蝶呤组合)对于实现银屑病面积严重程度指数(PASI)75响应(对于具有至少3%的BSA银屑病作为基线的参与者)是有效的。
在一些实施方案中,根据如本文所描述的方法和包括化合物I的组合物的患有中度至重度活动性银屑病关节炎的受试者的治疗(单独或者与甲氨蝶呤组合)对于实现最低疾病活动度(MDA)(基于满足7项结果测量值中的5项确定:TJC≤1;SJC≤1;PASI≤1或BSA-Ps≤3%;患者的疼痛评估NRS≤1.5;PtGA-疾病活动度NRS≤2.0;HAQ-DI评分≤0.5;和皮肤压痛点(tender entheseal point)≤1)是有效的。
在一些实施方案中,根据如本文所描述的方法和包括化合物I的组合物的患有中度至重度活动性银屑病关节炎的受试者的治疗(单独或者与甲氨蝶呤组合)对于显著降低TNF-α、IL-1β、IL-6、IL-8、IFNγ、IL 17、IL-18、IL-1α和MIP1β中的一种或更多种的给药前水平是有效的。在任意实施方案中,在施用包括化合物I的组合物后,上述细胞因子水平降低至少10%、至少15%、至少20%、至少30%、至少40%、至少50%、至少60%、至少70%或大于75%。
在一些实施方案中,根据如本文所描述的方法和包括化合物I的组合物的患有中度至重度化脓性汗腺炎(HC)的受试者的治疗(单独或者与抗生素组合)对于实现在治疗期间的化脓性汗腺炎临床响应(HiSCR)是有效的,其中HiSCR被定义为总脓肿和炎症结节(AN)数自基线至少50%的降低,而无脓肿或引流瘘管数增加。
在一些实施方案中,根据如本文所描述的方法和包括化合物I的组合物的患有中度至重度化脓性汗腺炎(HC)的受试者的治疗(单独或者与抗生素组合)对于实现在治疗期间患者的皮肤疼痛的整体评估(PGA皮肤疼痛)的数字分级标度(NRS30)的自基线(治疗前)水平至少30%的降低是有效的。
在一些实施方案中,根据如本文所描述的方法和包括化合物I的组合物的患有中度至重度化脓性汗腺炎(HC)的受试者的治疗(单独或者与抗生素组合)对于实现在治疗期内相对于基线具有至少2的增加的AN计数的至少25%的减少是有效的。
在一些实施方案中,根据如本文所描述的方法和包括化合物I的组合物的患有中度至重度化脓性汗腺炎(HC)的受试者的治疗(单独或者与抗生素组合)对于实现下列中至少一项或更多项是有效的:通过治疗,皮肤病生活质量指数(DLQI)自基线的增加(DLQI是由用于评估HS疾病症状和治疗对生活质量(QoL)的影响的10项组成的有效问卷);通过治疗,基于化脓性汗腺炎症状评估(HSSA)评估的HS相关的肿胀自基线的减少(HSSA是由开发以0至11分的NRS评估HS症状的9项组成的患者报告结果(PRO)问卷,其中0代表无症状,并且10代表极端症状经历);在12周内基于HSSA评估的HS相关的气味自基线的减少;基于通过治疗的HSSA评估的HS相关的最严重排泄自基线的变化。
在一些实施方案中,根据如本文所描述的方法和包括化合物I的组合物的患有中度至重度化脓性汗腺炎(HC)的受试者的治疗(单独或者与抗生素组合)对于实现选自TNF-α、IL-1β、IL-6、IL-8、IFNγ、IL 17、IL-18、MIP1β和IL-1α的一种或更多种内源性细胞因子自基线的减少是有效的。在任意实施方案中,在施用包括化合物I的组合物后,上述细胞因子水平降低至少10%、至少15%、至少20%、至少30%、至少40%、至少50%、至少60%、至少70%或大于75%。
在一些实施方案中,根据如本文所描述的方法和包括化合物I的组合物的患有冷凝比林相关的周期性综合征(CAPS)(例如,家族性寒冷性自身炎性综合征、Muckle-Wells综合征或新生儿起病多系统炎性疾病)的受试者的治疗对于引起由下列中的一项或两项定义的疾病缓解是有效的:(i)医师整体评估(PGA)评分为无或最低,以及(ii)在正常范围内(≤10mg/L)或在基线值的30%内的高敏性C反应蛋白(hsCRP)和血清淀粉样蛋白A(SAA)值。
在一些实施方案中,根据如本文所描述的方法和包括化合物I的组合物的患有冷凝比林相关的周期性综合征(CAPS)(例如,家族性寒冷性自身炎性综合征、Muckle-Wells综合征或新生儿起病多系统炎性疾病)的受试者的治疗对于引起由医师整体评估(PGA)评分为无或最低定义的临床缓解是有效的。
在一些实施方案中,根据如本文所描述的方法和包括化合物I的组合物的患有冷凝比林相关的周期性综合征(CAPS)(例如,家族性寒冷性自身炎性综合征、Muckle-Wells综合征或新生儿起病多系统炎性疾病)的受试者的治疗对于维持通过治疗的相比于基线高不超过2分的平均关键症状评分(KSS)是有效的。KSS来源于患者每日施用健康评估表(DHAF),并且是5个单独的标度(皮疹、发热和发冷的感觉、关节疼痛、眼睛发红和疼痛以及疲劳)的0至10标度(0=无,10=非常严重)的平均值。
在一些实施方案中,根据如本文所描述的方法和包括化合物I的组合物的患有冷凝比林相关的周期性综合征(CAPS)(例如,家族性寒冷性自身炎性综合征、Muckle-Wells综合征或新生儿起病多系统炎性疾病)的受试者的治疗对于显著减低TNF-α、IL-6、IL-8、IL-1β和MIP1的一种或更多种的给药前水平是有效的。在任意实施方案中,在施用包括化合物I的组合物后,上述细胞因子水平降低至少10%、至少15%、至少20%、至少30%、至少40%、至少50%、至少60%、至少70%或大于75%。
在任意实施方案中,本文所描述的化合物I的口服组合物可以以第一剂量施用以防止疾病的进展、以第二剂量施用以诱导疾病的缓解、和/或以第三剂量施用以预防疾病和/或维持疾病的缓解。这样的剂量可以是相同的剂量、较低的剂量或较高的剂量。可以更频繁、更不频繁或以相同频率施用剂量。在一些实施方案中,可以与另外的疗法、治疗剂、佐药等组合施用剂量。
如上文所描述的,适用于根据本文所描述的方法的治疗的受试者包含人类和非人类脊椎动物,如野生动物、家养动物和农场动物。在一些实施方案中,本文所描述的受试者是动物。在一些实施方案中,受试者是哺乳动物。在一些实施方案中,受试者是人类。在一些实施方案中,受试者是非人类动物。在一些实施方案中,受试者是非人类哺乳动物。
剂量方案
化合物I(即以如上文所描述的任意摩尔比和以如上文所描述的任意形式(例如,游离形式、结晶形式)的化合物(P)-I和化合物(M)-I)或包括化合物I的口服药物组合物以有效抑制有需要的受试者中p38 MAP激酶-介导的炎性信号传导的量被施用。需要这样的疗法的受试者在上文被公开。在一些实施方案中,受试者是患有炎性病况(例如,慢性炎性病况、急性炎性病况、免疫炎性病况、自身免疫病况或炎症小体病)的受试者。根据本文公开的方法,治疗有效的量包括5mg/天至300mg/天的化合物I。待施用至特定受试者的剂量将取决于正在被治疗的受试者的特征(例如被治疗的特定受试者、年龄、体重、健康状况、同期治疗(如果有的话)的类型以及治疗的频率),并且可以由本领域的技术人员(例如,由临床医生)容易地确定。
在任意实施方案中,向患有本文公开的病况的受试者施用10mg/天的化合物I。在任意实施方案中,向患有本文公开的病况的受试者施用40mg/天的化合物I。在任意实施方案中,向患有本文公开的病况的受试者施用50mg/天的化合物I。在任意实施方案中,向患有本文公开的病况的受试者施用60mg/天的化合物I。在任意实施方案中,向患有本文公开的病况的受试者施用80mg/天的化合物I。在任意实施方案中,向患有本文公开的病况的受试者施用100mg/天的化合物I。在任意实施方案中,向患有本文公开的病况的受试者施用120mg/天的化合物I。在任意实施方案中,向患有本文公开的病况的受试者施用160mg/天的化合物I。在任意实施方案中,向患有本文公开的病况的受试者施用200mg/天的化合物I。在任意实施方案中,向患有本文公开的病况的受试者施用240mg/天的化合物I。
在任意实施方案中,化合物I的治疗有效的量为5mg/天至300mg/天、约7.5mg/天至约mg/天、约10mg/天至约300mg/天、约12.5mg/天至约300mg/天、约15mg/天至约300mg/天、约17.5mg/天至约300mg/天、约20mg/天至约300mg/天、约22.5mg/天至约300mg/天、约25mg/天至约300mg/天、约27.5mg/天至约300mg/天、约30mg/天至约300mg/天、约32.5mg/天至约300mg/天、约35mg/天至约300mg/天、约37.5mg/天至约300mg/天、约40mg/天至约300mg/天、约42.5mg/天至约300mg/天、约45mg/天至约300mg/天、约47.5mg/天至约300mg/天、约50mg/天至约300mg/天、约50mg/天至约290mg/天、约50mg/天至约280mg/天、约50mg/天至约270mg/天、约50mg/天至约260mg/天、约50mg/天至约250mg/天、约50mg/天至约240mg/天、约50mg/天至约230mg/天、约50mg/天至约220mg/天、约50mg/天至约210mg/天、约50mg/天至约200mg/天、约50mg/天至约190mg/天、约50mg/天至约180mg/天、约50mg/天至约170mg/天、约50mg/天至约160mg/天、约50mg/天至约150mg/天、约50mg/天至约140mg/天、约50mg/天至约130mg/天、约50mg/天至约120mg/天、约50mg/天至约110mg/天、约50mg/天至约100mg/天、约50mg/天至约90mg/天、约50mg/天至约80mg/天、约50mg/天至约70mg/天、约50mg/天至约60mg/天、约40mg/天至约50mg/天、约30mg/天至约60mg/天、约20mg/天至约70mg/天、约15mg/天至约80mg/天、约10mg/天至约90mg/天、约5mg/天至约100mg/天或之间的任意量。在优选的实施方案中,化合物I的治疗有效的量为约100mg/天至约240mg/天。
在一些实施方案中,化合物I的治疗有效的量包括5mg/天、7.5mg/天、10mg/天、12.5mg/天、15mg/天17.5mg/天、20mg/天、22.5mg/天、25mg/天、27.5mg/天、30mg/天、32.5mg/天、35mg/天、37.5mg/天、40mg/天、42.5mg/天、45mg/天、47.5mg/天、50mg/天、52.5mg/天、55mg/天、57.5mg/天、60mg/天、62.5mg/天、65mg/天、67.5mg/天、70mg/天、72.5mg/天、75mg/天、77.5mg/天、80mg/天、82.5mg/天、85mg/天、87.5mg/天、90mg/天、92.5mg/天、95mg/天、97.5mg/天、100mg/天、105mg/天、107.5mg/天、110mg/天、112.5mg/天、115mg/天、117.5mg/天、120mg/天、122.5mg/天、125mg/天、127.5mg/天、130mg/天、132.5mg/天、135mg/天、137.5mg/天、140mg/天、142.5mg/天、145mg/天、147.5mg/天、150mg/天、152.5mg/天、155mg/天157.5mg/天、160mg/天、162.5mg/天、165mg/天、167.5mg/天、170mg/天、172.5mg/天、175mg/天、177.5mg/天、180mg/天、182.5mg/天、185mg/天、187.5mg/天、190mg/天、192.5mg/天、195mg/天、197.5mg/天、200mg/天、205mg/天、207.5mg/天、210mg/天、212.5mg/天、215mg/天、217.5mg/天、220mg/天、222.5mg/天、225mg/天、227.5mg/天、230mg/天、232.5mg/天、235mg/天、237.5mg/天、240mg/天、242.5mg/天、245mg/天、247.5mg/天、250mg/天、252.5mg/天、255mg/天257.5mg/天、260mg/天、262.5mg/天、265mg/天、267.5mg/天、270mg/天、272.5mg/天、275mg/天、277.5mg/天、280mg/天、282.5mg/天、285mg/天、287.5mg/天、290mg/天、292.5mg/天、295mg/天、297.5mg/天、300mg/天。在优选的实施方案中,化合物I的治疗有效的量包括100mg/天、160mg/天或240mg/天。
在任意实施方案中,根据本文公开的方法向受试者施用的化合物I的口服剂量包括以约4:1((P)-I:(M)-I)的摩尔比的化合物(P)-I和化合物(M)-I。在任意实施方案中,(P)-I和(M)-I的摩尔比为约4.3:1、约4.6:1、约4.9:1、约5.25:1、约5.7:1、约6.1:1、约6.7:1、约7.3:1、约8.1:1、约9:1、约10:1、约11.5:1、约13.3:1、约15.7:1、约19:1、约24:1、约32.3:1、约49:1、约91:1、约110.1:1、约124:1、约141.9:1、约165.7:1、约199:1、约249:1、约332.3:1、约399:1、约499:1和约999:1。化合物I的特别有用的口服剂量包括(P)-I和(M)-I的摩尔比为约399:1。
在任意实施方案中,根据本文公开的方法向受试者施用的化合物I的口服剂量包括至少80mol%的化合物(P)-I。在任意实施方案中,如本文公开的口服组合物包括至少81mol%的化合物(P)-I、至少82mol%的化合物(P)-I、至少83mol%的化合物(P)-I、至少84mol%的化合物(P)-I、至少85mol%的化合物(P)-I、至少86mol%的化合物(P)-I、至少87mol%的化合物(P)-I、至少88mol%的化合物(P)-I、至少89mol%的化合物(P)-I、至少90mol%的化合物(P)-I、至少91mol%的化合物(P)-I、至少92mol%的化合物(P)-I、至少93mol%的化合物(P)-I、至少94mol%的化合物(P)-I、至少95mol%的化合物(P)-I、至少96mol%的化合物(P)-I、至少97mol%的化合物(P)-I、至少98mol%的化合物(P)-I、至少99mol%的化合物(P)-I、至少99.1mol%的化合物(P)-I、至少99.2mol%的化合物(P)-I、至少99.3mol%的化合物(P)-I、至少99.4mol%的化合物(P)-I、至少99.5mol%的化合物(P)-I、至少99.6mol%的化合物(P)-I、至少99.7mol%的化合物(P)-I、至少99.8mol%的化合物(P)-I、至少99.9mol%的化合物(P)-I。化合物I的特别有用的口服剂量包括至少99.75mol%的化合物(P)-I。
在任意实施方案中,根据本文公开的方法向受试者施用的化合物I的口服剂量包括基本上不含化合物(M)-I的化合物(P)-I。
在任意实施方案中,根据本文公开的方法向受试者施用的化合物I的口服剂量包括游离碱。在任意实施方案中,根据本文公开的方法向受试者施用的化合物I的口服剂量包括药学上可接受的盐。
在任意实施方案中,根据本文公开的方法向受试者施用的化合物I的口服剂量包括以游离碱形式的如本文公开的化合物(P)-I和化合物(M)-I。在任意实施方案中,口服组合物的化合物I包括以药学上可接受的盐的形式的如本文公开的化合物(P)-I和化合物(M)-I。
在任意实施方案中,根据本文公开的方法向受试者施用的化合物I的口服剂量包括以结晶形式的化合物(P)-I(游离碱)。在任意实施方案中,化合物(P)-I的结晶形式是如本文公开的和表征的结晶形式A。
例如,口服组合物的化合物(P)-I的结晶形式A可以通过其PXRD图谱表征。因此,在任意实施方案中,化合物(P)-I的结晶形式A的特征在于具有以在约9.78±0.2的角度2θ表示的峰的PXRD图谱。在任意实施方案中,化合物(P)-I的结晶形式A的特征在于具有以在9.78±0.2和15.51±0.2的角度2θ表示的峰的PXRD图谱。在任意实施方案中,化合物(P)-I的结晶形式A的特征在于具有以在9.78±0.2、15.51±0.2、19.6±0.2和25.92±0.2的角度2θ表示的峰的PXRD图谱。在任意实施方案中,化合物(P)-I的结晶形式A的特征在于具有以在9.78±0.2、15.34±0.2、15.51±0.2、19.6±0.2、20.57±0.2、21.01±0.2、25.92±0.2、29.05±0.2和29.48±0.2的角度2θ表示的峰的PXRD图谱。在任意实施方案中,化合物(P)-I的结晶形式A的特征在于图27的PXRD图谱。
(P)-I的结晶形式A可以另外地或可替代地通过差示扫描量热法(DSC)表征。因此,在任意实施方案中,化合物(P)-I的结晶形式A的特征在于DSC曲线,DSC曲线包括具有约188℃的起始温度的初始吸热熔融事件,随后在约196℃的放热再结晶事件以及在约254℃的最终急剧吸热熔融事件。
在一些实施方案中,根据如本文所描述的方法和包括化合物I的组合物的患有炎性病况的受试者的治疗对于实现化合物I的39±10.4ng/mL的最高血药浓度(C
在另外的实施方案中,向患有炎性病况的受试者施用包括化合物I的组合物对于实现122.0±33.4的C
在另外的实施方案中,向患有炎性病况的受试者施用包括化合物I的组合物对于实现160.7±20.4的C
在另外的实施方案中,向患有炎性病况的受试者施用包括化合物I的组合物对于实现426.0±110.61的C
在另外的实施方案中,向患有炎性病况的受试者施用包括化合物I的组合物对于实现51.8±15.8的C
在另外的实施方案中,向患有炎性病况的受试者施用包括化合物I的组合物对于实现146.5±33.6的C
优选地,向患有炎性病况的受试者施用包括化合物I的组合物对于实现219.0±77.8的C
优选地,向患有炎性病况的受试者施用包括化合物I的组合物对于实现389±101的C
优选地,向患有炎性病况的受试者施用包括化合物I的组合物对于实现417±123的C
在任意实施方案中,患有炎性病况的受试者被施用5mg/天至300mg/天的化合物I。根据该实施方案,受试者患有选自下列的炎性病况:类风湿性关节炎、化脓性汗腺炎、痛风、斑块状银屑病、银屑病关节炎、强直性脊柱炎、心包炎(包含急性心包炎、复发性心包炎和慢性心包炎)、冷凝比林相关的周期性综合征(CAPS)(包含Muckle Wells综合征和家族性寒冷性自身炎性综合征)、坏疽性脓皮病、肠易激病(包含克罗恩病和溃疡性结肠炎)、斯蒂尔病(也称作幼年特发性关节炎)、特异性皮炎、急性冠状动脉综合征、心力衰竭和癌症(包含(但不限于)乳腺癌、胰腺癌、结肠直肠癌和肺癌)。
在任意实施方案中,患有炎性关节炎(如类风湿性关节炎(RA))、脊柱关节炎(如强直性脊柱炎和银屑病关节炎)、幼年型类风湿性关节炎(JIA)或特发性关节炎(JIA)(包含全身型(SJIA))和痛风);冷凝比林相关的自身炎性综合征(CAPS)(包含Muckle-Wells综合征(MWS)、新生儿起病多系统炎性疾病(NOMID)和家族性寒冷性自身炎性综合征(FCAS));慢性阻塞性肺病(COPD)、化脓性汗腺炎(HS);银屑病(如斑块状银屑病);炎性肠病(IBD)引起的结肠炎(如克罗恩病或溃疡性结肠炎);心包炎;转移性乳腺癌和胰腺癌的受试者被每天一次施用包括100mg的化合物I的口服组合物。
在任意实施方案中,患有炎性关节炎(如类风湿性关节炎(RA))、脊柱关节炎(如强直性脊柱炎和银屑病关节炎)、幼年型类风湿性关节炎(JIA)或特发性关节炎(JIA)(包含全身型(SJIA))和痛风);冷凝比林相关的自身炎性综合征(CAPS)(包含Muckle-Wells综合征(MWS)、新生儿起病多系统炎性疾病(NOMID)和家族性寒冷性自身炎性综合征(FCAS));慢性阻塞性肺病(COPD)、化脓性汗腺炎(HS);银屑病(如斑块状银屑病);炎性肠病(IBD)引起的结肠炎(如克罗恩病或溃疡性结肠炎);心包炎;转移性乳腺癌和胰腺癌的受试者被每天一次施用包括160mg的化合物I的口服组合物。
在任意实施方案中,患有炎性关节炎(如类风湿性关节炎(RA))、脊柱关节炎(如强直性脊柱炎和银屑病关节炎)、幼年型类风湿性关节炎(JIA)或特发性关节炎(JIA)(包含全身型(SJIA))和痛风);冷凝比林相关的自身炎性综合征(CAPS)(包含Muckle-Wells综合征(MWS)、新生儿起病多系统炎性疾病(NOMID)和家族性寒冷性自身炎性综合征(FCAS));慢性阻塞性肺病(COPD)、化脓性汗腺炎(HS);银屑病(如斑块状银屑病);炎性肠病(IBD)引起的结肠炎(如克罗恩病或溃疡性结肠炎);心包炎;转移性乳腺癌和胰腺癌的受试者被每天一次施用包括200mg的化合物I的口服组合物。
在任意实施方案中,患有炎性关节炎(如类风湿性关节炎(RA))、脊柱关节炎(如强直性脊柱炎和银屑病关节炎)、幼年型类风湿性关节炎(JIA)或特发性关节炎(JIA)(包含全身型(SJIA))和痛风);冷凝比林相关的自身炎性综合征(CAPS)(包含Muckle-Wells综合征(MWS)、新生儿起病多系统炎性疾病(NOMID)和家族性寒冷性自身炎性综合征(FCAS));慢性阻塞性肺病(COPD)、化脓性汗腺炎(HS);银屑病(如斑块状银屑病);炎性肠病(IBD)引起的结肠炎(如克罗恩病或溃疡性结肠炎);心包炎;转移性乳腺癌和胰腺癌的受试者被每天一次施用包括240mg的化合物I的口服组合物。
在任意实施方案中,患有炎性关节炎(如类风湿性关节炎(RA))、脊柱关节炎(如强直性脊柱炎和银屑病关节炎)、幼年型类风湿性关节炎(JIA)或特发性关节炎(JIA)(包含全身型(SJIA))和痛风);冷凝比林相关的自身炎性综合征(CAPS)(包含Muckle-Wells综合征(MWS)、新生儿起病多系统炎性疾病(NOMID)和家族性寒冷性自身炎性综合征(FCAS));慢性阻塞性肺病(COPD)、化脓性汗腺炎(HS);银屑病(如斑块状银屑病);炎性肠病(IBD)引起的结肠炎(如克罗恩病或溃疡性结肠炎);心包炎;转移性乳腺癌和胰腺癌的受试者被每天两次施用包括50mg的化合物I的口服组合物。
在任意实施方案中,患有炎性关节炎(如类风湿性关节炎(RA))、脊柱关节炎(如强直性脊柱炎和银屑病关节炎)、幼年型类风湿性关节炎(JIA)或特发性关节炎(JIA)(包含全身型(SJIA))和痛风);冷凝比林相关的自身炎性综合征(CAPS)(包含Muckle-Wells综合征(MWS)、新生儿起病多系统炎性疾病(NOMID)和家族性寒冷性自身炎性综合征(FCAS));慢性阻塞性肺病(COPD)、化脓性汗腺炎(HS);银屑病(如斑块状银屑病);炎性肠病(IBD)引起的结肠炎(如克罗恩病或溃疡性结肠炎);心包炎;转移性乳腺癌和胰腺癌的受试者被每天两次施用包括80mg的化合物I的口服组合物。
在任意实施方案中,患有炎性关节炎(如类风湿性关节炎(RA))、脊柱关节炎(如强直性脊柱炎和银屑病关节炎)、幼年型类风湿性关节炎(JIA)或特发性关节炎(JIA)(包含全身型(SJIA))和痛风);冷凝比林相关的自身炎性综合征(CAPS)(包含Muckle-Wells综合征(MWS)、新生儿起病多系统炎性疾病(NOMID)和家族性寒冷性自身炎性综合征(FCAS));慢性阻塞性肺病(COPD)、化脓性汗腺炎(HS);银屑病(如斑块状银屑病);炎性肠病(IBD)引起的结肠炎(如克罗恩病或溃疡性结肠炎);心包炎;转移性乳腺癌和胰腺癌的受试者被每天两次施用包括100mg的化合物I的口服组合物。
在任意实施方案中,患有炎性关节炎(如类风湿性关节炎(RA))、脊柱关节炎(如强直性脊柱炎和银屑病关节炎)、幼年型类风湿性关节炎(JIA)或特发性关节炎(JIA)(包含全身型(SJIA))和痛风);冷凝比林相关的自身炎性综合征(CAPS)(包含Muckle-Wells综合征(MWS)、新生儿起病多系统炎性疾病(NOMID)和家族性寒冷性自身炎性综合征(FCAS));慢性阻塞性肺病(COPD)、化脓性汗腺炎(HS);银屑病(如斑块状银屑病);炎性肠病(IBD)引起的结肠炎(如克罗恩病或溃疡性结肠炎);心包炎;转移性乳腺癌和胰腺癌的受试者被每天两次施用包括120mg的化合物I的口服组合物。
在任意实施方案中,患有类风湿性关节炎的受试者被口服施用50mg/天至300mg/天的化合物I。在一些实施方案中,患有类风湿性关节炎的受试者被每天一次施用包括化合物I的口服组合物。在一些实施方案中,患有类风湿性关节炎的受试者被每天两次包括化合物I的组合物。在一些实施方案中,患有类风湿性关节炎的受试者被每天一次施用包括10mg、30mg、50mg、80mg、100mg、120mg、160mg、200mg或240mg的化合物I的口服组合物。
在任意实施方案中,患有类风湿性关节炎的受试者被每天一次施用包括100mg的化合物I的口服组合物。
在任意实施方案中,患有类风湿性关节炎的受试者被每天一次施用包括160mg的化合物I的口服组合物。
在任意实施方案中,患有类风湿性关节炎的受试者被每天一次施用包括200mg的化合物I的口服组合物。
在任意实施方案中,患有类风湿性关节炎的受试者被每天一次施用包括240mg的化合物I的口服组合物。
在任意实施方案中,患有类风湿性关节炎的受试者被每天两次施用包括10mg、30mg、50mg、80mg、100mg或120mg的化合物I的口服组合物。
在任意实施方案中,患有类风湿性关节炎的受试者被每天两次施用包括50mg的化合物I的口服组合物。
在任意实施方案中,患有类风湿性关节炎的受试者被每天两次施用包括80mg的化合物I的口服组合物。
在任意实施方案中,患有类风湿性关节炎的受试者被每天两次施用包括100mg的化合物I的口服组合物。
在任意实施方案中,患有类风湿性关节炎的受试者被每天两次施用包括120mg的化合物I的口服组合物。
施用的剂量是足以导致症状或多项症状的减轻的治疗有效量的组合物,并且可以根据已知的因素(如活性成分的药效动力学特征及其施用模式;受者的年龄、性别、健康和体重;症状的性质和程度;同期治疗的种类、治疗的频率和期望的效果)变化。
在一些实施方案中,可以向受试者施用一次(例如,以单次剂量或单次施加)如本文所描述的包括化合物I的口服组合物。在一些实施方案中,至少每天一次(如,至少每天两次、三次或四次)施用本文实施方案的口服组合物。在一些实施方案中,可以根据需要或另外地根据医师的指导每天、每天两次、每天三次、每周、每周两次、每两周、每三周、每月施用本文实施方案的口服组合物。本文实施方案的口服组合物可以以任意间隔被施用以实现治疗上期望的效果,例如诱导或维持症状或多项症状的缓解、预防或减轻。在一些实施方案中,可以向受试者施用本文实施方案的口服组合物为期1天、2天、3天、4天、5天、6天、约一周、约两周、约三周、约四周、约五周、约六周、约两个月、约三个月、约四个月、约五个月、约六个月或这些数值中的任何两个的范围内。在一些实施方案中,治疗可以持续至少一周、一个月、一年或另外地根据医师的指导进行。在一些实施方案中,治疗可以延续多年、疾病的持续时间或者受试者的终生。在一些实施方案中,可以每天一次或两次向有需要的受试者施用本文实施方案的口服组合物为期约二至约二十八天、或从约七至约十天。也可以每天一次、两次或三次向受试者施用本文实施方案的口服组合物为期每年1次、2次、3次、4次、5次、6次、7次、8次、9次、10次、11次、12次或其组合。
在一些实施方案中,在餐前、餐后或用餐时施用如本文所描述的包括化合物I的口服组合物。在一些实施方案中,在高脂餐前、高脂餐后或用高脂餐时施用本文所描述的口服组合物。在一些实施方案中,在标准化高脂餐前、标准化高脂餐后或用标准化高脂餐时施用本文所描述的口服组合物。在一些实施方案中,高脂餐是高热量、高脂肪餐食。在一些实施方案中,高脂餐遵循FDA关于高脂肪餐食的指南。在一些实施方案中,高热量、高脂肪餐遵循FDA关于高脂肪和高热量餐食的指南。在一些实施方案中,高脂餐包括约为餐食的总热量的50%或更高的脂肪含量。参见美国卫生和公共服务部食品和药品管理局药物评价和研究中心(2002),Guidance for Industry:Food-Effect Bioavailability and FedBioequivalence Studies.Office of Training and Communications Division of DrugInformation,HFD-240。在一些实施方案中,高热量、高脂肪餐食包括餐食的总热量的至少50%的脂肪含量和约800至约1000千卡含量的总热量。
在一些实施方案中,本文所描述的口服组合物在过夜禁食后被施用。在一些实施方案中,过夜禁食至少约6小时、至少约7小时、至少约8小时、至少约9小时或至少约10小时。例如,本文所描述的口服组合物可以在过夜禁食至少10小时后在高脂肪餐食、高热量餐食后被施用。
组合疗法
化合物I(即以如上文所描述的任意摩尔比和以如上文所描述的任意形式(例如,游离形式、结晶形式)的化合物(P)-I和化合物(M)-I)或包括化合物I的口服药物组合物可以单独地或者与其他药物活性化合物组合地用于治疗病况(如上文所描述的那些病况)。本发明的一种或多种化合物和其他一种或多种药物活性化合物可以同时施用(以相同的剂型或者以单独的剂型)或者顺序施用。因此,在一实施方案中,本发明包括通过向受试者施用治疗有效量的本文所描述的口服药物组合物和一种或更多种附加的药物活性化合物来治疗病况的方法。
在某些情况下,将本文所描述的组合物与另外的药物药剂组合施用可以是适当的。仅作为示例,如果患者在接受本文组合物时经历的副作用之一是高血压,则将抗高血压药剂与初始药物药剂组合施用可以是适当的。或者,仅作为示例,可以通过佐剂的施用来增强本文所描述的组合物的治疗效果(例如,佐剂本身仅可以具有最小的治疗益处,但在与另外的药物药剂组合时,增强了对患者的整体治疗益处)。或者,仅作为示例,可以通过将本文所描述的组合物与也具有治疗益处的另外的药物药剂(其也包含治疗方案)一起施用来增加患者所经历的益处。仅作为示例,在涉及本文所描述的组合物的施用的针对类风湿性关节炎的治疗中,也可以通过向患者提供针对风湿性关节炎的另外的药物药剂来产生增加的治疗益处。在任何情况下,无论正在治疗的疾病、紊乱或病况,患者所经历的整体益处可以仅是两种药物组合物的累加,或者患者可以经历协同益处。
组合方法、组合物和制剂不限于仅使用两种药剂,还预期多种治疗组合的使用。应当理解的是,用于治疗、预防或减轻寻求缓解的一种或多种病况的剂量方案根据多种因素可选地被改变。这些因素包含受试者患有的紊乱、以及受试者的年龄、体重、性别、饮食和身体状况。因此,在一些实施方案中,实际采用的剂量方案在很大范围内变化,并且因此与本文阐述的剂量方案不同。
在任何情况下,可以以任何顺序或者甚至同时地附随施用多种治疗组合物(其中至少一种为本文公开的组合物)。如果同时施用,则可以以单一统一的形式或者以多种形式(仅作为示例,作为单个丸剂或作为两个单独的丸剂)提供多种治疗组合物。可以以多剂量给予治疗组合物之一,或者可以以多剂量给予两种药物组合物。如果不同时施用,则多剂量之间的时间可以是从几分钟至八周的任何持续时间,或者在适合于维持期望的治疗功效的任何间隔。在一些实施方案中,多剂量之间的时间可以是一分钟、一小时、六小时、一天、两天、三天、四天、五天、六天、一周、两周、三周、四周、五周、六周、七周或八周。
因此,在另一方面,某些实施方案提供用于在需要这样的治疗的人受试者或动物受试者中治疗炎性病况的方法,方法包括向所述受试者施用5mg/天至300mg/天的量的如本文公开的包括化合物I的口服组合物与本领域已知的用于治疗所述紊乱的至少一种附加药剂的组合,以降低或预防受试者中的所述病况。在相关方面,某些实施方案提供本文公开的治疗组合物与用于治疗炎性病况的一种或更多种附加的药物活性化合物、以及药学上可接受的载体的组合。
本文所描述的包括化合物I的口服组合物还可选地与其他治疗试剂组合使用,根据待治疗的病况的治疗价值来选择其他治疗试剂。通常,在采用组合疗法的实施方案中,本文所描述的组合物和其他药剂不必在同一药物组合物中施用,并且由于不同的物理特性和化学特性而可选地通过不同途径施用。通常根据已确立的方案进行初始施用,并且然后基于观察到的效果来随后改变剂量、施用模式和施用时间。
在一些实施方案中,一种或更多种附加的药物活性化合物选自由抗炎药物、抗动脉硬化药物、免疫抑制药物、免疫调控药物、细胞稳定药物(cytostatic drug)、抗增殖剂、血管生成抑制剂、激酶抑制剂、细胞因子阻断剂和细胞粘附分子的抑制剂组成的组。
在一些实施方案中,包括化合物I的口服组合物与已知用于治疗炎性病况的一种或更多种剂或组合物以任意组合一起施用给患有炎性病况或处于患炎性病况的风险的受试者。
用于上文列出的任意炎性病况的可能的组合疗法的具体非限制性实例包含如本文所描述的包括化合物I的口服组合物与下列物质的组合:(1)皮质类固醇,包含但不限于可的松、地塞米松、和甲泼尼龙;(2)非甾体类抗炎药(NSAID),包含但不限于布洛芬、萘普生、对乙酰氨基酚、阿司匹林、非诺洛芬(NALFON
当受试者患有类风湿性关节炎或与类风湿性关节炎相关的病况或处于患类风湿性关节炎或与类风湿性关节炎相关的病况的风险的情况下,如本文所描述的包括化合物I的口服组合物可选地与适用于治疗类风湿性关节炎或与类风湿性关节炎相关的病况的一种或多种剂以任意组合一起施用。可以与化合物I组合施用的用于炎性病况(如类风湿性关节炎或与类风湿性关节炎相关的病况)的治疗剂/治疗的实例包含(但不限于)非甾体抗炎性药物(NSAID)、类固醇(例如,泼尼松)、皮质类固醇和疾病修饰药物(disease modifyingdrugs,DMARD)(如甲氨蝶呤、来氟米特、羟氯喹、柳氮磺吡啶)、詹纳斯激酶(JAK)抑制剂(例如,托法替尼、乌帕替尼(upadacitinib)、巴瑞替尼(baricitinib)、非戈替尼(filgotinib)、鲁索利替尼(ruxolitinib)、奥拉替尼(oclacitinib)、培非替尼(peficitinib)、菲卓替尼(fedratinib)、赛度替尼(cerdulatinib)、更度替尼(gandotinib)、来他替尼(lesarturtinib)、莫洛替尼(momelotinib)、帕西替尼(pacritinib)、阿布昔替尼(abrocitinib)和BMS-986165)、肿瘤坏死因子抑制剂(例如,阿达木单抗(adalimumab)、依那西普(etanercept)、戈利木单抗(golimumab)、英利昔单抗(infliximab)、赛妥珠单抗(certolizumab))、抗B细胞抗体(例如,利妥昔单抗)、抗IL-6抗体(例如,沙利鲁单抗(sarilumab)、托珠单抗)、白介素-1受体(IL-1)拮抗剂(例如,阿那白滞素)和T细胞激活抑制剂(例如,阿巴西普(abatacept))。
在任意实施方案中,化合物I与甲氨蝶呤被组合施用用于类风湿性关节炎或如本文所描述的其他炎性病况的治疗。
在任意实施方案中,化合物I与JAK抑制剂被组合施用用于类风湿性关节炎或如本文所描述的其他炎性病况的治疗。与化合物I组合施用用于类风湿性关节炎或本文公开的其他炎性病况中的任意炎性病况的治疗的适合的JAK抑制剂包含(不限于)托法替尼、乌帕替尼、巴瑞替尼、非戈替尼、鲁索利替尼、奥拉替尼、培非替尼、菲卓替尼、赛度替尼、更度替尼、来他替尼、莫洛替尼、帕西替尼、阿布昔替尼和BMS-986165。
在任意实施方案中,化合物I与TYK2抑制剂被组合施用用于类风湿性关节炎或如本文所描述的其他炎性病况的治疗。与化合物I组合施用用于类风湿性关节炎或本文公开的其他炎性病况中的任意炎性病况的治疗的适合的TYK2抑制剂包含(不限于)PF-06826647(1r,3r)-3-(氰甲基)-3-(4-(6-(1-甲基-1H-吡唑-4-基)吡唑并[1,5-a]吡嗪-4-基)-1H-吡唑-1-基)环丁烷-1-甲腈)、3-哒嗪羧胺、托法替尼、乌帕替尼、氘可来昔替尼(6-(环丙甲酰氨基)-4-[2-甲氧基-3-(1-甲基-1,2,4-三唑-3-基)苯胺]-N-(三氘甲基)哒嗪-3-羧胺)、赛度替尼、AT9283(1-环丙基-3-(3-(5-(吗啉甲基)-1H-苯并[d]咪唑-2-基)-1H-吡唑-4-基)脲)、Nvp-bsk805·2HCl(4-(2,6-二氟-4-(3-(1-(哌啶-4-基)-1H-吡唑-4-基)喹喔啉-5-基)苄基)吗啉二盐酸盐)、S-鲁索利替尼、更度替尼、帕克替尼、巴瑞替尼、非戈替尼、伊森昔替尼、NDI-031301(Akahane et al.,Blood 128:1596,2016)和XL019((2R)-N-[4-[2-(4-吗啉-4-基苯胺)嘧啶-4-基]苯基]吡咯啶-2-酰胺)、brepocitinib([(1S)-2,2-二氟环丙基]-[(1S,5R)-3-[2-[(1-甲基吡唑-4-基)氨基]嘧啶-4-基]-3,8-二氮杂二环[3.2.1]辛-8-基]甲酮)、VTX-958(由Ventyx Biosciences开发的选择性变构TYK2抑制剂)和NDI-031407(由Nimbus Therapeutics开发并且在Gracey,E.,et al.J.Clin.Invest.;130(4):1863-1878(2020)(其通过引用被整体并入本文)中公开的选择性TYK2抑制剂)。
在任意实施方案中,化合物I与BTK抑制剂被组合施用用于类风湿性关节炎或如本文所描述的其他炎性病况的治疗。与化合物I组合施用用于类风湿性关节炎或本文公开的其他炎性病况中的任意炎性病况的治疗的适合的BTK抑制剂包含(不限于)依鲁替尼、阿卡替尼、非奈替尼和泽布替尼。
在任意实施方案中,化合物I与IRAK4抑制剂、IKKi抑制剂、tpl2抑制剂或CTLA4抑制剂被组合施用用于类风湿性关节炎或如本文所描述的其他炎性病况的治疗。与化合物I组合施用用于类风湿性关节炎或本文公开的其他炎性病况中的任意炎性病况的治疗的适合的IRAK4抑制剂、IKKi抑制剂、tpl2抑制剂和CTLA4抑制剂包含(不限于)N-[1-(2-吗啉-4-基乙基)苯并咪唑-2-基]-3-硝基苯甲酰胺、N-(1-苯基-5-(哌啶-1-基甲基)-1H-苯并[d]咪唑-2-基)-3-(三氟甲基)、苯甲酰胺、依鲁替尼、(R)-6-((1,6-萘并吡啶-2-基)氨基)-4-(环丙基氨基)-N-(2-氟-3-羟基-3-甲基丁基)烟酰胺、N-(4-吗啉-4-基环己基)-5-(四氢吡喃-4-基)-7H-吡咯并[2,3-d]嘧啶-4-胺、N-(反式-4-吗啉代环己基)-5-(四氢-2H-吡喃-4-基)-7H-吡咯并[2,3-d]嘧啶-4-胺、4-[(4-吗啉-4-基环己基)氨基]喹唑啉-6-甲腈、1-[[(2S,3S,4S)-3-乙基-4-氟-5-氧代-2-吡咯烷基]甲氧基]-7-甲氧基-6-异喹啉酰胺、2-(4-氟苯基)-6-甲基-4-(3-(三氟甲基)苯基)-1,2-二氢二吡唑并[3,4-b:3',4'-d]吡啶-3(6H)-酮、伊匹单抗、阿巴西普、4-(3-氯-4-氟苯胺)-6-(吡啶-3-基甲基氨基)-1,7-萘并吡啶-3-甲腈、6-[7-[3-氰基-4-(四氢吡喃-4-基氧基)苯基]呋喃并[3,2-b]吡啶-2-基]-5-甲氧基-N,N-二甲基吡啶-3-酰胺。
用于癌症的治疗的可能的组合疗法的具体非限制性实例包含如本文所描述的包括化合物I的口服组合物与下列物质的组合:(1)烷化剂,包含但不限于顺铂(PLATIN
在一些实施方案中,本文所描述的口服组合物与附加的治疗剂组合施用,附加的治疗剂选自化学治疗剂或抗增殖剂、抗病毒剂、抗生素、抗组胺剂、润滑剂、全身性光线疗法、补骨脂素光化学疗法、激光疗法、激素替代疗法、抗炎药剂、免疫调控剂或免疫抑制剂、用于治疗心血管疾病的药剂、用于治疗糖尿病的药剂、用于治疗免疫缺陷紊乱的药剂或其任何组合。
在任意实施方案中,化合物I与一种或更多种JAK抑制剂或BTK抑制剂可以被组合施用至患有银屑病关节炎的受试者,JAK抑制剂包含(但不限于)托法替尼、乌帕替尼、巴瑞替尼、非戈替尼、鲁索利替尼、奥拉替尼、培非替尼、菲卓替尼、赛度替尼、更度替尼、来他替尼、莫洛替尼、帕西替尼、阿布昔替尼和BMS-986165,BTK抑制剂包含(但不限于)依鲁替尼、阿卡替尼、非奈替尼和泽布替尼。在任意实施方案中,化合物I与下列中的一种或更多种可以被组合施用至患有银屑病关节炎的受试者:NSAID;一种或更多种DMARD(如甲氨蝶呤、柳氮磺吡啶、来氟米特、阿巴西普、阿达木单抗、赛妥珠单抗、依那西普、戈利木单抗、英利昔单抗、依奇珠单抗、司库奇尤单抗、托珠单抗、托法替尼、乌司奴单抗;一种或更多种免疫抑制剂(如硫唑嘌呤或环孢菌素);阿普斯特或类固醇(注射)。
在任意实施方案中,化合物I与一种或更多种JAK抑制剂或BTK抑制剂可以被组合施用至患有银屑病的受试者,JAK抑制剂包含(但不限于)托法替尼、乌帕替尼、巴瑞替尼、非戈替尼、鲁索利替尼、奥拉替尼、培非替尼、菲卓替尼、赛度替尼、更度替尼、来他替尼、莫洛替尼、帕西替尼、阿布昔替尼和BMS-986165,BTK抑制剂包含(但不限于)依鲁替尼、阿卡替尼、非奈替尼和泽布替尼。在任意实施方案中,化合物I与下列中的一种或更多种可以被组合施用至患有银屑病的受试者:皮质类固醇;维生素D类似物;视黄醇;钙调磷酸酶抑制剂;水杨酸;地蒽酚;环孢菌素;一种或更多种DMARD(如甲氨蝶呤、依那西普、英利昔单抗、阿达木单抗、乌司奴单抗、司库奇尤单抗、依奇珠单抗);硫鸟嘌呤、羟基脲、阿普斯特、可选地与光疗法组合的煤焦油(例如,格克曼疗法)或者任何天然或替代治疗(如芦荟精华霜、鱼油、俄勒冈葡萄或精油)。
在任意实施方案中,化合物I与一种或更多种JAK抑制剂、肿瘤坏死因子抑制剂、IL-1β抑制剂和IL-1α抑制剂可以被组合施用至患有化脓性汗腺炎(HS)的受试者,JAK抑制剂包含(但不限于)托法替尼、乌帕替尼、巴瑞替尼、非戈替尼、鲁索利替尼、奥拉替尼、培非替尼、菲卓替尼、赛度替尼、更度替尼、来他替尼、莫洛替尼、帕西替尼、阿布昔替尼和BMS-986165;肿瘤坏死因子抑制剂包含(但不限于)阿达木单抗、依那西普、戈利木单抗、英利昔单抗、赛妥珠单抗;IL-1β抑制剂和IL-1α抑制剂包含(但不限于)阿那白滞素、利那西普、卡那单抗、吉伏组单抗、LY2189102(Bihorel et al.,AAPS J.16(5):1009-1117,2014)和贝迈奇单抗。在任意实施方案中,化合物I与下列中的一种或更多种可以被组合施用至患有化脓性汗腺炎(HS)的受试者:抗生素(如克林霉素、庆大霉素、利福平、多西环素、米诺环素);一种或更多种DMARD(如甲氨蝶呤、阿达木单抗、英利昔单抗、阿那白滞素、卡那单抗、乌司奴单抗);一种或更多种NSAID;一种或更多种视黄醇(如异维甲酸和阿维A脂(acetretin));雷琐酚(resorcinol)(例如,局部);一种或更多种激素(如螺内酯、非那雄胺);或二甲双胍。
在任意实施方案中,化合物I与一种或更多种白介素-1受体拮抗剂可以被组合施用至患有冷凝比林相关的自身炎性综合征(CAPS)的受试者,白介素-1受体拮抗剂包含(但不限于)阿那白滞素(KINERET
用于与本文所描述的口服组合物组合使用的这些多个药剂/组合物可以根据其标准或常用剂量(如药物的商业可获得形式所附的处方信息中所规定的)使用(还参见2006版The Physician's Desk Reference中的处方信息)。在一些实施方案中,当与本文所描述的口服组合物组合使用时,这些药剂的标准计量可以被降低。在不限制本公开内容的范围的情况下,据信这样的组合可以产生具有更好疗效、更少毒性、更长作用时间或更快治疗反应的协同结果。在一些实施方案中,本文实施方案中的组合疗法可以以本文所描述的口服组合物或附加的药物药剂或两者的亚治疗量施用。
尽管本文已经详细描绘和描述了优选的实施方案,但对于相关领域的技术人员显而易见的是,可以在不背离本发明精神的情况下进行各种修改、添加、替换等,并且因此这些被认为是在所附的权利要求书中所限定的本发明的范围内。
公开的实施方案
本发明还提供下列非限制性实施方案。
实施方案1是用于治疗炎性病况的方法,方法涉及向患有炎性病况的人类受试者施用5mg/天至300mg/天的口服剂量的化合物I
实施方案2是实施方案1所述的方法,其中向所述受试者施用50mg/天的化合物。
实施方案3是实施方案1所述的方法,其中向所述受试者施用100mg/天的化合物。
实施方案4是实施方案1所述的方法,其中向所述受试者施用160mg/天的化合物。
实施方案5是实施方案1所述的方法,其中向所述受试者施用200mg/天的化合物。
实施方案6是实施方案1所述的方法,其中向所述受试者施用240mg/天的化合物。
实施方案7是实施方案1-6中任一项所述的方法,其中一天一次施用化合物。
实施方案8是实施方案1-6中任一项所述的方法,其中一天两次施用化合物。
实施方案9是权利要求1所述的方法,其中向受试者每天两次施用50mg的化合物。
实施方案10是权利要求1所述的方法,其中向受试者每天两次施用80mg的化合物。
实施方案11是实施方案1所述的方法,其中向受试者每天两次施用120mg的化合物。
实施方案12是实施方案1-11中任一项所述的方法,其中化合物I是氘化的。
实施方案13是实施方案1-12中任一项所述的方法,其中化合物I包括(P)-I与(M)-I的摩尔比为约4:1的化合物(P)-I和化合物(M)-I:
实施方案14是实施方案13所述的方法,其中(P)-I与(M)-I的摩尔比为约9:1。
实施方案15是实施方案13所述的方法,其中(P)-I与(M)-I的摩尔比为约99:1。
实施方案16是实施方案13所述的方法,其中(P)-I与(M)-I的摩尔比为约199:1。
实施方案17是实施方案13所述的方法,其中(P)-I与(M)-I的摩尔比为约399:1。
实施方案18是实施方案13所述的方法,其中(P)-I与(M)-I的摩尔比为约999:1。
实施方案19是权利要求1-12中任一项所述的方法,其中化合物I包括基本上不含化合物(M)-I的化合物(P)-I。
实施方案20是权利要求1-12中任一项所述的方法,其中化合物I包括至少80mol%的化合物(P)-I
实施方案21是实施方案20所述的方法,其中化合物I包括至少90mol%的化合物(P)-I。
实施方案22是实施方案20所述的方法,其中化合物I包括至少95mol%的化合物(P)-I。
实施方案23是实施方案20所述的方法,其中化合物I包括至少99mol%的化合物(P)-I。
实施方案24是实施方案1-23中任一项所述的方法,其中化合物(P)-I是游离碱。
实施方案25是实施方案1-23中任一项所述的方法,其中化合物(P)-I是药学上可接受的盐。
实施方案26是实施方案1-23中任一项所述的方法,其中化合物(P)-I是结晶形式。
实施方案27是实施方案26所述的方法,其中化合物(P)-I的结晶形式是结晶形式A,结晶形式A的特征在于具有在约9.78±0.2的角度2θ表示的峰的PXRD图谱。
实施方案28是实施方案26所述的方法,其中化合物(P)-I的结晶形式是结晶形式A,结晶形式A的特征在于具有在9.78±0.2和15.51±0.2的角度2θ表示的峰的PXRD图谱。
实施方案29是实施方案26所述的方法,其中化合物(P)-I的结晶形式是结晶形式A,结晶形式A的特征在于具有在9.78±0.2、15.51±0.2、19.6±0.2和25.92±0.2的角度2θ表示的峰的PXRD图谱。
实施方案30是实施方案26所述的方法,其中化合物(P)-I的结晶形式是结晶形式A,结晶形式A的特征在于具有在9.78±0.2、15.34±0.2、15.51±0.2、19.6±0.2、20.57±0.2、21.01±0.2、25.92±0.2、29.05±0.2和29.48±0.2的角度2θ表示的峰的PXRD图谱。
实施方案31是实施方案1-30中任一项所述的方法,其中炎性病况是类风湿性关节炎。
实施方案32是实施方案1-30中任一项所述的方法,其中炎性病况是银屑病关节炎。
实施方案33是实施方案1-30中任一项所述的方法,其中炎性病况是冷凝比林相关周期性综合征。
实施方案34是实施方案1-30中任一项所述的方法,其中炎性病况是化脓性汗腺炎。
实施方案35是实施方案1-30中任一项所述的方法,其中炎性病况选自银屑病、幼年特发性关节炎、溃疡性结肠炎、克罗恩病、强直性脊柱炎、胰腺癌、转移性乳腺癌、痛风、复发性心包炎和特发性肺纤维化。
实施方案36是实施方案1-30中任一项所述的方法,其中炎性病况选自脊柱关节炎(如强直性脊柱炎)、银屑病关节炎、反应性关节炎和赖特综合征、幼年型类风湿性关节炎(JIA)、全身起病型幼年型类风湿性关节炎、特发性关节炎(JIA)(包含全身型(SJIA))和痛风;冷凝比林相关的自身炎性综合征(CAPS)(包含Muckle-Wells综合征(MWS))、新生儿起病多系统炎性疾病(NOMID)和家族性寒冷性自身炎性综合征(FCAS);慢性阻塞性肺病(COPD)(包含肺气肿、慢性支气管炎和哮喘(过敏性和非过敏性))、化脓性汗腺炎(HS);银屑病(如斑块状银屑病);炎性肠病(IBD)引起的结肠炎(如克罗恩病或溃疡性结肠炎和炎性肠病相关关节炎);心包炎(包含急性心包炎、复发性心包炎和慢性心包炎);肺部炎症或纤维化(包含特发性肺纤维化);转移性乳腺癌和胰腺癌。
实施方案37是实施方案1-30中任一项所述的方法,其中炎性病况选自家族性地中海热(FMF);肿瘤坏死因子受体相关周期性综合征(TRAPS);成年起病型斯蒂尔病;坏疽性脓皮病;骨吸收紊乱(如与癌症(例如,乳腺癌)相关的紊乱);转移性黑色素瘤;卡斯尔门病;和慢性非典型中性粒细胞性皮肤病伴脂肪代谢障碍(CANDLE)。
实施方案38是实施方案1-30中任一项所述的方法,其中炎性病况是可能与任意其他病况相关的瘙痒,例如与化脓性汗腺炎相关的瘙痒、与炎症相关的瘙痒、与类风湿性关节炎相关的瘙痒、与银屑病相关的瘙痒和与TH17相关性炎症相关的瘙痒。
实施方案39是实施方案1-30中任一项所述的方法,其中炎性病况选自莱姆病;细胞因子释放综合征(CRS);急性呼吸窘迫综合征(ARDS);慢性或急性支气管炎;大疱性表皮松解症(EB);大疱性类天疱疮;幼年型皮肌炎;炎性白癜风(包含边缘性);寻常型天疱疮;小肠结肠炎;多发性肌炎;肌炎,骨癌;肺癌;炎性骨紊乱(如慢性复发性多发性骨髓炎(CRMO)、滑膜炎、痤疮、脓疱病、骨质增生和骨炎(SAPHO)综合征、马吉德综合征、白介素-1受体拮抗剂缺乏症(DIRA)和家族性巨颌症;骨吸收(如与自身免疫性疾病有关);神经炎性疾病(如阿尔茨海默病(AD)、帕金森病(PD)、多发性硬化(MS)、急性播散性脑脊髓炎(ADEM)、急性视神经炎(AON)、横贯性脊髓炎和视神经脊髓炎(NMO);白塞病;内毒素性休克(例如,中毒性休克综合征(TSS)和其他全身性革兰氏阴性细菌感染);起止点炎;结节性多动脉炎(PAN);慢性疼痛;风湿性多肌痛;慢性同种异体移植排斥;斯耶格伦综合征;和施尼茨勒(Schnitzler)综合征(SchS)。
实施方案40是实施方案1-30中任一项所述的方法,其中向患有类风湿性关节炎的受试者每天两次施用50mg的化合物。
实施方案41是实施方案1-30中任一项所述的方法,其中向患有类风湿性关节炎的受试者每天两次施用80mg的化合物。
实施方案42是实施方案1-30中任一项所述的方法,其中向患有类风湿性关节炎的受试者每天两次施用120mg的化合物。
实施方案43是实施方案36-38中任一项所述的方法,其中所述施用步骤在通过磁共振成像(MRI)评估的在所述受试者中有效抑制关节损伤的进展、有效改善滑膜炎或两者均有效的条件下进行。
实施方案44是实施方案1-43中任一项所述的方法,其中所述施用步骤在相比于被施用安慰剂的受试者中的一种或更多种炎性细胞因子的体内血清水平,有效显著降低一种或更多种炎性细胞因子的体内血清水平的条件下进行。
实施方案45是实施方案44所述的方法,其中一种或更多种炎性细胞因子选自由TNF-α、IL-1β、IL-6、IL-8、IFNγ、IL-17、IL-18、IL-1α和MIP1β组成的组。
实施方案46是实施方案1-45中任一项所述的方法,其中所述施用步骤被进行超过12周而无快速耐受性。
实施方案47是实施方案1-45中任一项所述的方法,其中所述施用步骤还包括:与化合物I连同施用一种或更多种附加的治疗剂。
实施方案48是实施方案47所述的方法,其中所述一种或更多种附加的治疗剂与化合物I被同时施用。
实施方案49是实施方案47所述的方法,其中所述一种或更多种附加的治疗剂与化合物I被顺序施用。
实施方案50是实施方案47-49中任一项所述的方法,其中一种或更多种附加的治疗剂选自由抗炎药物、抗动脉硬化药物、免疫抑制药物、免疫调控药物、细胞稳定药物(cytostatic drug)、血管生成抑制剂、激酶抑制剂、细胞因子阻断剂和细胞粘附分子的抑制剂组成的组。
实施方案51是实施方案47-49中任一项所述的方法,其中如实施方案2-30中任一项中所限定的化合物I与JAK抑制剂被连同施用至患有类风湿性关节炎的受试者。
实施方案52是实施方案47-49中任一项所述的方法,其中如实施方案2-30中任一项中所限定的化合物I与TYK2抑制剂被连同施用至患有类风湿性关节炎的受试者。
实施方案53是实施方案47-49中任一项所述的方法,其中如实施方案2-30中任一项中所限定的化合物I与BTK抑制剂被连同施用至患有类风湿性关节炎的受试者。
实施方案54是实施方案47-49中任一项所述的方法,其中如实施方案2-30中任一项中所限定的化合物I与IRAK4抑制剂被连同施用至患有类风湿性关节炎的受试者。
实施方案55是实施方案47-49中任一项所述的方法,其中如实施方案2-30中任一项中所限定的化合物I与IKKi抑制剂被连同施用至患有类风湿性关节炎的受试者。
实施方案56是实施方案47-49中任一项所述的方法,其中如实施方案2-30中任一项中所限定的化合物I与tpl2抑制剂被连同施用至患有类风湿性关节炎的受试者。
实施方案57是实施方案47-49中任一项所述的方法,其中如实施方案2-30中任一项中所限定的化合物I与CTLA4抑制剂被连同施用至患有类风湿性关节炎的受试者。
实施方案58是实施方案1-57中任一项所述的方法,其中化合物被配制成固体剂型,固体剂型选自片剂、胶囊剂、锭剂、囊袋剂、粉剂、颗粒剂和口服分散膜。
实施方案59是实施方案58所述的方法,其中固体剂型是片剂。
实施方案60是实施方案1-59中任一项所述的方法,其中化合物作为立即释放剂型被施用。
实施方案61是实施方案1-59中任一项所述的方法,其中化合物作为控制释放剂型被施用。
实施方案62是口服药物组合物,口服药物组合物包括:以5mg至300mg的量的化合物I
实施方案63是实施方案62所述的口服组合物,其中所述组合物包括50mg的化合物I。
实施方案64是实施方案62所述的口服组合物,其中所述组合物包括80mg的化合物I。
实施方案65是实施方案62所述的口服组合物,其中所述组合物包括100mg的化合物I。
实施方案66是实施方案62所述的口服组合物,其中所述组合物包括120mg的化合物I。
实施方案67是实施方案62所述的口服组合物,其中所述组合物包括160mg的化合物I。
实施方案68是实施方案62所述的口服组合物,其中所述组合物包括200mg的化合物I。
实施方案69是实施方案62所述的口服组合物,其中所述组合物包括240mg的化合物I。
实施方案70是实施方案62-69中任一项所述的口服组合物,其中化合物I是氘化的。
实施方案71是实施方案62-69中任一项所述的口服组合物,其中化合物I包括(P)-I与(M)-I的摩尔比为约4:1的化合物(P)-I和化合物(M)-I:
实施方案72是实施方案71所述的口服组合物,其中(P)-I与(M)-I的摩尔比为约9:1。
实施方案73是实施方案71所述的口服组合物,其中(P)-I与(M)-I的摩尔比为约99:1。
实施方案74是实施方案71所述的口服组合物,其中(P)-I与(M)-I的摩尔比为约199:1。
实施方案75是实施方案71所述的口服组合物,其中(P)-I与(M)-I的摩尔比为约399:1。
实施方案76是实施方案62-69中任一项所述的口服组合物,其中化合物I包括基本上不含化合物(M)-I的化合物(P)-I。
实施方案77是实施方案62-69中任一项所述的口服组合物,其中化合物I包括至少80mol%的化合物(P)-I
实施方案78是实施方案62-69中任一项所述的口服组合物,其中化合物I包括至少90mol%的化合物(P)-I。
实施方案79是实施方案62-69中任一项所述的口服组合物,其中化合物I包括至少95mol%的化合物(P)-I。
实施方案80是实施方案62-69中任一项所述的口服组合物,其中化合物I包括至少99mol%的化合物(P)-I。
实施方案81是实施方案62-80中任一项所述的口服组合物,其中化合物I(P)-I是游离碱。
实施方案82是实施方案62-80中任一项所述的口服组合物,其中化合物I(P)-I是药学上可接受的盐。
实施方案83是实施方案62-80中任一项所述的口服组合物,其中化合物I(P)-I是结晶形式。
实施方案84是实施方案83所述的口服组合物,其中化合物(P)-I的结晶形式是结晶形式A,结晶形式A的特征在于具有在约9.78±0.2的角度2θ表示的峰的PXRD图谱。
实施方案85是实施方案83所述的口服组合物,其中化合物(P)-I的结晶形式是结晶形式A,结晶形式A的特征在于具有在9.78±0.2和15.51±0.2的角度2θ表示的峰的PXRD图谱。
实施方案86是实施方案83所述的口服组合物,其中化合物(P)-I的结晶形式是结晶形式A,结晶形式A的特征在于具有在9.78±0.2、15.51±0.2、19.6±0.2和25.92±0.2的角度2θ表示的峰的PXRD图谱。
实施方案87是实施方案83所述的口服组合物,其中化合物(P)-I的结晶形式是结晶形式A,结晶形式A的特征在于具有在9.78±0.2、15.34±0.2、15.51±0.2、19.6±0.2、20.57±0.2、21.01±0.2、25.92±0.2、29.05±0.2和29.48±0.2的角度2θ表示的峰的PXRD图谱。
实施方案88是实施方案62-87中任一项所述的口服组合物,其中所述组合物被配制成固体剂型,固体剂型选自片剂、胶囊剂、锭剂、囊袋剂、粉剂、颗粒剂和口服分散膜。
实施方案89是实施方案62-87中任一项所述的口服组合物,其中所述组合物被配制成片剂。
实施方案90是实施方案62-87中任一项所述的口服组合物,其中组合物是立即释放剂型。
实施方案91是实施方案62-87中任一项所述的口服组合物,其中组合物是控制释放剂型。
实施例
如上文所详细描述的,3-氯-4-((3,5-二氟吡啶-2-基)甲氧基)-2'-(2-(2-羟基丙烷-2-基)嘧啶-4-基)-5',6-二甲基-2H-[1,4'-联吡啶]-2-酮(即,化合物I)以P阻转异构体和M阻转异构体存在。在本公开的实施例和附图中,“ATI-450”指含有约99.75mol%的P阻转异构体和0.25mol%的M阻转异构体的上文所述的化合物。
实施例1:口服施用的ATI-450的安全性、药代动力学和药效动力学
ATI-450是p38α丝裂原激活蛋白激酶(MAPK)/MAPK激活蛋白激酶2(MK2)炎性信号传导途径的口服小分子抑制剂。这项1期、单次和多次递增剂量(SAD、MAD)的研究评价了ATI-450的安全性、耐受性、药代动力学和药效动力学。健康成人被随机分配到ATI-450的SAD(10mg、30mg、50mg、100mg;n=24)组和MAD(10mg、30mg、50mg,每天两次(BID),持续7天;n=24)组或安慰剂(n=14)。通过临床和实验室评估,对安全性和耐受性进行评价。测定血液PK参数,并且对体外内毒素诱导的肿瘤坏死因子α(TNF-α)、白介素(IL)-1β、IL-6、IL-8以及MK2底物的磷酸化作用、磷酸化热休克蛋白27(p-HSP27)的药效动力学抑制进行定量。最常见的不良事件是头痛(10/48,20.8%)、头晕(6/48,12.5%)、上呼吸道感染(3/48,6.3%)和便秘(3/48,6.3%)。ATI-450具有与剂量成比例的药代动力学,其中在第7天,MAD组的终末半衰期为9-12小时。观察到体外刺激的细胞因子和靶标生物标志物的剂量和浓度依赖性抑制。在第7天,50mg BID剂量组中的患者记录的平均谷值药物水平分别比TNF-α、IL-1β、IL-8和p-HSP27的IC
由于p38丝裂原激活蛋白激酶(p38MAPK)信号传导途径参与多种细胞因子(例如,肿瘤坏死因子α[TNF-α]、白介素[IL]1β和IL-6)和其他炎性信号的调节和表达,其已成为炎性疾病的治疗性干预的靶标。p38MAPK的小分子抑制剂已在用于炎性疾病的治疗的临床试验中被评价;然而,这些化合物的临床开发已受到毒性和缺乏持续疗效的限制。整体p38MAPK抑制的关键挑战是其无处不在的表达和其对细胞生理学的广泛影响(这是其调节超过60种底物的结果)。通过这些底物的磷酸化,p38MAPK调节负反馈回路和抗炎性途径,这两者均参与下调炎症;因此,p38MAPK途径的整体阻断可以抑制促炎性途径和抗炎性途径两者,限制其在某些疾病状态中的潜在疗效。此外,数个p38MAPK底物参与细胞功能的调节,并且这些蛋白的抑制可能导致毒性。
用p38MAPK抑制剂实现的临床疗效通常是令人失望的。值得注意的是,在某些自身免疫疾病中有证据表明p38MAPK抑制剂在多次治疗后表现出快速耐受性,使得在这些治疗适应症中难以实现炎性反应的持续抑制。快速耐受性可以由上文所描述的负反馈和/或抗炎性靶标底物所解释。因此,靶向p38MAPK的关键下游激酶底物特异性调节炎性细胞因子,以潜在地实现针对病理性炎症的阻断的更大特异性的方法已经获得关注。MAPK激活蛋白激酶2(MK2)是p38MAPK的直接下游底物,p38MAPK已被认为是炎症的关键驱动。通过p38MAPK/MK2途径激活的炎性细胞因子包含TNF-α、IL-1β、IL-6和IL-8。
选择性地抑制p38MAPK/MK2生物分子复合物而不是单独抑制p38MAPK的新颖方法导致有效地阻断促炎性轴,同时避开抗炎症途径、负反馈底物和调节一般细胞功能的蛋白质。这种方法有潜力生成与整体p38MAPK抑制剂相比具有更大和更持久的抗炎性疗效的更安全的化合物。不幸的是,直接靶向MK2的方法在很大程度上并未成功。
ATI-450(最近开发的MK2抑制剂)具有新的作用机制,其通过该作用机制靶向在p38MAPK与MK2之间相互作用时形成的经修饰的p38MAPK ATP结合袋(binding pocket)和并列的MK2。MK2上的p38MAPK结合位点位于C末端内;热力学研究已经表明,复合物中的2个蛋白紧密结合,并且MK2中结合序列的缺失会废除p38依赖性磷酸化作用和MK2的激活。在p38MAPK-MK2生物分子复合物形成后,ATI-450以显著高于与任意激酶单独结合的亲和力与复合物的界面结合,从而选择性地抑制MK2的p38MAPK磷酸化,并且将MK2锁定在非活性构象。MK2通过下游效应物(包含腺苷酸-尿苷酸富集元件结合蛋白(例如,三四脯氨酸(tristetraprolin)))的磷酸化作用调节炎性细胞因子mRNA的稳定性和翻译。因此,p38MAPK-MK2的抑制阻断下游MK2介导的炎性作用。
临床前动物疾病模型已经被用于预测ATI-450作为数种免疫介导的炎性疾病的治疗潜力。在类风湿性关节炎的大鼠链球菌细胞壁关节炎模型中,ATI-450显示出关节保护作用并保留了骨质密度(bone mineral density)。还在新生儿起病多系统炎性疾病(NOMID)(其是冷凝比林相关的周期性综合征的重度形式)的小鼠模型中研究了ATI-450;NOMID和其他冷凝比林相关的周期性综合征包括其中IL-1β和IL-18被过度产生的一系列罕见的遗传性自身炎性紊乱。在该转基因动物模型中观察到疾病的急剧减毒,包含相对于用安慰剂治疗的小鼠,用ATI-450治疗的小鼠中IL-1β的骨髓水平的降低。这些临床前结果支持ATI-450在人类中的临床开发,以用于细胞因子TNF-α、IL-1β和IL-6驱动的自身免疫疾病和自身炎性疾病。
如本文所描述的,在健康受试者的单次和多次递增剂量(SAD和MAD)组中,进行了首次人体1期、随机、观察者单盲(observer-blind)、安慰剂对照研究,以评价ATI-450的安全性、耐受性、PK和药效动力学(PD)。此外,研究包含评价进食与禁食施用的PK以及与甲氨蝶呤共同施用的PK的单独组。在此报告的是SAD组和MAD组的数据。
结果
研究受试者。在SAD组中,32名受试者被招募、随机分配并被包含在安全性集(set)中。总计31名受试者完成了研究;30mg的ATI-450组中的1名受试者在第1天接受单剂量后由于非安全性原因退出研究,并且因此没有完成所有研究评估。PK和PD分析包含24名接受ATI-450的受试者。SAD组中的大多数受试者是女性(84%;表1),并且年龄在18至51岁的范围内。
在MAD组中,30名受试者被招募、随机分配并完成研究,并且所有受试者都被包含在安全性集中;接受ATI-450的24名受试者被包含在PK组和PD集中。这些组中女性受试者和男性受试者分配更平均(57%女性,43%男性),并且年龄在21至53岁的范围内。
安全性和耐受性。没有观察到生命体征、心电图或临床实验室值方面的临床上有意义的发现。所有研究参与者中最常见的治疗突发不良事件(TEAE)在表2中示出。在SAD组中,24名接受ATI-450的受试者中的7名受试者(29%)报告了15例TEAE,并且8名接受安慰剂的受试者中的4名受试者(50%)报告了6例TEAE。没有发生死亡或严重不良事件(SAE),并且没有发生由于TEAE的中止。所有的TEAE都是温和的、短暂的并且在研究结束时消退而无后遗症。24名接受ATI 450的受试者中的4名(17%)报告了研究者认为与研究药物有关的TEAE(10mg组中0名受试者;30mg组中2名[33%]受试者[1次恶心事件和头痛和头晕事件各2次];50mg组中1名[17%]受试者[1次头痛事件];以及100mg组中1名受试者[2次头晕事件])。
在MAD组中,24名接受ATI-450的受试者中的12名(50%)报告了24例TEAE,并且6名接受安慰剂的受试者中的3名(50%)报告了3例TEAE。与SAD组一样,没有死亡或SAE,并且没有受试者由于TEAE而中止研究;所有TEAE都是温和的、短暂的,并在下次访视时消退而无后遗症。24名接受ATI-450的受试者中的9名(38%)报告了研究者认为与研究药物有关的TEAE(10mg BID组中8名受试者中的1名[13%][1次头痛事件],30mg BID组中8名受试者中的5名[63%][视力模糊、便秘、腹痛、腹泻、关节痛、头晕和口咽痛事件各1次],以及50mg BID组中8名受试者中的3名[38%][1次头痛事件和2次头晕事件])。在安慰剂组中,6名受试者中的2名(33%)出现了研究者认为与研究药物有关的TEAE(呕吐和头痛事件各1次)。
实验室的变化一般来说是不明显的。观察到中性粒细胞的剂量依赖性降低,其中在第2天至第5天在MAD组中观察到最大效果。50mg BID组中第7天的平均降低为24%(基线处3318.4个细胞/μL相对于第7天2514.4个细胞/μL)。
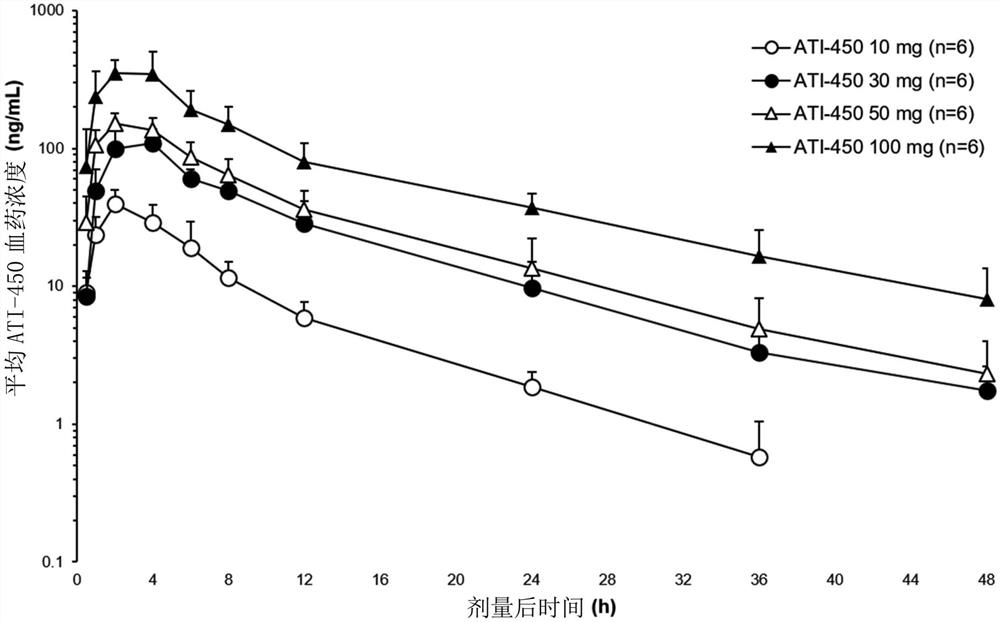
药代动力学-SAD组。如图1中所示,在禁食条件下,以10mg、30mg、50mg和100mg的单剂量施用ATI-450的结果是,在以10mg给药后24小时内,以及在以所有其他剂量水平给药后48小时内,ATI-450的剂量后平均血药浓度高于定量的下限。ATI-450的平均血药浓度随着剂量增加而增加(图1)。遍及ATI-450的从10mg至100mg的剂量范围,全身性暴露以与剂量成比例的方式增加,其中C
下文示出的表1概括了SAD组进食受试者和禁食受试者对比的药代动力学数据。
表1.在单次口服剂量至处于禁食或进食状态的健康受试者后ATI-450的平均(SD)药代动力学参数(n=6/剂量组)
在禁食状态下,ATI-450被快速吸收(T
药代动力学-MAD组。图2示出了在第7天最后一次剂量后从0小时至72小时ATI-450的平均(SD)血药浓度-时间曲线,代表了MAD组的与剂量成比例的PK,并且表3示出了在多次剂量后第7天的ATI-450PK参数。在MAD组中在第1天第一剂量后获得的结果与在SAD组中单次剂量后获得的结果一致(数据未示出)。T
下文表2概括了在以10mg BID、30mg BID或50mg BID给药的第1和第7天ATI-450的PK参数。
表2.在每日两次口服剂量至处于禁食状态的健康受试者后ATI-450的平均(SD)药代动力学参数(n=8/剂量组)
MAD组中第1天的PK与SAD组的PK一致。观察到多次给药后的线性(剂量无关和时间无关)PK。还观察到中度缓慢消除(末端t
下文表3概括了如所示的使用甲氨蝶呤单独给药或与ATI-450组合给药的受试者中的PK参数。
表3在单独口服施用或者组合施用至处于禁食状态的健康男性受试者后ATI-450和甲氨蝶呤的平均(SD)药代动力学参数(n=15)
具有ATI-450暴露或不具有ATI-450暴露,MTX的药代动力学都类似。基于与来自研究中MAD部分的50mg的ATI-450数据的组间比较,在存在MTX的情况下,ATI-450的全身性暴露似乎略有减少(C
药效动力学
建立了每种分析物的浓度与ATI-450血浆水平之间的关系,并且然后将数据拟合到非线性抑制性E
对于50mg BID剂量MAD组,针对靶标生物标志物p-HSP27和针对4种细胞因子中的3种(TNF-α、IL-1β和IL-8)在C
图3的图表示出从用50mg BID的ATI-450给药的受试者(MAD组)收集的第7天的血液样品中LPS刺激的细胞因子和趋化因子产生的抑制。
讨论
在7天单次剂量高达100mg和多次剂量高达50mg BID的范围内,ATI-450的施用普遍安全且健康受试者耐受性良好。所有的TEAE都消退而无后遗症,并且没有由于不良事件的中止。头晕在主动组(active arms)中更频繁发生,但没有明显的剂量反应;这些事件通常本质上是短暂的,并且几乎全部在服用研究药物期间消退。观察到中性粒细胞的剂量依赖性降低,并且中性粒细胞的剂量依赖性降低与ATI-450的作用机制一致。PK参数证明,ATI-450的血药浓度在单次或多次递增剂量施用后以与剂量成比例的方式增加,这也通过SAD组和MAD组两者用幂模型的统计分析确认。在全部SAD组和MAD组中,中值T
ATI-450呈现出新的机制,通过该机制抑制MK2和由p38MAPK途径激活的促炎性细胞因子。作为口服施用的药物,ATI-450为治疗免疫介导的炎性疾病的注射用生物制剂(biologic)药物提供了潜在的替代方案。ATI-450在各种免疫介导的炎性疾病和某些类型的癌症的动物模型中显示出疗效。
本研究中的PD反应已经表示为ATI-450的血药浓度与每种细胞因子的IC
ATI-450在所研究的剂量下耐受性良好,表现出剂量和时间无关的(即线性)PK,以及剂量相关的PD效应。该研究的结果支持ATI-450的进展进入2期开发,用于治疗类风湿性关节炎和其他免疫介导的炎性疾病。
表7. 50mg BID剂量组中整个给药间隔的细胞因子和生物标志物IC
BID,每天两次;C
方法
研究设计。PRA Health Sciences在堪萨斯州Lenexa的临床点领导了研究,并且为所有组准备了随机化方案,同时维持与研究相关的观察者盲法。PRA在健康受试者招募到研究中后有资格进行随机分配时,依次分配随机化代码。对于每个SAD剂量组,受试者以总体6:2的比例被随机分配到ATI-450或安慰剂。对于每个MAD剂量组,受试者以总体的8:2比例被随机分配到ATI-450或安慰剂。
单次递增剂量(SAD)组。32名受试者被随机分配到4个组中的1组,以接受10mg、30mg、50mg或100mg的ATI-450剂量(n=6每个组)或安慰剂(n=2每个组)。每个受试者在早晨空腹接受单次口服剂量的ATI-450或安慰剂。所有受试者在第1天被许可进入临床研究机构,并且在ATI-450或安慰剂施用后48小时内入住。
多次递增剂量(MAD)组。30名受试者被随机分配到3个组中的1组,以接受10mgBID、30mg BID或50mg BID的ATI-450剂量(n=8每个组)或安慰剂(n=2每个组)。受试者空腹接受ATI-450的各个剂量或安慰剂持续7天,早晚各一次,最后的剂量在第7天早上施用。受试者在第1天被许可进入临床机构,并留在该地点直至第7天早上剂量后72小时。
受试者。受试者为年龄18至55岁(含)之间的健康男性或女性;具有在18至32kg/m2(含)之间的体质指数,其中最低体重为50kg;并且筛查时HIV-1和HIV-2抗体、乙型肝炎表面抗原和丙型肝炎病毒抗体测试阴性。女性受试者没有怀孕或哺乳,并且如果是异性恋活跃的和有生育潜力的,则同意使用2种有效的避孕方法。
如果受试者目前患有急性或慢性疾病、或者在入住2周内使用可能影响其安全或其他研究评估的药物,则被从研究排除。
该研究是根据《赫尔辛基宣言》的原则和依照国际协调委员会E6良好临床实践指南以及任何适用的国家和地方法律和法规进行的。临床研究方案、知情同意书和所有文件的修正案均由堪萨斯州奥弗兰帕克市的米德兰独立审查委员会审查和批准。在任何与研究有关的程序开始之前,已获得所有受试者的知情同意。
安全性和耐受性评估。在研究的两部分中,对每个受试者的不良事件、临床实验室检测、生命体征、心电图、霍尔特氏心电动态监测(Holter monitoring)和体检结果进行了评价。
血液采样和生物分析。收集两毫升血液样品用于PK分析。对于SAD组,在给药前和给药后0.5小时、1小时、2小时、4小时、6小时、8小时、12小时、24小时、36小时和48小时收集血液。对于MAD组,在第1天早晨剂量前和给药后0.5小时、1小时、2小时、4小时、6小时、8小时和12小时(在晚上剂量前)收集血液;在第2至6天早晨剂量前收集血液;以及在第7天早晨剂量前和早晨剂量后0.5小时、1小时、2小时、4小时、6小时、8小时、12小时、13小时、14小时、24小时、36小时、48小时和72小时收集血液。
收集10mL的血液样品用于PD分析。对于SAD组,在给药前和给药后1小时、12小时和24小时收集血液。对于MAD组,第1天在早晨剂量前和早晨剂量后4小时和12小时(在晚上剂量前)收集血液;第7天,在早晨剂量前和早晨剂量后4小时和12小时收集血液。
血浆样品中的ATI-450浓度由PRA健康科学-生物分析实验室(Lenexa,Kansas)使用经过验证的超高效液相色谱与串联质谱方法测定(详见补充信息)。定量的下限是0.500ng/mL。
血液样品中的磷酸化热休克蛋白27(p-HSP27)、IL-1β、IL-6、IL-8和TNF-α水平的PD生物分析(由Confluence Discovery Technologies,Inc.[St.Louis,Missouri]进行)在3种条件下进行分析:(1)未刺激,(2)体外脂多糖刺激,以及(3)在存在外源添加10μM的ATI-450的情况下的体外脂多糖刺激。历史数据显示,针对规定分析物实现的ATI-450的最大抑制百分比是一致的,并且因此对不同样品的标准化有用。用于分析p-HSP27和细胞因子的测定的附加详情包含在补充信息中。
针对每个受试者集(第1天-给药前、第1天-给药后1小时、第1天-给药后12小时和第2天-给药后24小时),SAD组数据计算为第1天给药前的%(使用体外脂多糖刺激作为最大信号并且使用第1天体外脂多糖加10μM的ATI-450作为最大抑制标准化样品集)。
针对每个受试者集(第1天-给药前,第1天-给药后4小时,第1天-给药后2小时,第7天-给药前,第7天-给药后4小时和第7天-给药后12小时),MAD组数据计算为第1天给药前的%(使用体外脂多糖刺激作为最大信号并且使用第1天体外脂多糖加10μM的ATI-450作为最大抑制标准化样品集)。
药代动力学。针对所有SAD组和MAD组测定ATI-450血药浓度和PK参数。使用Phoenix WinNonlin
药效动力学。使用Microsoft Excel 2010(Microsoft,Redmond,WA)、Meso ScaleDiscovery
样本量计算。没有对样本量进行统计能力的前瞻性计算。针对SAD组和MAD组分别选择了32个受试者和30个受试者的样本量,并且这对首次在人类中的研究是典型的。
ATI-450的生物分析测定。通过使用50.0-μL样品体积的蛋白质沉淀进行样品处理。使用Waters Acquity UPLC BEH C18柱(2.1×50mm,1.7μm颗粒尺寸),在40℃使用10mM的甲酸铵和0.3%的甲酸作为流动相A和50:50:0.3的乙腈:甲醇:甲酸作为流动相B,在等度条件下操作(洗脱后梯度冲洗),流速0.700mL/min,通过液相色谱串联质谱法实现潜在代谢物和干扰性内源化合物之间的分离。配备有涡轮离子喷雾源的三重四极杆6500质谱仪被用于正离子模式的检测。基于对ATI-450的m/z 514.3→297.2和内标ATI-450-
p-HSP27分析的样品制备。针对p-HSP27分析,将每个样品的1mL等分试样转移到6个2-mL的微管中,其中后续处理命名:‘A’为仅脂多糖刺激(最大信号),‘B’为接受脂多糖刺激的体外添加ATI-450样品(最大抑制),‘C’为未刺激。处理组由3管组‘A’、2管组‘B’和1管组‘C’组成。
向组‘A’和组‘C’中的所有管中添加二甲基亚砜(DMSO)至最终浓度为0.1%(在含有10%胎牛血清(FBS)和1%青霉素/链霉素/谷氨酰胺的达尔伯克氏改良伊格尔氏培养基(Dulbecco's Modified Eagle Medium,DMEM)中,每mL10%DMSO溶液10μL)。然后在室温下轻轻摇晃样品。将100%DMSO中10mM的ATI-450的储备溶液稀释至1mM(在含有10%FBS和1%青霉素/链霉素的DMEM中),并且在组‘B’样品中每mL血液添加10μL该工作溶液,以提供0.1%DMSO中10μM的最终浓度。然后将样品在室温下轻轻摇晃至少1小时。
在室温下以100ng/mL的最终浓度用脂多糖刺激组‘A’和组‘B’中的样品22分钟。在含有10%FBS和1%青霉素/链霉素的DMEM中,将1mg/mL的脂多糖储备溶液稀释至在含有10%FBS和1%青霉素/链霉素的DMEM中10μg/mL,并且每mL血液添加10μL该工作溶液。将样品在室温下轻轻摇晃11分钟,然后在分离外周血单核细胞(PBMC)前直立放置11分钟。
所有患者样品(未受刺激的样品)的组‘C’中每mL血液添加10μL仅含10%FBS的DMEM。将样品在室温下轻轻摇晃11分钟,然后在分离PBMC之前直立放置11分钟。
每个样品的处理(脂多糖刺激或未刺激)完成后,从所有处理组中的每个人类全血等分试样分离PBMC。在2-mL微离心管中,将来自每个样品的1mL血液轻轻铺在0.75mL的人淋巴细胞分离液(Histopaque-1077)上。Histopaque-1077被保持在室温下。将样品在Eppendorf微型离心机中以16000×g离心2分钟。去除界面和上层,并且添加至含有1mL冷的达尔伯克氏磷酸缓冲盐溶液(DPBS)的管中。然后将这些样品在Eppendorf微型离心机中以16000×g离心30秒使细胞颗粒化。通过抽吸去除缓冲液上清液,并且将颗粒重悬于1mL的冷DPBS中。然后,如上所述,将来自每个样品的颗粒重颗粒化。通过抽吸去除缓冲液,并且将最后的颗粒在100μL完全裂解缓冲液(MSD Tris裂解缓冲液,1×Halt
使用Luminex技术的p-HSP27(Ser78)水平分析。采用来自Millipore Sigma(Burlington,MA)的定制
用50μL/孔的测定缓冲液2预洗试剂盒中提供的96孔平底透明底黑板10分钟。倾析清洗液,并且以25μL/孔向每孔添加测定缓冲液2中的1X HSP27磁珠。然后以25μL/孔添加来自子板的经稀释裂解物。将板密封并在4℃黑暗中摇晃孵育过夜。
过夜孵育后,使用手持式磁分离块,倾析样品,并且用测定缓冲液2洗涤板2次。将生物素标记的检测抗体(测定缓冲液2中1×)以25μL/孔添加至所有板中,并且孵育1小时。所有孵育都在室温下在黑暗中摇晃。然后使用手持式磁分离块倾析检测抗体,并且将链霉亲和素-藻红素(PE;测定缓冲液2中1×)以25μL/孔添加至每个孔中。然后将板孵育15分钟。将扩增缓冲液(测定缓冲液2中1×)以25μL/孔添加至含有链霉亲和素-PE的孔中,并且孵育附加的15分钟。使用手持式磁分离块,通过倾析去除链霉亲和素-PE/扩增缓冲液。以150μL/孔向每孔添加测定缓冲液2。将板放在平板摇床上至少5分钟,然后使用
细胞因子分析的样品制备。对于每个样品,将180μL的等分试样转移到圆底96孔组织培养聚苯乙烯低蒸发板的7个孔中。在每个样品的7个孔中,3个被指定为‘A’,用于仅脂多糖刺激(最大信号),2个被指定为‘B’,用于接受脂多糖刺激的体外添加ATI-450的样品(最大抑制),并且2个被指定为‘C’,用于未刺激的脂多糖。没有使用板上的外侧孔,并且样品周围的所有孔都含有200μL的磷酸盐缓冲盐水。
在组‘A’和组‘C’的所有孔中添加DMSO至最终浓度为0.1%(在含有10%FBS和1%青霉素/链霉素/谷氨酰胺的DMEM中10L/孔的2%DMSO溶液)。在含有10%FBS和1%青霉素/链霉素的DMEM中,将100%DMSO中的10mM的ATI-450储备溶液稀释到200mM,并且在组‘B’的所有孔中每孔添加10mL该工作溶液,以提供0.1%DMSO中10mM的最终浓度。使用针状工具将样品在平板摇床上轻轻混合30秒,然后在37℃/5%CO
用100ng/mL最终浓度的脂多糖刺激组‘A’和组‘B’中的样品。含有10%FBS和1%青霉素/链霉素的DMEM中1mg/mL的脂多糖的储备液被稀释至含有10%FBS和1%青霉素/链霉素的DMEM中2mg/mL,并且对于组‘A’和组‘B’中的所有样品每孔血液添加10mL该工作溶液。对于组‘C’中的所有样品(未受刺激的样品),每孔血液添加10mL仅含有10%FBS的DMEM。使用针状工具将样品在平板摇床上轻轻混合30秒,然后在37℃/5%CO
孵育后,将板从培养箱中取出,并且用粘性透明板密封条密封。然后板在室温下以1800×g离心10分钟。从每个孔去除75mL血浆并转移至96孔聚丙烯圆底母板中,然后将其密封并冷冻在-80℃,直到生成各自组的所有样品。一旦生成给定组的所有样品,使用MesoScale Discovery技术分析这些样品的细胞因子(TNF-α、IL-1b、IL-6和IL-8)产生。
使用Meso Scale Discovery技术的细胞因子水平分析。以Meso Scale Discovery(Rockville,MD)的V-plex人类促炎性板II(4-plex)试剂盒(V-plex HumanProinflammatory Panel II(4-Plex)Kit)分析所有脂多糖血浆样品的细胞因子产生。含有血浆样品的母板在冰上解冻,并且针对每个母板生成由每个血浆样品的25倍稀释液(3μL血浆加72μL Meso Scale Discovery稀释液2)组成的子板。来自子板的经稀释的血浆样品被用于细胞因子分析。用150μL/孔的洗涤缓冲液(SeraCare[Milford,MA]的1×KPL磷酸盐缓冲盐水和吐温20)洗涤Meso Scale Discovery 4-plex板3次。将准备好的校准物和来自子板的样品以50μL/孔添加至经洗涤的Meso Scale Discovery 4-plex板中。将4-plex板密封并在4℃下摇晃过夜。
过夜孵育后,通过倾析去除样品。然后用150μL/孔的洗涤缓冲液洗涤4-plex板3次。向每个孔以25μL/孔添加Meso Scale Discovery稀释液3中检测抗体混合物(由硫标记的抗人IL-1β、硫标记的抗人IL-6、硫标记的抗人IL-8和硫标记的抗人TNF-α抗体组成),并且将板密封并在室温下摇晃孵育2小时。孵育后,通过倾析去除检测抗体。用150μL/孔的洗涤缓冲液洗涤4-plex板3次,然后以150μL/孔向所有孔添加2×读取缓冲液。然后使用MesoScale Discovery Sector S 600仪器读取板。使用Meso Scale Discovery Workbench分析软件进行计算以建立细胞因子校准曲线并测定样品中的分析物浓度。
表8.SAD组中ATI-450的剂量比例性的统计学分析
使用幂模型进行剂量比例性分析。In(PK)=截距+斜率*In(剂量)+e,其中PK是PK参数,并且e是误差项。斜率=1的值表明剂量比例性。在表8中,使用了下列术语:AUC,浓度-时间曲线下的面积;C
表9.MAD组中ATI-450的剂量比例性的统计学分析
使用幂模型进行剂量比例性分析。In(PK)=截距+斜率*In(剂量)+e,其中PK是PK参数,并且e是误差项。斜率=1的值表明剂量比例性。在表9中,使用了下列术语:AUC,浓度-时间曲线下的面积;C
表10.抑制性E
在表10中,使用了下列术语:IC
实施例2:口服施用的ATI-450的较高剂量的延伸研究
进行实施例1中描述的I期临床研究的该扩展以建立健康志愿者中ATI-450的更高剂量(80mg和120mg,每天两次)的安全性和耐受性。
方法:在随机、观察者单盲、安慰剂对照研究中,对两组受试者进行了安全性、药代动力学(PK)和药效动力学(PD)的评估,其中男性和女性健康受试者年龄在18-55岁(n=77)。研究由二十(20)名受试者组成,被招募到2个组。在每个剂量水平/组中,总计10名受试者被随机分配以接受ATI-450的多种口服剂量(即,80mg或120mg)(n=8)或安慰剂(n=2),每天两次(BID),持续7天。在第7天早上施用最后的剂量。针对每个受试者,在第1天-给药前、第1天-给药后4小时和第1天-给药后12小时以及第7天-给药前、第7天-给药后4小时、第7天-给药后12小时和第7天-给药后24小时获得血液样品。
结果:图4A-4E的图表示出从用10mg、30mg、50mg、80mg和120mg的ATI-450给药的受试者采集的第7天的血液样品的LPS刺激的pHSP27(图4A,上部)、TNF-α(图4B,上部)、IL-1β(图4C,上部)、IL-8(图4D,上部)、IL-6(图4E,上部)的持续剂量依赖性抑制。图4A-4E还示出每天两次施用10mg、30mg、50mg、80mg和120mg的受试者中pHSP27(图4A,下部)、TNF-α(图4B,下部)、IL-1β(图4C,下部)、IL-8(图4D,下部)、IL-6(图4E,下部)的平均(±SEM)水平,比较了第1天给药前的数值(设定为100%)与第7天给药后4小时(大约C
较高剂量的ATI-450不会以与促炎性细胞因子相同的程度抑制调节性抗炎细胞因子IL-10。图5是示出从用安慰剂或80mg或120mg的ATI-450给药的受试者采集的血液样品中,体外刺激的IL-10与TNF-α和IL-1β的差分调控的图表。在对促炎性细胞因子(TNF-α和IL-1β)生成接近最大抑制的药物剂量下,IL-10仅被调控了25-30%。
ATI-450有效地抑制体外IL-1β诱导的促炎性细胞因子TNF-α、IL-6和IL-8(无论通过LPS还是IL-1β刺激)。图6描绘了比较ATI-450调控的LPS和IL-1β刺激的TNF-α(最左边)、IL-6(中间)和IL-8(最右边)产生的三个图表。
图7A-7C和下文表8概括了80mg和120mg BID给药后ATI-450的药代动力学数据。图7A描绘了BID给药第1天(上部)和第7天(中间和下部)后ATI-450的平均血药浓度-时间曲线的图表,半对数标度。BID,每天两次。图7B示出了以120mg BID给药的ATI-450的平均(±标准偏差)血药浓度-时间曲线。图7C描绘了示出120mg BID给药后第1天(左)和第7天(右)单独的受试者中的ATI-450的血药浓度-时间曲线的图表。
下文表11中提供了ATI-450 80mg和120mg BID给药的平均药代动力学参数的概述。
针对50mg BID组的对应的参数被包含用于对比。
表12和图4A-4E中概述了相对于在五个MAD组中的ATI-450剂量的分析物浓度变化的定量。
表12
表12中的数据概括了相对于5个MAD剂量组(10mg、30mg、50mg、80mg和120mg)中的C
ATI-450表现出靶生物标志物pHSP27的浓度依赖性和剂量依赖性调控以及分析的四种细胞因子(TNF-α、IL-1β、IL-6和IL-8)的产生的抑制。针对pHSP27、TNF-α、 IL-1β和IL-8的ATI-450降低的浓度响应参数是相当的,而针对IL-6的参数更高。在50mg BID剂量下,针对pHSP27的ATI-450降低以及针对TNF-α、IL-1 β和IL-8的产生的ATI-450抑制,给药7天后C
较高的给药方案(即80mg和120mg BID)耐受性良好,而没有报告严重的副作用。下文表13中提供了安全性数据的概述。
表13.施用80mg BID或120mg BID的患者中的初步安全性数据
#仅第1天或第2天
+7例通过药物消退
*在停药后
实施例3:ATI-450的13周口服施用的非临床安全性评估
研究设计。在该研究中,在大鼠和小型猪中测试了ATI-450的13周口服施用。大鼠被施用0mg/kg/天(溶媒)、3mg/kg/天、10mg/kg/天和30mg/kg/天,并且小型猪被施用0mg/kg/天(溶媒)10mg/kg/天、30mg/kg/天和60mg/kg/天。
结果:针对大鼠,未观察到的有害效应的水平(NOAEL)为30mg/kg/天,并且针对小型猪为60mg/kg/天。未观察到与研究药物相关的死亡。
大鼠无不良发现:在中高剂量下,在数只动物的鼻区周围观察到最小的溃疡形成/混合细胞炎症。包含对照动物在内,所有剂量下的肌细胞均出现轻度至中度退化。然而,观察到在28天的非给药期后完全恢复。
小型猪无不良发现:在≥30mg/kg/天下临床观察。在≥10mg/kg/天下的雄性和60mg/kg/天的雌性中观察到中性粒细胞、淋巴细胞和血小板计数略有增加,同时红细胞量减少。
表14.相比于给予每天两次口服剂量50mg、80mg和120mg的研究药物6.5天的健康人类受试者中的初步平均全身性暴露(C
实施例4:用于类风湿性关节炎的治疗的ATI-450的口服组合物
进行2a期、随机、研究者和患者双盲、申办者非盲、平行组、安慰剂对照研究,以研究患有中度至重度RA的患者中ATI-450加MTX与单独MTX的安全性、耐受性、PK和PD。
招募大约19名患者,预计至少15名患者将完成12周的治疗。研究由长达28天的筛查期、12周的治疗期和4周的随访期组成。持续直到患者的最终随访评估的针对患者的研究总持续时间为20周。
主要目标是评价ATI-450加MTX在患有中度至重度类风湿性关节炎(RA)的患者中的安全性和耐受性。研究的次要目标是评估(i)ATI-450加MTX在患有中度至重度RA的患者中的PD曲线,以及(ii)ATI-450在同时接受MTX的患有中度至重度RA的患者中的药代动力学。除了主要和次要目标外,还评估了ATI-450加MTX在患有中度至重度RA的患者中的PD曲线。
基于下列标准评估患者资格:(i)成人起病型RA的诊断(ACR/EULAR分类标准);(ii)DAS28-CRP≥3.2限定为中度至高度疾病活动度;(iii)通过至少4/28关节压痛和4/28肿胀关节限定的中度至重度活动性RA;(iv)筛查时hsCRP≥5mg/L;(v)限定为手腕MRI(使用RAMRIS)得分为1或更高的明确的关节内滑膜炎或骨炎;(vi)筛查访视前至少4周的稳定MTX剂量(限定为每周7.5mg至25mg)。在基线处确认资格的患者以3:1的比例随机分配以接受ATI-450片剂(50mg每天两次[BID])加MTX,或匹配的安慰剂片剂加MTX。研究药物口服施用12周。在研究期间,患者需要保持稳定剂量的MTX(7.5mg至25mg/周)和稳定剂量的叶酸或亚叶酸(≥5mg/周)。
患者在第7天、第14天、第28天、第42天、第56天和第84天(±1天)参加临床访视,进行安全性、疗效、PK和PD评估。在每个研究访视日在诊所施用研究药物的早晨剂量。
在完成4周的治疗(第28天)时,将审查每位患者的安全数据(例如,AE、实验室值、生命体征和ECG)以确保患者耐受治疗方案并被视为适合继续接下来的8周的治疗。
在第84天(第12周),患者将完成研究结束评估。最后一次剂量的研究药物后30天(+7)将进行安全性随访。
疗效评估和结果
下文表15中提供了患者人口统计的概述。
表15.患者人口统计
总体而言,患者群体表现出高的疾病活动度(如通过针对治疗组(5.65)和安慰剂组(6.0)的中值28个关节的疾病活动度评分(DAS-28)所指示的)(参见Smolen et al.,“Validity and reliability of the twenty-eight-joint count for the assessmentof rheumatoid arthritis activity.”Arthritis Rheum 38(1):38-43(1995),其特此通过引用被整体并入)。此外,患者群体表现出范围广的病程(0.3-34年),其中尽管病史较长且有多种治疗选择,但许多患者具有高hsCRP。这表明了一组相对难治的患者。
28个关节的疾病活动度评分。在研究开始前和研究第1天、第7天、第14天、第28天、第42天、第56天、第84天和后续访视时,进行通过疾病活动度评分的RA评估(根据Smolen etal.,“Validity and reliability of the twenty-eight-joint count for theassessment of rheumatoid arthritis activity.”Arthritis Rheum 38(1):38-43(1995)(其特此通过引用被整体并入)修改为包含28个关节数(DAS28))。DAS28由下列变量的综合评分组成:关节压痛数、肿胀关节数、CRP和患者的疾病活动度评分的整体评估(Prevoo et al.,“Modified Disease Activity Scores that include twenty-eightjoints,”Arthritis&Rheumatism 38(1):44-48(1995),其特此通过引用被整体并入)。
下列等式用于计算DAS28(CRP):
DAS28(CRP)=0.56√TJC28+0.28√SJC28+0.36·ln(CRP+1)+0.014×(患者的疾病活动度的整体评估)+0.96,其中:
·TJC28=28个关节中关节压痛的数量
·SJC28=28个关节中肿胀关节的数量
·CRP=C-反应性蛋白
·患者记录的100mm可视模拟标度(VAS)的患者的疾病活动度的整体评估
DAS28(CRP)疾病活动度测量的解释是基于0到9.4的标度,其中:<2.6被认为是缓解,≥2.6至<3.2被认为是低度/最低,≥3.2至≤5.1被认为是中度,并且>5.1被认为是高度/重度(Anderson et al.,“Rheumatoid arthritis disease activity measures:American College of Rheumatology recommendations for use in clinicalpractice,”Arthritis Care Res(Hoboken)64(5):640-7(2012),其特此通过引用被整体并入)。
图8和图9中提供该评估的结果。图8是示出截至评估日期针对所有患者的自基线的DAS28-CRP的中值变化的图表。该数据示出了快速且持续发生的疗效,这与其他p38抑制剂明显不同。在随访患者(即第112天)中观察到的DAS28-CRP增加是意料之中的,因为治疗已停止并且表明药物特异性作用。总体而言,在所有患者中观察到的直至第84天的DAS28-CRP的持续减少表明疾病活动度的临床相关的减少,而没有任何快速耐受性的证据。图9是示出在第1天、第7天、第24天、第28天、第42天、第56天和第84天的每一天具有低于2.6和低于3.2的DAS28-CRP的患者百分比的图表。
ACR20/50/70。ACR20(50/70)响应标准是指示肿胀和压痛关节数量(66/68关节数)中至少20%(50%/70%)的改善且下列ACR核心测量值中的至少3项或更多项的至少20%(50%/70%)的改善的终点:
为了满足ACR20标准,患者必须具有下列ACR核心集中至少20%的改善:
·肿胀关节数(66个关节数)
·关节压痛数(68个关节数)
下列5个测量的至少3个中至少20%的改善:
·患者的疾病活动度的整体评估(VAS)
·患者的关节炎疼痛的评估(VAS)
·患者的身体功能/健康评估问卷–功能障碍指数(HAQ-DI)的评估
·医师的疾病活动度整体评估(VAS)
·通过hsCRP测量的急性期反应物
ACR50=肿胀关节数和关节压痛数的50%的改善和5项测量中的至少3项的至少50%的改善。
ACR70=肿胀关节数和关节压痛数的70%的改善和5项测量中的至少3项的至少70%的改善。
该评估的结果在图11-12中示出。图11的图表示出了肿胀关节数自基线的中值变化,并且图12的图表示出了截至评估日期所有患者的关节压痛数自基线的中值变化。在整个治疗期(84天)的肿胀关节数和关节压痛数的持续减少显示疾病活动度的临床上显著的减少,而没有任何快速耐受性的证据。在第112天随访患者中观察到的肿胀关节数和关节压痛数的增加(或自基线的变化的减少)是意料之中的,因为治疗已经停止。
图14的图表示出了在治疗的第1天、第28天、第56天、第84天和在治疗后第112天的响应者分析。如上所述,ACR值代表每组中满足如上所述的ACR20、ACR50和ACR70标准的患者的比例。
RAMRIS和CARLOS手部MRI评估。手腕MRI RAMRIS是在RA试验中被验证和接受良好的客观影像测量。RAMRIS具有针对滑膜炎(得分0-3)、骨炎(0至3)和骨侵蚀(0至10)的分项评分。CARLOS是已经成功用于4个RA的随机分配的对照临床试验的9分制的软骨损失标度(Perterfy et al.,“Monitoring cartilage loss in the hands and wrists inrheumatoid arthritis with magnetic resonance imaging in a multi-centerclinical trial:IMPRESS(NCT00425932),”Arthritis Research&Therapy,15:R44(2013),其特此通过引用被整体并入)。RAMRIS滑膜炎评分随着有效的RA疗法而迅速变化,并且即使在小规模组中也可以检测到变化。在随机分配的对照试验中,已经观察到每组大约30名患者在有效治疗开始后2周内滑膜炎评分在统计上显著减少(Beals et al.“Magneticresonance imaging of the hand and wrist in a randomized,double-blind,multicenter,placebo-controlled trial of infliximab for rheumatoid arthritis:Comparison of dynamic contrast enhanced assessments with semi-quantitativescoring,”PLoS ONE 12(12):e0187297(2017),其特此通过引用被整体并入)。
这些研究还显示,仅在12周内证明关节损伤(RAMRIS骨侵蚀和CARLOS软骨损失)的进展的显著抑制。手腕MRI扫描将由研究机构(或研究机构指定的合适的当地机构)完成,并且将被集中读取。将在一份来自MRI中心实验室的单独的现场指导手册中提供概述扫描收集参数提交要求的说明。
图15-17以及下文表格中提供了目前研究的手腕MRI RAMRIS评估的结果。图15是示出通过CARLOS(软骨损失)、侵蚀、骨炎、滑膜炎评估的对ATI-450治疗有响应的患者百分比的图表。图15的图表是示出通过治疗和手部的这些终点的百分比响应性。如果观察到任何一只手≥1分的改善,则认为患者是有响应的。
图16是示出安慰剂和ATI-450(50mg BID)治疗的患者的双手的CARLOS、侵蚀、骨炎和滑膜炎在第84天的基线变化的图表。数据代表了通过手腕MRI RAMRIS评估的双手的平均值。图17是示出安慰剂患者和ATI-450(50mg BID)患者中每只手的CARLOS、侵蚀、骨炎和滑膜炎在第84天的基线变化的类似的图表。菱形代表平均值。
下文表16和17提供了基线处和用ATI-450(50mg BID)或安慰剂治疗12周后的CARLOS、侵蚀、骨炎和滑膜炎的MRI RAMIS评估得分。该分析结果共同表明,ATI 450治疗防止关节破坏的进展,并且提供滑膜炎的显著改善。
表16.针对最重度手的手腕MRI RAMIS评估
表17.针对其他手的手腕MRI RAMIS评估
患者的疾病活动度可视模拟标度的整体评估。患者的疾病活动度VAS的整体评估被用于测量患者的疾病活动度的整体评估。VAS的长度为100mm,其中线的左端为“0”(完全没有活动度,非常好),并且线的右端为“100”(可以想象的最差的活动度,非常差)。参与者在横线上画一条竖线,以指示他们的疾病活动度水平。研究人员从线的左端测量到参与者标记的线,并且以mm记录这个长度(0到100)。图13A的图表中示出了治疗组(n=13)和安慰剂组(n=2)在研究第84天的该评估结果。
患者的关节炎疼痛可视模拟标度的评估。患者的关节炎疼痛VAS评估被用于测量患者的关节炎疼痛水平。VAS的长度为100mm,其中线的左端为“0”(完全没有疼痛,无疼痛),并且线的右端为“100”(可以想象的最严重的疼痛,可能的最严重的疼痛)。参与者在横线上画一条竖线,以指示他们当天经历的疼痛程度。研究人员从线的左端测量到参与者标记的线,并且以mm记录这个长度(0到100)。图13B的图表中示出了治疗组(n=13)和安慰剂组(n=2)在研究第84天的该评估结果。
患者的身体功能/健康评估问卷-功能障碍指数的评估。患者的身体功能/HAQ-DI评估被用于评估参与者的身体功能或功能障碍。HAQ-DI询问一个人在8个功能领域(穿衣、起床、吃饭、走路、卫生、伸手、抓握和活动[差事和家务])完成任务的困难程度。每个功能领域的回答被评分为从“0”(指示没有困难)到“3”(指示无法完成该领域的任务)。研究人员不应该向参与者说明任何问题。图13C中示出了治疗组(n=13)和安慰剂组(n=2)到研究第84天的该评估结果。
医师完成的疗效问卷。当这个评估在研究的第1天、第28天、第56天、第84天和随访时完成,其将是访视时做的第一批评估之一,并且必须在给药前完成。医师的疾病活动度VAS的整体评估是由研究者或指定人员完成的测量。VAS的长度为100mm,其中线的左端为“0”(完全无活动度,无关节炎活动度),并且线的另一端为“100”(可以想象的最严重的活动度,非常活跃的关节炎)。研究者或指定人员在横线上画一条竖线,以指示患者的疾病活动度。研究人员从线的左端测量到研究者(或指定人员)的线,并且以mm记录这个长度(0到100mm)。图13D的图表中示出了治疗组(n=13)和安慰剂组(n=2)在研究第84天的该评估结果。
高敏性C反应蛋白(hsCRP)。在研究开始前,以及在研究的第1天、第7天、第14天、第28天、第42天、第56天、第84天和随访时(第112天),收集用于评价hsCRP的血液样品。样品被运送到中心实验室。在单独的实验室手册中提供了针对hsCRP的样品的收集、处理、储存和运输的具体说明。
图10中提供了该评估的结果。图10的图表示出了针对所有患者第84天以及针对一些患者第112天(即治疗停止后)自基线的hsCRP的中值变化。在随访患者中观察到hsCRP的轻微增加是意料之中的,因为治疗已经停止,并且指示了药物特异性治疗效果。总的而言,在所有患者中观察到的第84天的自基线的hsCRP的快速且持续的降低,指示疾病活动度的临床相关的减少,而没有任何快速耐受性的证据。
药效动力学。在研究日期1、7、14、28、42、56、84和随访时,收集大约10mL的静脉血液样品,用于测量体外刺激的细胞因子水平(例如,TNF-α、IL-1β、IL-6、IL-8、IFNγ、IL-17、IL-18、IL-10、IL-1RA和IL-1α)以及磷蛋白PD参数。对于内源性细胞因子水平(如TNF-α、IL-1β、IL-6、IL-8、IFNγ、IL-17、IL-18、IL-10、IL-1α和IL-1RA),将在每个确定的时间点抽取1mL的血清。
p38抑制剂化合物作为一个类别,已经表明在RA研究中缺乏持久的疗效。据认为,用p38抑制剂给药超过12周会导致随着时间的途径的重新编程(reprogramming)和抑制剂诱导的p38独立的细胞因子的产生。相反,如图18A和18B的图表中所示,RA患者中施用ATI-450持续12周导致促炎性细胞因子的持续抑制。在用LPS体外刺激后,对RA患者给药第1天和给药第84天的血液进行分析,并且评价ATI-450与安慰剂相比抑制TNF-α、IL-1β、IL-6和IL-8的能力。如通过图18A和18B中示出的每个细胞因子组的最后两个柱所证明的,给药84天后每个细胞因子的ATI-450抑制与给药1天后每个细胞因子的ATI-450抑制非常相似。该数据显示使用MK2抑制剂ATI-450没有观察到抑制剂依赖性的途径重新编程和快速耐受性,这与已经与p38抑制剂进行的假设相反。
在给药ATI-450持续12周的过程中,对来自RA患者的血浆中的内源性细胞因子进行评价。图19A和19B中分别提供了TNF-α和IL-1RA的数据。这些数据表示为相对于药物治疗的患者和安慰剂治疗的患者两者的给药前的中值%变化。在第84天的谷值药物水平对样品进行评价,并且最后点是第84天最后一次剂量后的C
图20A-20D提供了针对在给药ATI-450持续12周的过程中,ATI-450对来自RA患者的血浆中的内源性细胞因子水平的影响的更广阔的视角。该图中示出了来自13-plex测试的三种促炎性细胞因子(TNF-α(图20A)、IL-6(图20C)、IL-8(图20B))连同趋化因子MIP1β(图20D)的血浆水平。数据表示为相对于用药物治疗的患者给药前前的中值%变化。在给药4周、8周和12周后以及在第84天最后剂量后4小时,评价样品的谷值药物水平。横线(––·––)描述了来自正常健康志愿者受试者的血浆中的细胞因子/趋化因子水平。给药4周后,TNF-α水平显著降低,并且这种降低在整个12周内一直保持,并接近于健康志愿者中存在的细胞因子的水平。类似地,IL-6、IL-8和MIP1β水平在第4周时降低,并且这种降低在研究的12周内一直保持。该分析进一步证明了以50mg BID给药ATI-450在RA患者中12周内的持久的抗炎活性。
安全性。总体而言,研究药物耐受性非常好,没有报告严重或重度不良事件。只有一名受试者因调查心悸和升高的CPK而退出。下文表18概括了报告的事件。
表18.研究受试者报告的不良事件
结果概述:
ATI-450通常耐受性良好。治疗组中的一名受试者由于心悸(与研究药物无关)和升高的CPK(通过现场研究者确定与药物有关)而退出。安慰剂组中的一名受试者因需要治疗肌肉拉伤的被禁止的药物而退出。没有报告严重不良事件,并且所有的不良事件都是轻度到中度的。最常见的不良事件(至少有两名受试者报告)是尿路感染和血脂升高。
在这项试验中,ATI-450显示了持久的临床活性(如由DAS28-CRP的明显和持续降低以及超过12周的ACR20/50/70响应的评价限定的)。在第12周,DAS28-CRP自基线的平均变化为治疗组中2.0的降低和安慰剂组中0.35的增加。第12周时具有DAS28-CRP评分≤至3.2(低疾病活动度或缓解)的受试者比例分别为在治疗组的15名受试者的40%和安慰剂组的2名受试者中的0%,其中具DAS28-CRP评分<2.6(缓解)的受试者比例分别为治疗组中的20%和安慰剂组中的0%。
在第12周观察到ACR20/50/70分别为治疗组中的15名受试者的60%、33%和20%,并且在安慰剂组中的2名受试者为0%。在试验的12周内,治疗组中的hsCRP自基线的中值减低>40%。来自从治疗组采集的血液样品的体外刺激的细胞因子的中期分析(11个治疗,2个安慰剂)显示出,在12周的给药时间段,TNF-α、IL-1β、IL-6和IL-8的明显且持久的抑制。类似地,内源性细胞因子的分析也表明,在12周的时间段,治疗组中的TNF-α、IL-6、IL-8和MIP1β的中值浓度的明显且持续的抑制。
ATI-450通常耐受性良好。没有报告严重不良事件,并且所有的不良事件都是轻度到中度的。最常见的不良事件(各自在2名受试者中报告)是尿路感染(UTI)、血脂升高和室性期前收缩,除了一例UTI外,所有这些都被确定为与治疗无关。两名受试者退出试验,一名在治疗组中,并且一名在安慰剂组中。
实施例5:在患有中度至重度RA的患者中研究多次剂量的ATI-450加MTX与仅MTX的疗效、安全性、耐受性、药代动力学和药效动力学的2B期、随机、双盲、平行组、安慰剂对照研究。
这是在患有中度至重度RA的患者(其已经对单独的甲氨蝶呤具有不充分的响应)中研究多次剂量的ATI-450加MTX与仅MTX的疗效、安全性、耐受性、药代动力学(PK)和药效动力学(PD)的2b期、随机、双盲、平行组、安慰剂对照研究。
在基线处确认资格的受试者将按1:1:1:1的比例随机分配以接受ATI-450片剂(20mg、50mg和80mg,每天两次[BID])加MTX,或匹配的安慰剂片剂BID加MTX。患者随机分配到治疗组将由两个独立的因素进行分层。第一个分层因素是筛查时类风湿因子(RF)和/或抗环瓜氨酸肽(CCP)检测阳性的患者相比于这两种抗体检测非阳性的患者。第二个分层因素是已经经历生物制剂(biologic)RA治疗或JAK RA治疗的那些患者相比于没有这样的经历的那些患者。招募人数将受到限制,使得不超过25%的研究群体将对RF或抗CPP或两者均测试阴性。同样,招募人数也将受到限制,使得不超过25%的群体具有生物制剂RA治疗或JAK RA治疗的先前经历。研究药物将被口服施用12周。在研究期间,患者将被要求保持稳定剂量的MTX(筛查前4周为15mg/周至25mg/周,或对于具有不耐受记录的患者,筛查前4周为10mg/周)。
患者将在第1天、第8天、第15天、第29天、第43天、第57天和第85天(±1天)参加门诊访视,进行安全性、疗效、谷值PK和PD评估。在每个研究访视日,将在诊所中施用研究药物的早晨剂量。
在第84天(第12周),患者将完成研究结束评估。最后一次剂量的研究药物后30天(+7)将进行安全性随访。
受试者的纳入标准包含:1)如根据2010年美国风湿病学会/欧洲抗风湿联盟的分类标准(the 2010American College of Rheumatology/European League AgainstRheumatism classification)限定的起病型类风湿性关节炎(RA)的诊断;2)使用28个关节C反应蛋白(CRP)(DAS28-CRP)的疾病活动度评分大于3.2(被限定为中度或高度疾病活动度);3)具有由最低疾病活动度标准限定的中度至重度RA(在筛查和基线访视时6个或更多的肿胀关节;在筛查和基线访视时6个或更多的压痛关节;以及在筛查访视时高敏性C反应蛋白(hsCRP)水平大于正常上限(ULN)或RF和/或抗环瓜氨酸肽(CCP)均为阳性);4)筛查前至少12周服用MTX,且MTX剂量稳定(为15mg/周至25mg/周,或针对具有不耐受记录的患者为10m/周);5)18-75(含)岁男性或非孕期、非哺乳期女性;和6)筛查实验室评价结果落在中心实验室参考范围的正常范围内。
疗效分析
所有疗效概述将在意向治疗(ITT)和符合方案(PP)群体中进行。
本研究评估的主要终点是在12周时达到美国风湿病学会(ACR)20的受试者比例。
本研究中待评估的次要终点包含:1)第12周时具有ACR 50/70的受试者的比例;2)随着时间的推移,具有ACR 20/50/70的受试者的比例;3)随着时间的推移,使用28个关节C反应蛋白(CRP)(DAS28-CRP)的疾病活动度评分自基线的平均变化;4)随着时间的推移且在第12周实现DAS 28缓解(得分<2.6)的患者的比例;5)临床疾病活动度指数(CDAI)随时间自基线的平均变化;6)高敏性C反应蛋白(hsCRP)水平随时间自基线的中值百分比变化;7)随时间的健康评估问卷功能障碍指数(HAQ-DI);8)在诊所访视时的ATI-450浓度(对所有患者的谷值和剂量后2小时采样;用于PK子研究中的患者的非房室PK分析的附加连续采样);9)内源性细胞因子水平(例如,TNF-α、IL-1β、IL-6、IL-9、IFNγ、IL-17、IL-18、IL-10、IL-1α和IL-1RA)自基线的平均变化。
ACR 20/50/70响应率以及具有DAS28-CRP<2.6的患者的比例将使用逻辑回归进行分析。缺少ACR值或缺少DAS28-CRP值的患者将被视为非响应者。具有负面的间歇性事件(如需要抢救)的患者也将被视为ACR20/50/70和DAS28-CRP<2.6的非响应者。
将使用混合模型重复测量法随时间(对于所有预定时间点)分析DAS28-CRP、CDAI、66/68肿胀关节数/关节压痛数、患者的疾病活动度的整体评估、患者的关节炎疼痛的评估和医师的疾病活动度的整体评估,以及其自基线的相应变化。针对治疗效果,提供基于模型的点估计和95%的置信区间。针对治疗与安慰剂的差异,提供点估计值、95%置信区间和p-值。
将使用连续的统计汇总测量概括随时间的推移的hsCRP水平、hsCRP自基线的变化和hsCRP自基线的百分比变化。hsCRP的持续治疗效果的测定将基于PP群体中hsCRP随时间自基线中值百分比变化。将分别对ACR 20/50/70响应率、DAS28-CRP的自基线的变化、66/68肿胀关节数/关节压痛数、患者的疾病活动度的整体评估、患者的关节炎疼痛的评估、医师的疾病活动度的整体评估以及hsCRP的自基线的百分比变化进行探索性剂量响应建模。
治疗的安全性将基于1)不良事件(AE)的数量和频率,2)生命体征,3)ECG,4)血清化学,5)血液学,6)尿液分析,和7)凝血功能。
将在每个评估日测量ATI-450及其主要循环代谢物(CDD-2164)的血药浓度。
实施例6:在患有中度至重度活动性银屑病关节炎的患者中研究多次剂量的ATI-450与安慰剂的疗效、安全性、耐受性、药代动力学和药效动力学的2B期、随机、双盲、平行组、安慰剂对照研究。
这是在患有中度至重度活动性银屑病关节炎(PSoA)的受试者(其已经对单独的甲氨蝶呤具有不充分的响应)中研究多次剂量的ATI-450与安慰剂的安全性、耐受性、PK和PD的2b期、随机、双盲、平行组、安慰剂对照研究。
满足资格标准的受试者将接受50mg的ATI-450每天两次或匹配的安慰剂片每天两次。治疗将持续12周,同时在研究前和研究期间保持稳定剂量的MTX(每周15mg至25mg)。PK和PD评估发生在第1天、第8天、第15天、第29天、第43天、第57天和85天。受试者组基于先前使用的DMARD(疾病修饰抗风湿药物(Disease Modifying Anti-Rheumatic Drug))进行分层。
受试者的纳入标准包含:1)在筛查访视前至少6个月出现症状并且符合PsA的分类标准(CASPAR标准)的PSoA的临床诊断;2)限定为在筛查和基线访视时至少3个压痛关节和至少3个肿胀关节的基线处有活动度的疾病;3)在筛查时存在下列情况之一:(a)如通过中心影像学审查测定的X射线上至少有一处侵蚀;或(b)hsCRP水平高于实验室定义的ULN;4)活动性斑块状银屑病的诊断或有记录的斑块状银屑病病史;5)对以最大耐受剂量的至少1种非生物制剂DMARD(MTX、柳氮磺吡啶、来氟米特、环孢菌素、阿普斯特、布西拉明或艾拉莫德)的先前或当前治疗反应不充分(最少12周持续时间后治疗后缺乏疗效),或对DMARD不耐受或有禁忌;和6)加入研究时正在同时用非生物制剂DMARD治疗的受试者必须使用少于或等于2种非生物制剂DMARD(MTX和来氟米特的组合除外)。允许使用下列DMARD:MTX、柳氮磺吡啶、来氟米特、阿普斯特、羟氯喹、布西拉明和艾拉莫德。治疗必须在基线访视前至少4周内以稳定的剂量持续进行至少12周。最后,筛查实验室评估(血液学、化学、凝血和尿液分析)必须落入中心实验室参考范围的正常范围内。
受试者的排除标准包含:1)当前治疗使用超过两种的非生物制剂DMARD;使用除甲氨蝶呤、柳氮磺吡啶、来氟米特、阿普斯特、羟氯喹、布西拉明和艾拉莫德以外的DMARD;或使用甲氨蝶呤与来氟米特的组合。2)纤维肌痛病史、17岁前发病的任何关节炎、或目前诊断为PsA以外的炎性关节疾病(包含(但不限于)类风湿性关节炎、痛风、重叠结缔组织病、硬皮病、多肌炎、皮肌炎、系统性红斑狼疮)。如果有PsA诊断变化或进行PsA的附加诊断的改变的证明文件,则反应性关节炎或轴性脊柱关节炎(包含强直性脊柱炎和非放射性轴性脊柱关节炎)的先前病史是允许的。如果有PsA的诊断变化的证明文件或纤维肌痛的诊断错误的证明文件,则纤维肌痛的先前病史是允许的。3)目前有除PsA以外的可能影响PSoA的病程或评估的急性或慢性免疫炎性疾病。4)可能使患者在研究过程中处于增加风险或影响结果的解释的未受控制的非免疫炎性疾病。5)有活动性或潜伏性结核病史或证据。6)酗酒/当前酒精中毒、酒精性肝病或其他慢性肝病病史;积极用抗生素治疗感染。7)HIV或乙型肝炎或丙型肝炎阳性。8)白细胞计数低于3.0×10
疗效分析:
本研究的主要目标是评估多次剂量的ATI-450在患有中度至重度PSoA的患者中的疗效。该目标将通过确定ITT群体中在第12周达到ACR(美国风湿病学会)20的受试者的比例来评估。响应限定为与基线相比,关节压痛数(TJC)、肿胀关节数(SJC)和5项剩余ACR核心集测量(患者的疼痛的评估、患者的疾病活动度的整体评估(PtGA);医生的疾病活动度的整体评估(PhGA)、健康评估问卷-功能障碍指数(HAQ-DI)以及高敏性C反应蛋白(hsCRP))中的至少3项的至少20%的降低(改善)。
本研究的次要目标是评估患有中度至重度PSoA的患者中ATI-450的药效动力学(PD)。该目标将通过确定下列进行评估:1)第12周具有ACR 50/70的患者的比例;2)随着时间的推移,具有ACR 20/50/70的患者的比例;3)银屑病静态研究者整体评估(sIGA)达到0或1且自基线至少改善2分的患者的比例;4)银屑病面积严重程度指数(PASI)75的响应(对于基线的具有至少有3%的BSA银屑病的参与者);5)实现最低疾病活动度(MDA)的参与者的比例,基于受试者满足7项结果测量中的5项来确定:TJC≤1;SJC≤1;PASI≤1或BSA-PS≤3%;患者的疼痛评估NRS≤1.5;PtGA-疾病活动度NRS≤2.0;HAQ-DI得分≤0.5;以及皮肤压痛点≤1;5)12周内HAQ-DI自基线的变化;6)短表(SF)-36躯体健康状况(Physical ComponentSummary)(PCS)自基线的变化;7)FACIT-疲劳问卷自基线的变化;8)银屑病症状自我评估(SAPS)问卷自基线的变化;以及9)安全性(不良反应、ECG、实验室值)。本研究的另一个次要目标是评估正在同时接受MTX的患有中度至重度PSoA的患者中的ATI-450的药代动力学(PK)。该目标将通过确定门诊访视时的ATI-450浓度(谷值PK分析)来评估。
本研究的一个探索性目标是评估患有中度至重度PSoA的患者中ATI-450加MTX的PD。该目标将通过确定内源性细胞因子水平(例如,肿瘤坏死因子-α[TNF-α]、白介素[IL]-1β、IL-6、IL-8、IFNγ、IL 17、IL-18、IL-10、IL-1α和IL-1RA)自基线的平均变化进行评估。
全部疗效概述将在ITT和PP群体两者中进行。
主要疗效分析将是在ITT群体中在第12周达到ACR20的患者百分比的治疗对比。该分析将在逻辑回归模型的背景下进行。治疗组将作为类别变量进入模型,并且基线严重程度将作为协变量包含在内。假设检验和相应的p-值将是具有0.05的α水平的单边检验。除p-值之外,还将提供针对比值比(odds ratio)的点估计值和90%的置信区间。
所有次要终点的治疗对比将使用如主要分析那样的逻辑回归进行。这将在ITT和PP群体两者中进行。
所有连续次要终点的治疗对比将使用混合模型重复测量(MMRM)进行。该模型将包含针对治疗组、时间、时间与治疗的交互作用以及基线协变量(如适用)的因素。受试者标识符将以允许给定的受试者内随时间的观察结果被视为重复测量的方式被包含在模型中。所有的治疗对比将基于具有0.05的α水平的单边假设检验。除p-值之外,还将提供针对最小平方平均数差异的点估计和90%的置信区间。这些分析将同时针对ITT和PP群体进行。
治疗的安全性将基于:1)AE的发生率和严重程度、生命体征、ECG测量、临床实验室值(血液学、血清化学、凝血、尿液分析)。
实施例7:在患有中度至重度化脓性汗腺炎(HS)的患者中研究ATI-450与安慰剂的疗效、安全性、耐受性、药代动力学和药效动力学的2a期、随机、双盲、平行组、安慰剂对照剂量范围研究。
这是在已经对口服抗生素的充分试验响应不充分或不耐受的患有中度至重度HS的受试者中研究ATI-450 50mg BID与安慰剂的疗效、安全性、耐受性、PK和PD的2b期、随机、双盲、平行组、安慰剂对照研究。满足资格标准的受试者将接受50mg或80mg ATI-450(口服片剂)每天两次、或匹配的安慰剂片剂每天两次。PK和PD评估发生在第1天、第8天、第15天、第29天、第43天、第57天和第85天。
纳入标准包含:1)受试者在基线访视前至少一年被诊断患有中度至重度化脓性汗腺炎(HS);2)受试者表现出至少在两个不同的解剖区存在HS病变(Hurley II期或III期);3)基线访视时引流瘘管数<=20;4)基线访视时总脓肿和结节数(AN计数)>=5;5)受试者同意每天使用杀菌剂清洗其HS病变部位;6)受试者必须有对口服抗生素治疗HS的充分试验响应不充分或不耐受的历史。
排除标准包含有HS以外的可能干扰HS的评估的活动性皮肤疾病病史;可能使患者在研究过程中处于增加风险或影响结果的解释的未受控制的非免疫炎性疾病;有活动性或潜伏性结核病史或证据;酗酒/当前酒精中毒、酒精性肝病或其他慢性肝病病史;积极用抗生素治疗感染;HIV或乙型肝炎或丙型肝炎阳性;白细胞计数低于3.0×103个细胞/mm
疗效分析
本研究的主要目标是评估患有中度至重度HS的患者中多次剂量的ATI-450的疗效。该目标将通过确定在第12周达到化脓性汗腺炎临床响应(HiSCR)的患者的百分比来评估(HiSCR被限定为总脓肿和炎性结节(AN)数自基线至少50%的降低,而没有脓肿或引流瘘管数增加)。
本研究的次要目标是评估患有中度至重度HS的患者中ATI-450的疗效和安全性。该目标将通过确定下列进行评估:1)在基线NRS30≥3的患者中,在第12周实现患者的皮肤疼痛的整体评估(PGA皮肤疼痛)的数字分级标度(NRS30)自基线降低至少30%的患者的百分比(NRS30是基于24小时回忆期最严重的皮肤疼痛(每天最大的疼痛)进行评价的);2)在基线NRS30≥3的患者中,在第12周实现PGA皮肤疼痛的NRS30自基线降低至少30%的患者的百分比;3)在12周的治疗期内,AN计数增加至少25%,其中相对于基线最少增加2的患者的百分比;4)12周内皮肤病学生活质量指数(DLQI)自基线的变化(DLQI是用于评估HS疾病症状和治疗对生活质量(QoL)的影响的10个项目的有效问卷。其由10个问题组成,评价皮肤病对参与者前一周的QoL的不同方面的影响,包含症状和感受、日常活动、休闲、工作或学习、个人关系和治疗的副作用);5)在12周内,基于化脓性汗腺炎症状评估(HSSA)评估的与HS相关的肿胀自基线的变化(HSSA是9项患者报告结果(PRO)的问卷,开发以0至11分的NRS来评估HS的症状,其中0代表没有症状,并且10代表极端症状经历);6)12周内基于HSSA评估的与HS相关的气味自基线的变化;7)12周内基于HSSA评估的与HS相关的最严重排泄物自基线的变化;以及8)安全性(AE、SAE、实验室值、ECG)。本研究的另一个次要目标是评估患有HS的患者中ATI-450的药代动力学(PK)和药效动力学(PD)。该目标将通过确定门诊访视时的ATI-450浓度(谷值PK分析)来评估。
患有中度至重度HS的患者中ATI-450的药效动力学也将通过确定内源性细胞因子水平(如TNF-α、IL-1β、IL-6、IL-8、IFNγ、IL 17、IL-18、IL-10、IL-1α和IL-1RA)自基线的平均变化来评估。
所有疗效概述将在ITT和PP群体两者中进行。
主要疗效分析将是ITT群体中第12周达到HiSCR的患者的百分比的治疗对比。该分析将在逻辑回归模型的背景下进行。治疗组将作为类别变量进入模型,并且基线严重程度将作为协变量被包含在内。假设检验和相应的p-值将是具有0.05的α水平的单边检验。除p-值之外,还将提供针对比值比(odds ratio)的点估计值和90%的置信区间。
所有次要响应者终点(实现PGA皮肤疼痛的NRS30自基线至少30%降低的患者的百分比和在12周治疗期内经历AN计数增加至少25%且相对于基线最少增加2的患者的百分比)的治疗对比将使用如主要分析那样的逻辑回归进行。这将在ITT和PP群体两者中进行。
所有连续次要终点的治疗对比将使用混合模型重复测量(MMRM)进行。该模型将包含针对治疗组、时间、时间与治疗的交互作用以及基线协变量(如适用)的因素。受试者标识符将以允许给定的受试者内随时间的观察结果被视为重复测量的方式被包含在模型中。所有的治疗对比将基于具有0.05的α水平的单边假设检验。除p-值之外,还将提供针对最小平方平均数差异的点估计和90%的置信区间。这些分析将同时针对ITT和PP群体进行。
实施例8:在先前用抗IL-1疗法处理的患有冷凝比林相关的周期性综合征(CAPS)的患者中研究ATI-450用于维持缓解的安全性和疗效的2a期、开放标签、单组研究。
CAPS是由编码冷凝比林的NLRP3基因的功能增益突变引起的罕见的遗传性自身炎性疾病,冷凝比林是NLRP3炎症小体的组成部分。NLRP3炎症小体的失调导致白介素-1(IL-1)的过度产生,并因此出现CAPS中的炎性症状。目前的抗IL-1疗法已被证明能在CAPS患者中诱导快速和持续的疾病缓解,并且通常耐受性良好。
在临床前和临床研究中,ATI-450已被证明引起IL-1的明显抑制。目前用于治疗CAPS的抗IL-1疗法是静脉注射或皮下注射施用的生物制剂。ATI-450是口服施用的小分子药物,与目前可用的疗法相比,有可能具有类似的安全性和疗效,具有一些CAPS患者可能倾向的不同的施用途径。
进行本研究以测定患有CAPS的患者中ATI-450的安全性、耐受性、药代动力学(PK)、药效动力学(PD)和初步疗效。
本研究的主要目标是评估先前用抗IL-1疗法处理的患有CAPS的患者中ATI-450维持缓解的安全性和耐受性。该目标将通过调查不良事件(AE)和严重不良事件(SAE)的数量和百分比;实验室值、生命体征和心电图(ECG)自基线的平均变化进行评估。
本研究的次要目标是评估先前用抗IL-1疗法处理的患者中ATI-450维持缓解的疗效。为了评估该目标,将评估数个终点。这些终点包含:(i)确定随时间疾病缓解的参与者的比例。疾病缓解被限定为具有不存在或最低的医生整体评估(PGA)得分,并且高敏性C反应蛋白(hsCRP)和血清淀粉样蛋白A(SAA)值在正常范围内(≤10mg/L)或在基线值的百分之30以内。(ii)确定随时间临床缓解的参与者的比例。临床缓解被限定为具有不存在或最低的PGA评分。(iii)确定复发的时间;其中复发被限定为PGA标度的2分恶化。(iv)确定停用ATI-450后经历疾病症状重新出现的参与者的比例。重新出现被限定为至少连续2天每日关键症状评分(KSS)比基线高≥3分。KSS来源于患者管理的每日健康评估表(DHAF)。(v)确定在治疗期内,8周中的至少6周的平均KSS比基线高不超过2分的参与者的比例。(vi)确定PGA自基线的变化。(vii)确定KSS自基线的变化。(viii)确定hsCRP和SAA自基线的变化。
除上述外,本研究还将调查下列两个探索性目标:(i)评估ATI-450在患有CAPS的患者中的药效动力学(PD),和(ii)评估ATI-450在患有CAPS的患者中的药代动力学(PK)。这些目标将通过确定血清细胞因子IL-1β、IL-1α、IL-6、IL-18和TNF-α自基线的变化以及ATI-450的谷值浓度来评估。
这是在先前用抗IL-1疗法处理的患有CAPS的患者中研究ATI-450维持缓解的安全性、耐受性、疗效、PK和PD的2a期、开放标签、单组研究。计划有多达10名患者被招募到研究中。研究将由长达8周的筛查期、12周的治疗期和4周的安全性随访期组成。直至最后的安全性随访评估,留在研究中的患者的总研究时间将长达24周。
在进行任何研究程序之前,研究者将从患者处获得签署的知情同意书。关于知情同意程序的进一步细节,参见第9.3节。在筛查访视期间,每名患者都将需要进行评估计划表(表13)中概述的所有评估项。
符合研究条件的患者包含下列患者:(1)诊断为家族性寒冷性自身炎性综合征、Muckle-Wells综合征或新生儿起病型多系统炎性疾病的患者。在加入研究时没有进行NLRP3突变的分子诊断的患者(未进行测试,或进行了测试但结果为阴性),针对研究资格,需要研究者和阿克拉瑞斯(Aclaris)之间事先同意。对于那些没有进行NLRP3突变分子测试的患者,应在研究期间进行分子测试;(2)具有“最低”或更小PGA评分并且被认为是由于成功的抗IL-1疗法而实现响应的患者;以及(3)用抗IL-1疗法连续治疗至少6个月的患者。
在基线处确认资格的患者将开始ATI-450片剂(50mg BID)给药。由于先前用抗IL-1生物制剂治疗,这样的患者将处于方案限定的缓解期。给药将从其抗IL-1疗法的下一次剂量安排的当天开始,届时将停止抗IL-1疗法。
ATI-450的第一剂量(50mg BID)将在下列时间服用(基于抗IL-1疗法):
阿那白滞素:最后一次施用后24小时(+/-1小时)或在常规阿那白滞素剂量的大致时间。
卡那单抗:最后一次施用后8周(+/-1周)。
利那西普:最后一次施用后1周(+/-1天)。
ATI-450将被口服施用12周。患者将在第14天、第28天、第56天和第84天(+/-1天)参加针对安全性、疗效、PK和PD评估的门诊访视。患者将在开始施用ATI-450前7天开始在其每日日记卡中记录疾病症状,如果第1天的访视被安排发生在筛查访视后不到7天,则立即在筛查访视时开始记录。患者将继续完成其日记,直到完成第7天的安全性访视。
在12周结束时,第84天(+/-1天),患者将停止ATI-450并进行研究结束评估。患者将在最后剂量的ATI-450后7天(+4天)现场完成安全性随访,并在最后剂量后30天(+/-3天)通过电话完成安全性随访。
本研究被设计以评估ATI-450在患有CAPS的患者中的安全性、耐受性、疗效、PK和PD。
许多患有CAPS的患者通过目前可用的抗IL-1疗法达到完全或接近完全的疾病缓解。因此,本研究被设计以最小化患者由于疾病症状的长期复发可能导致的潜在有害的长期并发症而停止抗IL-1疗法的时间。
本研究是开放标签的,并且没有针对抗IL-1疗法的洗脱期(washout period);招募到试验中的患者将在其下一次安排剂量的抗IL-1疗法的当天开始用研究药物治疗(第1天)。在12周治疗期结束时,将停止ATI-450施用,并且患者将推迟抗IL-1疗法的施用,直到最后剂量的ATI-450后7天(+4天)。
选择12周的治疗期以允许有足够的时间探索ATI-450在维持疾病缓解方面的疗效,并充分排除由最后剂量的抗IL-1疗法引起的延长的疾病缓解的可能性。由于其t
研究的开始将是第一位患者提供知情同意书的日期,并且研究的结束将是最后一位患者最后一次评估的日期。预计总持续时间可能长达24周。
表19
ECG=心电图,Hep=肝炎,PD=药效动力学,TB=结核病,EOS=研究结束。
统计方法:
确定样本量——本研究的样本量是基于可行性限制而非正式的幂计算确定的。计划最多招募10名患者。
分析群体。意向治疗(ITT)群体将包含所有已接受至少一次剂量的研究药物的患者。符合方案(PP)群体将包含所有完成12周治疗和4周安全性随访并已经完成研究评估的患者。
疗效分析——所有疗效概述将在ITT和PP群体两者中进行。
·随时间疾病缓解的参与者的比例;其中疾病缓解被限定为具有最低或更好的PGA评分,并且hsCRP和SAA值在正常范围内或在基线值的30%内(ITT和PP群体)。
·随时间临床缓解的参与者的比例;其中临床缓解被限定为具有最低或更好的PGA评分(ITT和PP群体)。
·随时间在停用ATI-450后至少两天内,经历每日KSS较基线增加≥3的参与者的比例(PP群体)。
·在ITT和PP群体中,8周中的至少有6周中维持平均KSS比基线高不超过2分的参与者的比例。
·PGA和KSS得分随时间自基线的变化将被描述性地报告(ITT和PP群体)。
·随时间hsCRP和SAA自基线的变化以及hsCRP和SAA自基线的变化百分比将使用连续的统计概括测量来总结。hsCRP和SAA的持续治疗效果的测定将基于ITT和PP群体中自基线的中值百分比变化。
疗效评估
医师完成的疗效问卷。医师的自身炎性疾病活动度的整体评估(PGA)是由研究者或指定者完成的测量。PGA使用5分分级标度:无、最低、轻度、中度和重度。研究者将基于患者在访视时的当前疾病活动度选择分级。
疾病缓解和临床缓解的维持。在治疗期间将评估疾病缓解的维持情况。疾病缓解被限定为具有不存在的或最低的PGA评分并且hsCRP和SAA值在正常范围内(≤10mg/L)或在基线值的30%以内。
在治疗期间将评估临床缓解的维持情况。临床缓解被限定为具有不存在的或最低的PGA评分。
复发时间在治疗期间将评估复发时间。复发被限定为PGA标度上2分的恶化。
患者完成的疗效问卷。当这些评估需要在表13中列出的时间进行时,其应该是在任意访视时以及在研究药物给药之前所做的第一项工作。
关键症状评分(KSS)来源于患者管理的每日健康评估表(DHAF),并且是5个独立标度(皮疹、发烧和发冷的感觉、关节疼痛、眼睛红肿和疼痛以及疲劳)的0至10标度的平均值(0=无,10=非常严重)。
DHAF被设计使用以半级为单位的圆圈的线性分级标度(例如,0.5、1.0、1.5、2.0等),标示为0(无,无严重性)至10(非常严重)。患者将选择其确定最准确地代表自己在过去24小时内所经历的症状严重程度的圆圈。
DHAF将由患者每天完成。每日KSS将通过对5个单独症状得分之和求平均值来计算。规定时间段期间的KSS将进一步通过对该时间段的每日KSS得分之和求平均值来计算。
将为患者提供安静、私密的地点来完成评估。将指导患者在没有他人(研究人员、家人或朋友)的帮助下,尽其所能回答所有问题。研究人员应在问卷完成后进行审查,并鼓励患者填写任何缺失的信息。患者可以不回答任何问题。研究人员将在源文件中记录患者拒绝回答任何问题的情况。
CAPS症状的重新出现在治疗期结束后,停用ATI-450后将评估CAPS症状的重新出现。重新出现被限定为:至少连续两天,每日KSS自基线增加≥3。
高敏性C反应蛋白和血清淀粉样蛋白A。将在表13中规定的时间收集用于评价hsCRP和SAA的血液样品。样品将被运到中心实验室。关于血液样品的收集、处理、储存和运输的具体说明将在单独的实验室手册中提供。血清hsCRP和SAA两者的正常范围被限定为≤10mg/L。
安全性评估
不良事件:将跟踪、记录和报告AE。
临床实验室评价:治疗期间的实验室评价将由中心实验室进行。将在表13所指示的时间收集血液和尿液样品。在给药日,将在研究药物施用前进行临床实验室参数分析的采样。
除非另有说明,否则所有的实验室样品都将按照中心实验室手册中所描述的进行处理并运送到中心实验室。中心实验室将按照手册中所指示的对样品进行分析或者将样品送往参考实验室(一个或多个)进行分析。在整个研究中,针对每位患者要采集的最大总血液体积,参考中心实验室手册。
将对下列参数进行评估:
血液学:血红蛋白、血细胞比容、红细胞、血小板、总WBC计数、分类WBC计数和ANC
凝血:INR、部分凝血活酶时间和凝血酶原时间
生物化学:白蛋白、碱性磷酸酶(ALP)、ALT、淀粉酶、AST、血尿素氮(BUN)、钙、肌酸磷酸激酶、hsCRP、肌酸酐、γ-谷氨酰转移酶、葡萄糖、无机磷酸酶、乳酸脱氢酶、脂肪酶、镁、钾、SAA、钠、氯、碳酸氢盐、总胆红素、总蛋白和尿酸
脂类:总胆固醇、高密度脂蛋白、低密度脂蛋白和甘油三酯
尿液分析:pH值、比重、肌酸酐、葡萄糖、胆红素、血液和蛋白质
潜在的药物引起的肝损伤。Hy’s法则病例有下列3个组成部分:(1)药物引起肝细胞损伤,通常表现为相比于安慰剂,ALT或AST的ULN提升≥3倍的更高发生率;(2)在显示出这样的转氨酶升高的研究患者中,通常转氨酶远大于3×ULN,一个或更多个患者还显示血清总胆红素升高至>2×ULN或INR>1.5,而没有初步发现胆汁淤积(升高的ALP);(3)未发现其他原因来解释增加的转氨酶和总胆红素的组合,如病毒性甲型肝炎、乙型肝炎或丙型肝炎;胆梗阻的证据;急性酒精性肝炎(近期喝酒和AST>2×ALT是支持性的);重度低血压或充血性心力衰竭的近期病史;其他潜在的病毒性疾病;原有的或急性的肝脏疾病;或其他能够引起观察到的损伤的药物(包含非处方产品,如草药补充剂)。
实施例9:ATI-450抑制涉及自身免疫病况的炎性细胞因子
ATI-450增加小鼠胶原蛋白诱导的关节炎模型中的调节性T(Treg)细胞。为了研究ATI-450用于治疗自身免疫病况的治疗的效用,在胶原蛋白诱导的关节炎(CIA)小鼠模型中评价ATI-450对T细胞亚群的影响。在该模型中,用牛胶原蛋白/CFA免疫DBA/1小鼠(12/组)以诱发关节炎。在第18天,给动物施用溶媒、ENBREL(10mg/kg QD)或ATI-450(1000ppm食物)。在第21天,用牛胶原蛋白/CFA增强小鼠,并且在第35天终止研究。从小鼠身上解剖腘窝淋巴结,清洗掉任何微量脂肪,机械分离并通过70微米的细胞滤网过滤。使用Moxi GO II细胞计数器(Orflo)对活淋巴细胞进行计数。将100万个淋巴细胞进行铺板,用Fc阻断处理,并且用靶向表面受体(包含CD45、CD3、CD4)的抗体和可固定的活/死染料进行染色。细胞被固定/透化(eBioscience转录因子染色试剂盒),并用抗Foxp3抗体染色。在Attune NxT仪器上经由流式细胞仪分析收集细胞。T调节性细胞被筛选(gated)为淋巴细胞尺寸、活体、CD45+CD3+CD4+Foxp3+细胞。
如图21中所示,在小鼠CIA模型中的ATI-450治疗显著增加了调节性T细胞(TREG)的数量。鉴于已知TREG细胞参与抑制免疫响应和自身免疫疾病的预防,该数据指示了ATI-450用于自身免疫病况(如炎性肠病、系统性狼疮肾炎(SLE)及其他)的治疗的效用。
ATI-450阻断人类全血中脂多糖(LPS)刺激的TNF-α、IL-1β和IL-6产生。在含有肝素钠的试管中收集来自健康人类志愿者的静脉血,并且将其等分(每孔180μL)到96孔圆底组织培养板中。将化合物在100%DMSO(Sigma-Aldrich,D2650)中连续稀释,然后在DMEM/10%FBS(Gibco,11965/26140)中进行50倍稀释。然后将经稀释后的化合物以一式两份10μL的等分试样添加至每个板中,最终浓度如图22A所指示的。溶媒由最终检测浓度为0.1%的DMSO组成。将针状工具(V&P Scientific,Inc.,VP246)置于孔中,并且将板置于37℃下,5%的CO
LPS刺激Toll样受体4(TLR4)释放一些促炎性细胞因子。如图22A中所示,ATI-450以在0.01-0.1μM之间的IC
人类全血中ATI-450抑制的Poly(I:C)刺激的IP10、IFNγ、IL-6、IL-8和TNF-α的产生。如上文所述进行人类全血测定,但下文列出的情况除外。将人类全血等分(每孔175μL)到96孔圆底组织培养板。将化合物在100%DMSO(Sigma-Aldrich,D2650)中连续稀释,然后在DMEM/10%FBS(Gibco,11965/26140)中进行25倍稀释。然后将经稀释的化合物以一式两份5μL的等分试样添加至每个板中,最终浓度如图22B中所指示的。在与化合物或溶媒孵育一小时后,向每个孔中添加20μL的DPBS(Gibco,14190-136)中的Poly(I:C)(InvivoGen,HMW,tlrl-pic),最终浓度100μg/mL,无刺激的孔除外。与poly(I:C)孵育二十四小时后,收获血浆并且根据试剂盒方案,使用Meso Scale Discovery Technology V-plex人类细胞因子试剂盒对IP10、IFNγ、IL-6、IL-8和TNF-α进行测定。
Poly(I:C)是诱导促炎性细胞因子、IL-6、TNF-α、干扰素-γ和炎性趋化因子、IL-8和干扰素-γ可诱导蛋白(IP10)的激活的TLR3激动剂。如图22B中所示,ATI-450能够以在0.01-0.1μM之间的IC
ATI-450抑制R848刺激的人类全血中TNF-α、IL-1β、IL-6和IL-8产生。如上文所述进行人类全血测定,但下文列出的情况除外。将人类全血等分(每孔200μL)到96孔圆底组织培养板中。将化合物在100%DMSO(Sigma-Aldrich,D2650)中连续稀释,然后在RPMI(Gibco,11875)中进行10倍稀释。然后将经稀释的化合物以一式两份2μL的等分试样添加至每个板中,最终浓度如图22C中所指示的。在与化合物或溶媒孵育一小时后,向每个孔中添加2μL的RPMI(Gibco,11875)中的R848(InvivoGen,tlrl-r848),最终浓度0.5μg/mL,无刺激的孔除外。与R848孵育5小时后,收获血浆并且根据试剂盒方案,使用Meso Scale DiscoveryTechnology V-plex人类细胞因子试剂盒对TNF-α、IL-1β、IL-6和IL-8进行测定。
R848(雷西莫特(resiquimod))是TLR7和TLR8的强效激动剂。TLR7和TLR8的激活诱导各种炎性细胞因子和趋化因子(包含IL-6、IL-8、TNF-α和IL-1β)的表达和活性。如图22C中所示,ATI-450能够以~0.1μM的IC
虽然鉴于ATI-450在促炎性信号传导途径中的MK2抑制作用,ATI-450介导的对TLR4刺激的细胞因子释放的抑制是意料之中的,但ATI-450是否会类似地阻断由其他多种刺激物(如TLR3和TLR7及TLR8受体激活)引发的细胞因子释放是不可预期的。这些数据指示了ATI-450对于治疗自身免疫疾病和具有强烈的抗核抗体驱动的其他紊乱(如狼疮、硬皮病、斯耶格伦综合征、青少年关节炎等)的治疗具有治疗性效用。
为了进一步评估ATI-450对各种细胞细胞因子的抑制作用,如下文所述,测试了多种细胞类型和各种刺激物。这些研究的结果在下文表14中呈现。
U937细胞分化和LPS刺激的细胞因子测定:使U937人类前单核细胞系(ATCC,CRL1593.2)在完全培养基:RPMI 1640(Gibco,A10491)与青霉素-链霉素(10U/mL)-谷氨酰胺(2mM)(Gibco,10378016)和10%FBS(Gibco,26140-079)中生长。添加PMA(Sigma Aldrich,P1585)(20ng/mL,24小时)使细胞分化为单核细胞/巨噬细胞表型,用DPBS(Gibco,14190-136)洗涤,并且在37℃、5%CO
经分化的U937细胞在经连续稀释的化合物或溶媒存在下预处理一小时,然后用最终浓度为100ng/mL的LPS刺激4小时。然后收集培养基,使用Meso Scale技术测定细胞因子水平。
A549 IL-1β刺激的IL-6测定—使A549细胞(美国典型培养物保藏中心(AmericanType Culture Collection),ATCC CRL-1593.2)在具有青霉素-链霉素(10U/ml)(Gibco,15140-122)和10%胎牛血清(Gibco,26140-079)的Ham’s F-12K(Kaighn's)(Gibco,21127-022)中生长。细胞生长至80%汇合,然后用胰蛋白酶移除细胞。将细胞在完全培养基中的96孔平底板中铺板,并允许其恢复过夜。在连续稀释的化合物或溶媒(0.5%DMSO)(Sigma-Aldrich,D2650)的存在下,将细胞预处理一小时,然后用IL-1β(1ng/mL)(R&D Systems,201-LB-005)刺激18小时。然后收集培养基,使用Meso Scale技术测定IL-6水平。
人类PBMC LPS刺激的IL-1测定—将冷冻的人类PBMC原料快速解冻,重悬于DMEM/10%FBS(Gibco,11965/26140)中,在130×g下进行造粒5分钟,然后重悬于DMEM/10%FBS中,并以每孔20万个细胞(180μL)在平底96孔板中进行铺板。在37℃和5%CO
猪LPS刺激的TNF-α测定—将猪全血收集到肝素化的真空采血管中,然后转移到锥形管中并在湿冰上运输过夜。将猪全血等分(160μL/孔)放入圆底96孔组织培养板。将化合物在100%DMSO(Sigma-Aldrich,D2650)中连续稀释,然后在RPMI/10%FBS(Gibco,11875/26140)中进行100倍稀释。然后将经稀释后的化合物以一式两份20μL的等分试样添加至每个板,最终浓度从10μM到1pM。DMSO最终浓度为0.1%。在37℃和5%CO
表20
实施例10:ATI-450抑制参与炎性皮肤病况的细胞因子
ATI-450在体外抑制TLR2刺激的细胞因子信号传导:重要文献表明,TLR2激动剂驱动TH17分化并抑制调节性T细胞(参见例如,Nyirenda et al.,“TLR2 Stimulation DrivesHuman
使用Meso Scale Discovery技术(类似于ELISA的形式,具有捕获和检测抗体,但使用电化学发光进行最后读取)测量释放的TNF-α。
如图23中所示,ATI-450剂量依赖性地抑制TLR2配体诱导的TNF-α产生。如上所述,TLR2信号传导驱动TH17细胞分化。
ATI-450体外抑制IL-17产生。为了研究ATI-450介导的对TLR2途径的抑制是否也起到了调控IL-17产生的作用,使用CD4干细胞分离试剂盒从人类PBMC中分离全部CD4+细胞。在TH17倾斜条件下(skewing condition)(抗CD3、抗CD28、抗IL-4、抗IFNγ、rIL-6、rIL-1β、rIL-23、rTGFβ)将CD4+细胞培养3天。在第3天,收获细胞、冲洗、重新铺板并且仅用化合物或DMSO处理。孵育1小时后,用抗CD3免疫磁珠(dynabeads)以1:1的细胞:珠比例重新刺激细胞过夜。18-20小时后,收集上清液,并且使用MSD测定IL-17A水平。如图24中所示,ATI-450部分地抑制了CD4+细胞中IL-17的产生。
Il-17的抑制已经证明在银屑病、银屑病关节炎和脊柱炎的治疗中的效用。观察到的ATI-450对IL-17的部分抑制也可能使ATI-450特别适用于IBD的治疗,其中仅部分的IL-17抑制被认为能提供疗效而不加重IBD。
ATI-450抑制人类全血中IL-1β刺激的TNF-α、IL-6和IL-8产生—如上文所述进行人类全血测定,但下文列出的情况除外。将人类全血等分(每孔200μL)到96孔圆底组织培养板。将化合物在100%DMSO(Sigma-Aldrich,D2650)中连续稀释,然后在RPMI(Gibco,11875)中进行10倍稀释。然后将经稀释的化合物以一式两份2μL的等分试样添加至每个板中,最终浓度如图25中所指示的。在与化合物或溶媒孵育一小时后,向每个孔中添加2μL的RPMI(Gibco,11875)中的IL-1β(R&D,201-LB-010),最终浓度为0.5μg/mL,无刺激的孔除外。与IL-1β孵育6小时后,收获血浆并且根据试剂盒方案,使用Meso Scale DiscoveryTechnology V-plex人类细胞因子试剂盒对TNF-α、IL-6和IL-8进行测定。
如图25中所示,ATI-450剂量依赖性地抑制人类全血中IL-1β刺激的TNF-α(IC
ATI-450抑制人类免疫细胞中IL-1α的生物合成和活性。用ATI-450预处理人类PBMC持续1小时,并且然后用LP刺激24小时。使用MSD技术对IL-1α的生物合成进行量化。在单独的研究中,从CD-1小鼠骨髓中分离出小鼠中性粒细胞、铺板并且在加入不同浓度的ATI-450前放置一小时,然后添加ATI-450并在37℃下孵育1小时。用小鼠IL-1α刺激细胞和化合物至最终浓度为10ng/mL并且在37℃下孵育4小时。收集上清液,并且使用MSD技术对TNF-α水平进行定量。
如图26A中所示,ATI-450以31.1nM的IC
实施例11:ATI-450片剂的配制。
下文表21中提供了本文实施例中使用的包括ATI-450的片剂和包括安慰剂的片剂的组合物。下文表22中提供了在药物片剂中使用的赋形剂及其功能。
表21.ATI-450片剂的成分
表22.包括ATI-450的片剂中的赋形剂
实施例12:化合物(P)-I的分离
可以根据美国专利号9,115,089(其特此通过引用被整体并入)中描述的方法制备外消旋的3-氯-4-((3,5-二氟吡啶-2-基)甲氧基)-2'-(2-(2-羟基丙烷-2-基)嘧啶-4-基)-5',6-二甲基-2H-[1,4'-联吡啶]-2-酮(250mg,0.49mmol)。如下文所述进行3-氯-4-((3,5-二氟吡啶-2-基)甲氧基)-2'-(2-(2-羟基丙烷-2-基)嘧啶-4-基)-5',6-二甲基-2H-[1,4'-联吡啶]-2-酮的手性拆分以获得P阻转异构体(即如本文公开的化合物(P)-I)。
使用具有二氧化碳和乙醇的流动相的超临界流体色谱法(Thar 80,制备型SFC,ChiralCel OD-H,250×30mm ID柱)分离外消旋的3-氯-4-((3,5-二氟吡啶-2-基)甲氧基)-2'-(2-(2-羟基丙烷-2-基)嘧啶-4-基)-5',6-二甲基-2H-[1,4'-联吡啶]-2-酮。分离方法使用具有50mL/min的流速和10min的循环时间的40%乙醇的等度法。使用WZZ-2S旋光仪测定旋光性。
在1.77分钟洗脱的较快的异构体((P)-I),产生在乙二醇中115mg的(-)-3-氯-4-((3,5-二氟吡啶-2-基)甲氧基)-2'-(2-(2-羟基丙烷-2-基)嘧啶-4-基)-5',6-二甲基-2H-[1,4'-联吡啶]-2-酮:[α]
在3.68分钟洗脱的较慢的异构体((M)-I),产生在乙二醇中112mg的(+)-3-氯-4-((3,5-二氟吡啶-2-基)甲氧基)-2'-(2-(2-羟基丙烷-2-基)嘧啶-4-基)-5',6-二甲基-2H-[1,4'-联吡啶]-2-酮:[α]
实施例13:化合物(P)-I的晶体形式筛选
晶体形式筛选研究涉及总共48种纯溶剂系统和二元溶剂系统,其解决了输入材料的中等溶解度并提供了极性、介电常数、偶极矩和氢键供体/受体属性的多样化集。还包含了具有多种水分活度(a
筛选研究包括以下结晶模式:
·API浆状物在5-40℃之间温度循环熟化4天(TC)
·将澄清的饱和溶液从40℃快速冷却至-20℃,并保持在-20℃三天(RC)
·澄清的溶液在室温下缓慢蒸发14天。在14天后,在减压下从在缓慢蒸发期间不产生固体的溶液中快速蒸发溶剂(EV)。
筛选研究的结果的概述在表23中示出。
表23晶体形式筛选的结果
实施例14:化合物(P)-I(形式A)的单晶结构测定
已经使用单晶X射线衍射相对于原子的空间排列的绝对立体化学构型表征了化合物(P)-I的结晶形式。在Stout&Jensen,X-Ray Structure Determination:A PracticalGuide,Macmillan Co.,New York(1968),Chapter 3(其通过引用并入本文)中提供了通过X射线衍射测定结构的详细说明。可替换地,可以通过X射线粉末衍射分析来表征晶格内三个维度上原子的独特空间排列。Cullity,B.D.Elements of X-ray Diffraction.Addison–Wesley,(1978)ISBN 0-201-01174-3Chapter 14(其通过引用并入本文)中提供了X射线粉末衍射的详细说明。PXRD数据由实验确定的2θ位置值、多次晶体学反射(也称为布拉格反射)的强度值及其峰形组成。PXRD数据可以被计算分析,包含通过Rietveld修正(Rietveldrefinement)的方法。Pecharsky,Vitalij K.;Zavalij,Peter Y.(2009)Fundamentals ofpowder diffraction and structural characterization of materials(2nd ed.).NewYork:Springer.ISBN 978-0-387-09579-0.OCLC 314182615(其通过引用并入本文)中提供了对X射线粉末衍射数据的Rietveld修正的详细描述。
可以在各种温度或压力下收集PXRD数据以便利Rietveld修正。可以使用如Macrae,Clare F.,et al.“Mercury 4.0:from visualization to analysis,design andprediction.”Journal of Applied Crystallography vol.53,226-235.1Feb.2020,doi:10.1107/S1600576719014092中描述的计算方法,将包含2θ值、d-间距、布拉格反射和强度值的实验PXRD数据与来源于代表理想化纯粉末的单晶结构测定的模拟PXRD图谱进行比较。
本领域普通技术人员将理解的是,可能获得具有取决于数据收集中采用的测量条件的测量误差的X射线粉末衍射图谱。通常认为,来源于X射线粉末衍射图谱的峰形、强度值和2θ位置可以根据所用仪器的类型、测量条件和执行的计算分析方法而波动。应进一步理解的是,2θ值及其相对强度也可能变化,因此不应考虑强度值的真正阶次。
此外,常规X射线粉末衍射图谱的衍射角测量的实验误差通常为约5%或更小。在描述2θ衍射峰的位置时,应考虑对测量误差程度的评估。因此,应当理解的是,本发明中描述的晶体形式不限于提供与本文公开的图27中描绘的X-射线粉末衍射图谱完全相同的X-射线粉末衍射图谱的晶体形式。提供与附图中公开的那些基本相同的X射线粉末衍射图谱的任何晶体形式都落入本发明的范围内。确定X-射线衍射图谱实质相同的能力在本领域普通技术人员的能力范围内。同样,应当理解,提供与图28中公开的那些基本相同的差示扫描量热法(DSC)和/或热重量分析(TGA)的任何晶体形式都落入本发明的范围内。确定这些图谱实质相同的能力在本领域普通技术人员的能力范围内。
化合物(P)-I的结晶形式A是无水的并且是采用各种有机溶剂和有机/水溶剂系统从实施例13中描述的结晶条件获得的。
X射线粉末衍射(PXRD)衍射图是在使用Ni-过滤的Cu Ka(45kV/40mA)辐射和0.02°2θ的步长的PANalytical X’Pert Pro衍射仪和X'celerator
相对于来自化合物(P)-I的结晶形式A的模拟PXRD数据的结果,显著的布拉格反射的值、其2θ位置和d-间距值在来源于图27中示出的PXRD谱图的表24中示出。
表24:实验和模拟PXRD数据
用配备有自动进样器和冷冻冷却系统的TA Instruments Q100差示扫描量热仪在40mL/min N
用TA Instruments Q500热重分析仪在40mL/min N2吹扫下以15℃/min在Pt或Al盘中获得的热重量分析(TGA)热谱图。
图28(上部轨迹)中描绘的DSC分析表明化合物(P)-I的结晶形式A表现出在187.92℃熔融/外消旋化事件,随后在195.8℃的再结晶事件,以及最后在253.5℃的急剧吸热(外消旋体熔融)。通过TGA在25℃和256℃之间观察到可忽略不计的重量损失(0.7%)。
表25.FT-IR吸光度数据
实施例15:化合物(P)-I的p38抑制效力和p38/MK2底物选择性
分别研究了化合物(P)-I和化合物(M)-I对MK2和PRAK的抑制,以了解化合物(P)-I和/或化合物(M)-I在降低炎性反应方面的选择性。在酶测定中评价化合物(P)-I和化合物(M)-I,酶测定对抑制剂在阻断p38/MK2方面的效力与在来源于HSP-27的肽底物的p38/PRAK诱导的磷酸化方面的效力进行比较。使用p38α/MK2和p38α/PRAK级联测定形式评价化合物(P)-I和化合物(M)-I抑制激活的磷酸化p38α的能力。通过p38α磷酸化GST-MK2或GST-PRAK的能力确定p38α的激酶活性。通过测量荧光标记的MK2/PRAK特异性肽底物Hsp27肽的磷酸化来定量p38α对MK2或PRAK的激活。使用IMAP技术(美谷分子(Molecular Devices),加利福尼亚州森尼韦尔)定量Hsp27肽的磷酸化。在384孔板(Greiner,781280)中在20mM HEPESpH7.5、10mM MgCl
根据上文描述的测定测试化合物(P)-I和化合物(M)-I,从而产生下文表26中所述的IC
表26
实施例16:化合物(P)-I在人类单核细胞中细胞因子的调节
p38途径已被证明对包含TNFα、IL-1β和IL-6的许多促炎性细胞因子的生物合成至关重要。因此,p38 MAP激酶途径的抑制将通过减少促炎性细胞因子的生物合成来降低炎性反应。该研究显示了将TNFα、IL-6和IL-1β(促炎性细胞因子)的生物合成抑制一半所需的化合物(P)-I和化合物(M)-I的量,从而证明各个化合物在降低炎症方面的有效性。使用人类U937细胞系对化合物(P)-I和化合物(M)-I阻断细胞因子产生的效力和功效进行了评估。U937人类前单核细胞系从美国典型培养物保藏中心(Rockville,MD)获得。这些细胞分化为如Burnette(Burnette et al,(2009).SD0006:a potent,selective and orallyavailable inhibitor of p38 MAP Kinase,Pharmacology 84(1):42-60)所述的单核细胞/巨噬细胞表型。将经分化的U937细胞(人外周血单核细胞(hPBMC))接种到完全培养基中的96孔组织培养板(20万个细胞/孔)中。在24小时后,在化合物(P)-I和化合物(M)-I存在或不存在的情况下对细胞进行60分钟预处理,并且然后用LPS(0.1μg/mL)刺激4小时。然后收集培养基用于通过ELISA测定TNFα、IL-6或IL-1β水平。使用四参数逻辑模型从重组蛋白标准曲线外推细胞因子浓度,并在迭代到最佳最小二乘拟合后求解IC
根据上文描述的测定测试ATI-450的两种阻转异构体(化合物(M)-I和化合物(P)-I)),从而产生下文表27中所述的IC
表27
尽管已参考本文的某些优选的实施方案相当详细地描述了本文的实施方案,但其他版本是可能的。因此,所附权利要求书的精神和范围不应当限于本说明书中含有的描述和优选的版本。
- 用于各种肿瘤免疫治疗的新型肽和肽组合物
- 包含抗-LAG3抗体、PD-1途径抑制剂和免疫治疗剂组合的组合物
- 含有MK2抑制剂肽的组合物用于治疗非小细胞肺癌的用途