占诺美林衍生物的苹果酸盐、A晶型及其制备方法和用途
文献发布时间:2023-06-19 19:28:50
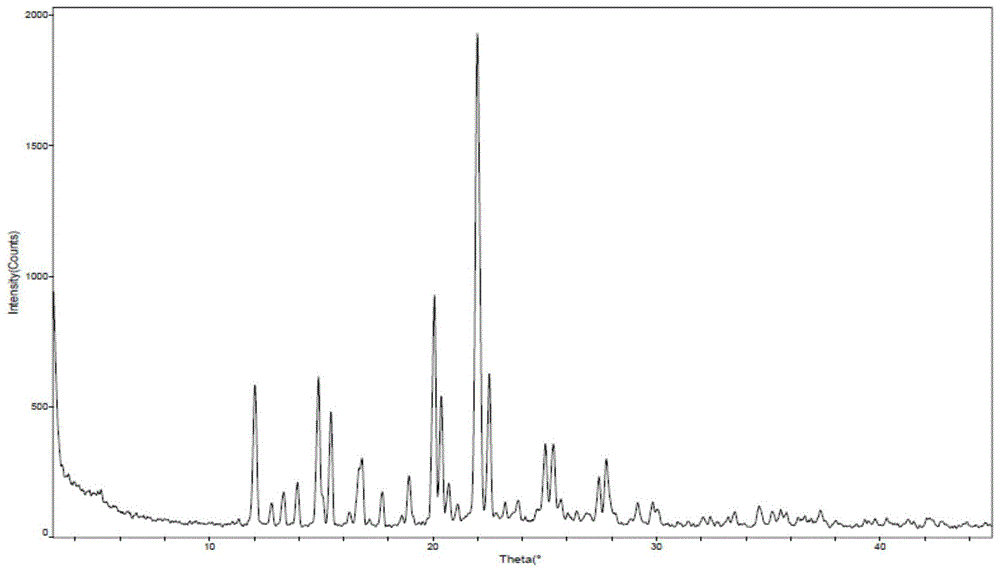
本申请要求以下在先申请的优先权:2021年10月14日向中国国家知识产权局提交的专利申请号为202111198168.4,发明名称为“占诺美林衍生物的苹果酸盐、A晶型及其制备方法和用途”的在先申请。所述在先申请的全文通过引用的方式结合于本申请中。
技术领域
本发明涉及药物化学领域,具体涉及一种占诺美林衍生物的苹果酸盐、A晶型及其制备方法,还包括所述占诺美林衍生物的苹果酸盐、A晶型在制备预防或治疗中枢神经系统紊乱疾病药物中的应用。
背景技术
神经递质是神经元分泌的化学信使,用以促进信息流动并与中枢神经系统和周围神经系统中的其它细胞(例如肌肉或类似神经细胞)进行通讯。乙酰胆碱是大脑中的关键神经递质之一,其有两种不同的受体类别:毒蕈碱型受体(M受体,G蛋白偶联受体)和烟碱型受体(N受体,离子通道受体)。
M受体家族包含M1至M5五种亚型,它们都在大脑和周围组织中表达,并在认知、行为、感觉、运动和自主神经过程中起着许多关键的生理作用。M型受体信号的破坏会导致记忆障碍和认知障碍,引发包括精神分裂症和阿尔茨海默症(AD)在内的多种疾病,并加剧精神病。相反,第三方的临床前和临床数据表明,M型受体信号传导的增强则会改善这些症状,此外M型受体尤其是M1、M2、M4受体也被认为与镇痛有关。
占诺美林(xanomeline)是一种毒蕈碱受体的部分激动剂,可对毒蕈碱受体的5个亚型都产生激动作用,不具备选择性。由美国礼来公司及Novo Nordisk公司共同开发并上市,临床主要用于阿尔茨海默症的治疗,占诺美林的化学名称为3-[(4-己氧基)-1,2,5-噻二唑-3-基]-1,2,5,6-四氢-1-甲基吡啶,化学结构式如下:
专利文献CN94192681.8公开了占诺美林可以转化为草酸盐,但草酸盐对病人的肾功能具有潜在的副作用,因而在药学上不宜,特别是在治疗老年人时尤为不宜。并进一步公开了在十二种可药用酸(未公开具体药用酸的名称)系列中,只有占诺美林酒石酸盐才具备好的生物利用率,好的处理性质和可重复的晶型。
目前,公知常识普遍教导,选择具有所需的性能组合的盐仍然是一个困难的半经验性的选择,需要盐形式的性质的折衷选择,但是仍然存在评估哪种盐形式为最适合筛选特定候选药物的困难。
药物盐型的筛选是一个困难的半经验性选择,药物吸湿性会严重影响药物的流动性,甚至会影响药物的稳定性。药物的溶解度对药剂的制备、药物溶出、吸收等都具有至关重要的影响。但如何提高药物的溶解度,不以药物的吸湿性为代价,得到稳定性、溶解度及吸湿性均合适的候选药物盐是困难的。
另外,Xanomeline作为一种M受体激活剂在临床上治疗精神分裂症和AD患者的精神病和相关行为症状方面具有令人鼓舞的治疗效果,但其潜力一直受到胆碱能副作用的限制,包括流涎、恶心、头晕等,这被认为是由于刺激外周神经组织中的M受体所导致的。占诺美林在人体内的代谢情况复杂且不可预测,且占诺美林对毒蕈碱受体亚型缺乏选择性,因此对占诺美林的药物开发造成了困难。
因此,需要进一步寻找具有良好疗效、副作用小、具有更好的药代动力学性质的适于成药的新型化合物;还需要开发出该化合物具有适合可靠的配制和制备特性的结晶盐及其多晶型物。
发明内容
本发明的目的在于提供一种式I所示的占诺美林衍生物的苹果酸盐,所述苹果酸盐能够用于制备预防或治疗中枢神经系统紊乱疾病的药物并且这种衍生物的溶解能力改善、储存稳定、给药方便、药物代谢、药物毒副作用改善,并且给药时患者的依从度高。
本发明的另一目的在于提供一种式I所示占诺美林衍生物的苹果酸盐的A晶型,所述A晶型能够具备优异的热力学稳定性和机械稳定性,重复性好,适于商业化大生产。
第一方面,本发明提供了式I化合物的苹果酸盐。
在本发明的一些方案中,上述式I化合物的苹果酸盐,如式Ⅱ所示,其中,x选自0.5~2。
本发明的一些方案中,上述x为0.5、1.0、1.5、2.0。
本发明的一些方案中,上述式Ⅱ化合物中x为1.0。
本发明的一些方案中,上述式Ⅱ化合物,其中苹果酸为L-苹果酸。
第二方面,本发明提供了式I化合物的苹果酸盐的A晶型,其X射线粉末衍射图谱在下列2θ角处具有特征衍射峰:12.007±0.2°、14.827±0.2°、16.796±0.2°、20.012±0.2°和21.942±0.2°。所述式I化合物的苹果酸盐的A晶型是x为1.0的式Ⅱ化合物,且其中的苹果酸为L-苹果酸。
本发明的一些方案中,上述A晶型的X射线粉末衍射图谱在下列2θ角处具有特征衍射峰:12.007±0.2°、14.827±0.2°、16.796±0.2°、20.012±0.2°、21.942±0.2°、22.473±0.2°和25.333±0.2°。
本发明的一些方案中,上述A晶型的X射线粉末衍射图谱在下列2θ角处具有特征衍射峰:12.007±0.2°、13.288±0.2°、14.827±0.2°、16.796±0.2°、20.012±0.2°、21.942±0.2°、22.473±0.2°和25.333±0.2°。
本发明的一些方案中,上述A晶型的X射线粉末衍射图谱在下列2θ角处具有特征衍射峰:12.007±0.2°、13.288±0.2°、14.827±0.2°、15.399±0.2°、16.796±0.2°、20.012±0.2°、20.327±0.2°、21.942±0.2°、22.473±0.2°和25.333±0.2°。
本发明的一些方案中,上述A晶型的X射线粉末衍射图谱在下列2θ角处具有特征衍射峰:12.007±0.2°、13.288±0.2°、14.827±0.2°、15.399±0.2°、16.796±0.2°、20.012±0.2°、20.327±0.2°、21.942±0.2°、22.473±0.2°、24.996±0.2°、25.333±0.2°和27.718±0.2°。
本发明的一些方案中,上述A晶型的X射线粉末衍射图谱在下列2θ角处具有特征衍射峰:12.007±0.2°、12.755±0.2°、13.288±0.2°、13.903±0.2°、14.827±0.2°、15.399±0.2°、16.796±0.2°、17.686±0.2°、18.906±0.2°、20.012±0.2°、20.327±0.2°、21.942±0.2°、22.473±0.2°、24.996±0.2°、25.333±0.2°和27.718±0.2°。
本发明的一些方案中,上述A晶型的X射线粉末衍射图谱在下列2θ角处具有特征衍射峰:12.007±0.2°、12.755±0.2°、13.288±0.2°、13.903±0.2°、14.827±0.2°、15.399±0.2°、16.796±0.2°、17.686±0.2°、18.906±0.2°、20.012±0.2°、20.327±0.2°、20.660±0.2°、21.942±0.2°、22.473±0.2°、24.996±0.2°、25.333±0.2°、27.382±0.2°和27.718±0.2°。
本发明的一些方案中,上述A晶型,其具有基本如图1所示的XRPD图谱。
本发明的一些方案中,上述A晶型,其具有基本如图2所示的DSC图谱。
本发明的一些方案中,上述A晶型的差示扫描量热曲线在111.03±3℃处具有吸热峰。
本发明的一些方案中,上述A晶型,其具有基本如图3所示的TGA曲线。
第三方面,本发明还提供式I化合物的苹果酸盐的制备方法,包括将式I化合物与苹果酸反应。
本发明的一些方案中,上述式I化合物苹果酸盐的A晶型的制备方法包括如下步骤:
将式I化合物、苹果酸与溶剂混合,加热至完全溶解,再冷却,析出晶体;其中,所述溶剂选自乙醇、异丙醇、正丙醇、丁醇、丙酮、乙酸乙酯、正庚烷、乙腈、四氢呋喃中的一种或一种以上的混合物;优选为乙醇、异丙醇、乙酸乙酯、正庚烷中的一种或一种以上的混合物;或者
将式I化合物、苹果酸与第一溶剂混合,加热至完全溶解,再加入第二溶剂,冷却,析晶。所述第一溶剂选自甲醇、乙醇、异丙醇、正丁醇中的一种或一种以上的混合物;所述第二溶剂选自甲基叔丁基醚、酯类、乙腈、烷烃类中的一种或一种以上的混合物,说是酯类选自乙酸乙酯和/或,所述脂类例如为醋酸异丙酯;所述烷烃类选自正庚烷和/或正己烷。
本发明的一些方案中,加热温度为50~80℃,例如60~70℃。
本发明的一些方案中,将式I化合物、苹果酸与溶解该化合物所用的溶剂或第一溶剂混合后进行搅拌,所述搅拌时间可以是1~5小时,优选2~3小时。
所述式I化合物与溶解该化合物所用的溶剂或第一溶剂的重量/体积比(g/mL)为1:4-10,优选地为1:6-8,例如1:6、1:7、1:8。
本发明的一些方案中,所述冷却可以是冷却至0~30℃,优选地,冷却至2~8℃,例如冷却至5℃。在冷却之前可以先进行一次过滤。
本发明的一些方案中,在冷却过程中进行搅拌,所述搅拌的时间可以是6~24小时,优选18小时。
根据本发明,在析出晶体后进行过滤,干燥;在本发明的一些方案中,干燥之前还包括任选地用溶剂洗涤的步骤,所述洗涤溶剂选自乙醇、异丙醇、正丙醇、丙酮、乙酸乙酯、乙腈、四氢呋喃中的一种或一种以上的混合物。
本发明的一些方案中,所述式I化合物与苹果酸的摩尔比为1:0.9-1.3,优选地,所述式I化合物与苹果酸的摩尔比为1:1-1.1,例如为1:1、1:1.05、1:1.1。
第四方面,本发明提供了一种药物组合物,所述药物组合物包含式I化合物的苹果酸盐或者式I化合物的苹果酸盐A晶型,和任选的药学上可接受的赋形剂。
第五方面,本发明提供了式I化合物的苹果酸盐、上述式I化合物的苹果酸盐A晶型、或包含式I化合物苹果酸盐的药物组合物、或包含式I化合物的苹果酸盐A晶型的药物组合物在制备用于治疗中枢神经系统紊乱疾病的药物中的用途。
本发明的一些方案中,所述中枢神经系统紊乱疾病包括但不限于精神分裂症、阿尔茨海默氏病、帕金森氏病、抑郁症、运动障碍、吸毒成瘾、疼痛和神经退行性变(例如陶氏病或突触核蛋白病)。
本发明的一些方案中,所述中枢神经系统紊乱疾病为精神分裂症。
第六方面,本发明提供了一种治疗或预防哺乳动物(例如人)的中枢神经系统紊乱疾病的方法,所述方法包括向哺乳动物(例如人)给予治疗有效量的式I化合物的苹果酸盐、式I化合物的苹果酸盐A晶型、包含式I化合物的苹果酸盐的药物组合物和包含式I化合物的苹果酸盐A晶型的药物组合物。
本发明的一些方案中,所述中枢神经系统紊乱疾病包括但不限于精神分裂症、阿尔茨海默氏病、帕金森氏病、抑郁症、运动障碍、吸毒成瘾、疼痛和神经退行性变(例如陶氏病或突触核蛋白病)。
本发明的一些方案中,所述中枢神经系统紊乱疾病为精神分裂症。
有益效果
1.本发明首次提供了式I化合物的苹果酸盐;式I化合物的苹果酸盐在物理稳定性、溶解性、吸湿性、生物活性、安全性、生物利用度、毒副作用等至少一方面具有优异的效果。
2.式I化合物苹果酸盐的A晶型的稳定性好,吸湿性小,水溶性好,批次间差异小、成药前景良好。
3.本发明的盐型、晶型制备工艺简单,适宜工业化生产。
附图说明
图1为式I化合物L-苹果酸盐的A晶型的XRPD图谱;
图2为式I化合物L-苹果酸盐的A晶型的DSC图谱;
图3为式I化合物L-苹果酸盐的A晶型的TGA图谱;
图4为式I化合物L-苹果酸盐的A晶型在高温(60℃)条件下0天、6天、14天、30天的XRPD图谱;
图5为式I化合物L-苹果酸盐的A晶型在高湿(RH92.5%)条件下0天、6天、14天、30天的XRPD图谱;
图6为式I化合物L-苹果酸盐的A晶型在光照条件(4500±500Lux)条件下0天、6天、14天、30天的XRPD图谱。
图7为口服给予式I化合物L-苹果酸盐的A晶型(实施例2样品)、式I化合物(实施例1样品)后大鼠血浆中式I化合物的药代动力曲线。
具体实施方式
下文将结合具体实施例对本发明的通式化合物及其制备方法和应用做更进一步的详细说明。应当理解,下列实施例仅为示例性地说明和解释本发明,而不应被解释为对本发明保护范围的限制。凡基于本发明上述内容所实现的技术均涵盖在本发明旨在保护的范围内。
除非另有说明,以下实施例中使用的原料和试剂均为市售商品,或者可以通过已知方法制备。
本发明采用下述缩略词:r.t.代表室温;aq代表水溶液;DCM代表二氯甲烷;THF代表四氢呋喃;MeOH代表甲醇;EtOH代表乙醇;IPA代表异丙醇;DSC代表差示扫描量热仪;TGA代表热重分析;
化合物经手工或者ChemDraw软件命名,市售化合物采用供应商目录名称。
下列实施例中未注明具体条件的实验方法,通常按照常规条件,或按照制造厂商所建议的条件。除非另外说明,否则百分比和份数按重量计算。
本发明实施例对盐和晶体进行表征所采用的测试方法和仪器如下:
粉末X-射线衍射(又称“X-射线粉末衍射”,X-ray powder diffractometer,XRPD)方法
仪器型号:布鲁克D8 advanceX-射线衍射仪
测试方法:大约10~20mg样品用于XRPD检测。
详细的XRPD参数如下:
光管:Cu,kα,
光管电压:40kV,光管电流:40mA
发散狭缝:0.60mm
探测器狭缝:10.50mm
防散射狭缝:7.10mm
扫描范围:4-40deg
步径:0.02deg
步长:0.12秒
需要说明的是,在X-射线粉末衍射光谱(XRPD)中,由结晶化合物得到的衍射谱图对于特定的结晶往往是特征性的,其中谱带(尤其是在低角度)的相对强度可能会因为结晶条件、粒径和其它测定条件的差异而产生的优势取向效果而变化。因此,衍射峰的相对强度对所针对的结晶并非是特征性的,判断是否与已知的结晶相同时,更应该注意的是峰的相对位置而不是它们的相对强度。此外,对任何给定的结晶而言,峰的位置可能存在轻微误差,这在结晶学领域中也是公知的。例如,由于分析样品时温度的变化、样品移动、或仪器的标定等,峰的位置可以移动,2θ值的测定误差有时约为±0.2°。因此,在确定每种结晶结构时,应该将此误差考虑在内。在XRPD图谱中通常用2θ角或晶面距d表示峰位置,两者之间具有简单的换算关系:d=λ/2sinθ,其中d代表晶面距(又称“面间距”),λ代表入射X射线的波长,θ为衍射角。对于同种化合物的同种结晶,其XRPD谱的峰位置在整体上具有相似性,相对强度误差可能较大。还应指出的是,在混合物的鉴定中,由于含量下降等因素会造成部分衍射线的缺失,此时,无需依赖高纯试样中观察到的全部谱带,甚至一条谱带也可能对给定的结晶是特征性的。
由包括以下的多种因素产生与这类X射线粉末衍射分析结果相关的测量差异:(a)样品制备物(例如样品高度)中的误差,(b)仪器误差,(c)校准差异,(d)操作人员误差(包括在测定峰位置时出现的误差),和(e)物质的性质(例如优选的定向误差)。校准误差和样品高度误差经常导致所有峰在相同方向中的位移。当使用平的支架时,样品高度的小差异将导致XRPD峰位置的大位移。系统研究显示1mm的样品高度差异可以导致高至1°的2θ的峰位移。可以从X射线衍射图鉴定这些位移,并且可以通过针对所述位移进行补偿(将系统校准因子用于所有峰位置值)或再校准仪器消除所述位移。如上所述,通过应用系统校准因子使峰位置一致,可校正来自不同仪器的测量误差。
差热分析(又称“差示扫描量热法”,Differential Scanning Calorimeter,DSC)方法
仪器型号:METTLER TOLEDO DSC3+差示扫描量热仪
测试方法:取样品(~1mg)置于DSC铝锅内进行测试,在50mL/minN
热重分析(Thermal Gravimetric Analyzer,TGA)方法
仪器型号:TAQ5000IR热重分析仪
测试方法:取样品(2~5mg)置于TGA铂金锅内进行测试,在25mL/minN
高效液相色谱(HPLC)分析方法:
所用仪器为Waters ARC 2998HPLC;色谱柱:Agilent Poroshell bonus-RP 4.6×100mm,2.7μm;
测定条件如下:
进样体积:10ul;
流速:1.0mL/min;
检测波长:275nm;
样品浓度:1.0mg/mL;
稀释液:20%乙腈水溶液;
柱温:40℃;
流动相A:0.2%磷酸水溶液;
流动相B:乙腈-甲醇(1:1)溶液;
洗脱梯度如表1所示:
表1洗脱梯度条件
目前,公知常识普遍教导,选择具有所需的性能组合的盐仍然是一个困难的半经验性的选择,需要盐形式的性质的折衷选择,但是仍然存在评估哪种盐形式为最适合筛选特定候选药物的困难。
在实验过程中,发明人发现药物吸湿性会严重影响药物的流动性,甚至会影响药物的稳定性。药物的溶解度对药剂的制备、药物溶出、吸收等都具有至关重要的影响。但如何提高药物的溶解度,不以药物的吸湿性为代价,得到药物稳定性、溶解度及吸湿性均合适的候选药物盐是困难的。
发明人经过广泛而深入的研究,出乎意料地发现式I化合物苹果酸盐经DSC测定具有较高的熔点且经过稳定性实验证实:式I化合物苹果酸盐具有较高的稳定性。另外,式I化合物苹果酸盐有较低的吸湿性,并且有适当的溶解度。式I化合物苹果酸盐的高稳定性、低吸湿度和适当溶解度的结合是不可预料的,这使得式I化合物苹果酸盐(尤其是式I化合物L-苹果酸盐)适用于制备口服固体制剂,特别是片剂的制备。
活性成份的溶解度对于剂型的选择具有重要意义,这是因为剂型对生物利用度具有直接影响。对于口服剂型而言,通常认为活性组分具有较高的溶解度是有利的,这是因为其使得生物利用度提高。
在此基础上,发明人发现式I化合物苹果酸盐的A晶型,所述A晶型在物理稳定性、热力学稳定性和机械稳定性等至少一个方面具备优势;制备得到的式I化合物苹果酸盐的A晶型,晶体大小合适,适于工业化生产制备。
一种式I化合物苹果酸盐A晶型,能够显著改善式I化合物的水溶性、且具有良好稳定性和极低的吸湿性,有利于制成口服固体制剂。该式I化合物苹果酸盐制成的制剂可以较长时间维持体内有效生理浓度的式I游离碱并且给药方便,从而具备提高患者的依从度等优点。
实施例1.式I化合物的合成
步骤1、2-羟基-2-(3-吡啶基)乙腈(中间体1)的制备
向100mL单口反应瓶中依次加入3-吡啶甲醛(6.00g,56.02mmol,1.0eq)、冰醋酸(3.36g,56.02mmol,1.0eq)和6mL纯水,在室温搅拌混匀。于2~8℃滴入三甲基氰硅烷(7.42g,74.42mmol,1.3eq)的水溶液中,搅拌反应,TLC(薄层色谱)板确认反应结束,冰盐浴降温至-5℃搅拌析晶体。过滤,用冰水(5mL*3)洗涤滤饼,得白色固体(6.72g,收率89%)。
1
步骤2、2-氨基-2-(3-吡啶基)乙腈(中间体2)的制备
向50mL二口反应瓶依次加入NH
步骤3、3-(4-氯-1,2,5-噻二唑-3-基)吡啶(中间体3)的制备
向250mL三口反应瓶依次加入S
1
步骤4、3-(4-己氧基-1,2,5-噻二唑-3-基)吡啶(中间体4)的制备
向100mL三口反应瓶依次加入60%NaH(6.21g,155.28mmol,9.0eq)、四氢呋喃(9.00mL),0~5℃滴入四氢呋喃(18.00mL)稀释的正己醇(6.27g,61.47mmol,3.0eq),室温搅拌2小时。将四氢呋喃(15.00mL)溶清的中间体3(3.41g,17.25mmol,1.0eq)于室温滴入体系,磁力搅拌3小时。反应液加饱和碳酸氢钠水溶液(30mL)洗涤,水相用二氯甲烷(30mL*3)萃取,减压浓缩。粗品柱层析(石油醚(60-90)/乙酸乙酯5:1-3:1)得类白色固体(3.61g,收率79%)。
1
步骤5、碘化-1-氘代甲基-3-(4-己氧基-1,2,5-噻二唑-3-基)吡啶(中间体5)的制备
向100mL三口反应瓶依次加入中间体4(3.47g,13.2mmol,1.0eq)、丙酮(50.00mL)和氘代碘甲烷(5.72g,39.5mmol,3.0eq)于室温搅拌24小时。中间体5从体系析出,过滤得亮黄色固体(5.18g,收率96%)。
1
步骤6、氘代占诺美林(式I化合物)的制备
向250mL三口反应瓶依次加入中间体5(2.00g,4.9mmol,1.0eq)、乙醇(24.00mL)搅拌溶清,-5~0℃滴加NaBH
1
实施例2.式I化合物L-苹果酸盐A晶型的制备
称取式I化合物(10.0g,35.2mmol)、L-苹果酸(4.81g,35.8mmol)于烧瓶中,加入异丙醇70mL,升温至60~70℃,搅拌混合,使得混合物完全溶解,趁热过滤,得澄清液,冷却、将温度降至2~8℃、搅拌析晶,过滤得产物,用冰异丙醇洗涤,得到湿产物,将湿产物置于40℃真空干燥,干燥后得到白色固体(13.83g,收率93.9%),熔点1 11.03℃。
将所得白色固体送检XRPD、DSC和TGA,经测试,发现所得白色固体以晶体形态存在,所得晶型命名为式I化合物L-苹果酸盐A晶型,简称A晶型。所得A晶型的XRPD图、DSC图和TGA图基本上分别如图1、图2和图3所示。所得晶体的X射线粉末衍射图谱中,特征峰的峰位置及强度如表2所示;所得晶型的XRPD图谱的衍射角数据基本如表3所示,其中2θ值误差范围为±0.2°。
表2.A晶型的X射线粉末衍射图谱的特征峰的峰位置及强度
表3.A晶型的XRPD解析数据
实施例3、式I化合物L-苹果酸盐A晶型的制备
向100mL烧瓶中加入式I化合物(10.0g,35.2mmol),L-苹果酸(4.72g,35.2mmol)和异丙醇40mL,加热混合物直至完全溶解,趁热过滤、得澄清液。在搅拌下缓慢冷却溶液至2~8℃析晶,过滤收集产物,冷异丙醇洗涤,在40℃真空干燥,得到白色固体(13.3g,收率90.3%)。经测试所得产物为式I化合物L-苹果酸盐A晶型。
实施例4.式I化合物L-苹果酸盐A晶型的制备
向100mL烧瓶中加入式I化合物(4.9g,17.2mmol),L-苹果酸(2.54g,18.9mmol)和甲醇5mL,加热混合物直至完全溶解,趁热过滤、得澄清液。然后向其加入34mL乙酸乙酯。在搅拌下缓慢冷却溶液至2~8℃析晶,过滤收集产物,冷乙酸乙酯洗涤,在40℃真空干燥,得到白色固体(6.6g,收率91.7%)。经测试所得产物为式I化合物L-苹果酸盐A晶型。
实施例5.式I化合物L-苹果酸盐A晶型的制备
向100mL烧瓶中加入式I化合物(5.0g,17.6mmol),L-苹果酸(2.83g,21.1mmol)和异丙醇12mL,加热混合物直至完全溶解,趁热过滤、得澄清液。然后向其加入38mL乙酸乙酯。在搅拌下缓慢冷却溶液至2~8℃析晶,过滤收集产物,冷异丙醇洗涤,在40℃真空干燥,得到白色固体(6.8g,收率92.4%)。经测试所得产物为式I化合物苹果酸盐A晶型。
实施例6.式I化合物L-苹果酸盐A晶型的制备
向50mL烧瓶中加入式I化合物(1.0g,3.52mmol),L-苹果酸(0.6g,4.47mmol)和甲醇0.5mL,甲基叔丁基醚0.5mL,加热混合物直至完全溶解,趁热过滤、得澄清液。然后向溶液中补加5mL甲基叔丁基醚。在搅拌下缓慢冷却溶液至2~8℃析晶,过滤收集产物,冷甲基叔丁基醚洗涤,在40℃真空干燥,得到白色固体(1.3g,收率88.4%)。经测试所得产物为式I化合物苹果酸盐A晶型。
实施例7.式I化合物硝酸盐的制备
向25mL烧瓶中加入式I化合物1085mg,乙酸乙酯3mL,搅拌混合物直至完全溶解,趁热过滤、得澄清液。在搅拌下向其加入硝酸至pH值介于3~4之间,然后向其补加甲基叔丁基醚12mL,搅拌析晶,过滤收集产物,冷甲基叔丁基醚洗涤,在40℃真空干燥,得到白色固体1000mg,收率75.5%。经测试所得产物为式I化合物硝酸盐。熔点:64.5℃(DSC)。
实施例8.式I化合物丁二酸盐的制备
向50mL烧瓶中加入式I化合物505mg,丁二酸210mg和异丙醇2mL,加热混合物直至完全溶解,趁热过滤、得澄清液,然后向其加入18mL乙酸乙酯。在搅拌下缓慢冷却溶液至2~8℃析晶,过滤收集产物,冷乙酸乙酯洗涤,在40℃真空干燥,得到白色固体630mg,收率88.2%。经测试所得产物为式I化合物丁二酸盐。熔点:93.4℃(DSC)。
实施例9.式I化合物对甲苯磺酸盐的制备
向50mL烧瓶中加入式I化合物501mg,对甲苯磺酸337mg和异丙醇2mL,加热混合物直至完全溶解,趁热过滤、得澄清液,然后向其加入18mL乙酸乙酯。在搅拌下缓慢冷却溶液至2~8℃析晶,过滤收集产物,冷乙酸乙酯洗涤,在40℃真空干燥,得到白色固体720mg,收率90%。经测试所得产物为式I化合物对甲苯磺酸盐。
实施例10.式I化合物酒石酸盐的制备
向100mL烧瓶中加入式I化合物3.0g,L-酒石酸1.6g和异丙醇15mL,加热混合物直至完全溶解,趁热过滤、得澄清液,然后向其加入45mL乙酸乙酯。在搅拌下缓慢冷却溶液至2~8℃析晶,过滤收集产物,冷乙酸乙酯洗涤,在40℃真空干燥,得到白色固体4.2g,收率91.5%。经测试所得产物为式I化合物酒石酸盐。
实施例11.式I化合物其他盐的制备
选取甲磺酸、苯磺酸、苯甲酸、丙二酸、己二酸,参照实施例2-10的制备方法与式I化合物进行成盐,但均未得到固体形式的式I化合物的盐。
测试例1实施例1-10合成的各化合物和盐的理化性质测定实验
1.熔点测定实验
通过差式扫描量热仪(DSC)测定实施例1-10合成的式I化合物(实施例1所得样品)、式I化合物L-苹果酸盐A晶型(实施例2所得样品)、式I化合物硝酸盐(实施例7所得样品)、式I化合物丁二酸盐(实施例8所得样品)、式I化合物对甲苯磺酸盐(实施例9所得样品)、式I化合物酒石酸盐(实施例10所得样品)的熔点,不同盐型的熔点见表4。
2.溶解度测定实验
取七份2mL纯化水分别逐次向其中加入式I化合物(实施例1所得样品)、式I化合物L-苹果酸盐A晶型(实施例2所得样品)、式I化合物硝酸盐(实施例7所得样品)、式I化合物丁二酸盐(实施例8所得样品)、式I化合物对甲苯磺酸盐(实施例9所得样品)、式I化合物酒石酸盐(实施例10所得样品)至不溶,并记录相应用量m,目测溶解度S=m/2mL。另搅拌24h后,取上层清液送HPLC分析、所测得溶液浓度即为HPLC分析方法测定溶解度。不同盐型在水中溶解度结果见表4。本实验溶解度均在环境温度(实测环境温度为10℃)中测定。
3、吸湿性考察
取上述实施例1、2、7-10合成的各样品适量,精密称定,称得重量为m1。将样品平摊放置于扁形称量瓶中,称得总重为m2,于25±1℃、80±1%RH条件下放置24h后,称重为m3。通过下式计算得到吸湿增重ΔW,ΔW=(m3-m2)/m1*100%,不同盐型吸湿增重结果见表4。
吸湿性评价指标参见表5:
表4.各样品的理化性质
表4中,a:使用HPLC法测定溶解度;b:使用目测法测定溶解度。NA表示未测定。
表5.吸湿性评价指标
熔点测定实验结果:式I化合物L-苹果酸盐的熔点为111.03℃,式I化合物L-苹果酸盐的稳定性较好。
溶解度测定实验结果:式I化合物L-苹果酸盐的溶解度为40.48mg/mL,式I化合物L-苹果酸盐的溶解度较好。
吸湿性实验结果:式I化合物L-苹果酸盐在25±1℃、80±1%RH条件下的吸湿增重为1.97%,说明式I化合物L-苹果酸盐为略有吸湿性。
实验结果分析:式I化合物L-苹果酸盐具有较高的熔点(即具备较高稳定性)、较低的吸湿性、并且有适当的溶解度,这使得式I化合物苹果酸盐适用于制备口服固体制剂,特别是片剂的制备。
测试例2.实施例2、7-10所制备的盐的物理稳定性研究
将式I化合物L-苹果酸盐A晶型(实施例2所得样品)、式I化合物硝酸盐(实施例7所得样品)、式I化合物丁二酸盐(实施例8所得样品)、式I化合物对甲苯磺酸盐(实施例9所得样品)、式I化合物酒石酸盐(实施例10所得样品)分别敞口平摊放置,在高温(60℃)、高湿(RH92.5%)、光照(4500±500Lux)条件下进行样品的稳定性试验,测定在不同取样时间(0天、6天、14天、30天)样品中活性物质(式I化合物或占诺美林)和有关物质的含量,具体如表6所示。
表6.盐型的稳定性实验
表6中,a:式1化合物的硝酸盐,在高温条件下为液态,物理性质不稳定。NA表示未测定。
由上表6中的结果可见,本发明的式I化合物L-苹果酸盐A晶型的杂质含量很低,且在高温、高湿及光照条件下稳定性非常好,具有良好的成药前景;出乎意料的,式I化合物苹果酸盐A晶型在光照条件下相对其他盐型更加稳定,便于原料药及药物制剂的储存。
测试例3.不同盐型的毒副作用研究
实验材料:SD大鼠(购自北京维通利华实验动物技术有限公司,生产许可证号:SCXK(京)2016-0006)、式I化合物(实施例1)、式I化合物苹果酸盐(实施例2)、式I化合物酒石酸盐(实施例10)、纯化水(自制)。
实验方法:将24只SD大鼠随机分为4组(每组6只),经口灌胃给药,分别按30mg/kg(以游离碱量计)的剂量给予式I化合物苹果酸盐(实施例2所得样品)水溶液、式I化合物酒石酸盐(实施例10所得样品)水溶液、占诺美林(实施例1所得样品)水溶液及适量的纯化水。给药前、后分别观察SD大鼠的精神状态,并观察给药后各组SD大鼠的流涎情况,记录流涎大鼠的只数。给药前及给药过程中如发生任何异常,需及时记录,具体结果见表7。
表7.不同盐型诱导的副反应
实验结果分析:占诺美林是一种M受体激活剂,其不仅能作用于中枢神经系统,其也可以刺激外周神经组织中的M受体,从而导致胆碱能副作用,如流涎、恶心、头晕等。本实验通过观察各组SD大鼠的流涎情况来评价各化合物的毒副作用。通过实验结果可知式I化合物L-苹果酸盐A晶型的胆碱能副作用小,具有良好的安全性。
测试例4式I化合物L-苹果酸盐的多晶型研究
将通过实施例2方法制备得到的式I化合物L-苹果酸盐A晶型在表8所示溶剂中悬浮打浆,在40℃下避光搅拌2天,将溶液离心去沉淀干燥后经XRPD检测,结果如表8所示:
表8.测试溶剂
经上表中的结果分析得出:晶型A稳定性良好,在不同溶剂体系下均能保持稳定。
测试例5.式I化合物L-苹果酸盐A晶型的稳定性加速实验研究
将实施例2方法制得的式I化合物L-苹果酸盐A晶型样品敞口平摊放置,考察在高温(60℃)、高湿(RH92.5%)和光照条件(4500±500Lux)下样品的化学稳定性,分别在0天、7天、14天和30天取一定量样品检测样品纯度(用HPLC检测)及样品晶型,来评价氘代占诺美林苹果酸盐A晶型稳定性,实验结果见表9。
表9.A晶型物理化学稳定性实验结果
经过实验及图4、图5及图6所示的XRPD图谱可知,式I化合物苹果酸盐A晶型在高温(60℃)、高湿(RH92.5%)和光照条件(4500±500Lux)条件下,0天、6天、14天、30天时刻点的XRPD图谱与图1中A晶型的XRPD图谱基本相同,A晶型的稳定性良好。在高温(60℃)、高湿(RH92.5%)和光照条件(4500±500Lux)下,经过30天的稳定性加速实验,A晶型的纯度基本保持不变,没有杂质产生,A晶型没有发生改变。
测试例6.不同盐型的药代动力学研究
实验材料:雄性SD大鼠(体重180-220g,购自北京维通利华实验动物技术有限公司,生产许可证号:SCXK(京)2016-0006)、式I化合物L-苹果酸盐A晶型(实施例2)、式I化合物(实施例1)、纯化水(自制)。
实验方法:将6只雄性SD大鼠随机分为2组(每组3只),试验期间自由饮水,给药前禁食12小时以上,给药后4小时喂食。经口灌胃给药,对两组SD大鼠分别按30mg/kg(以游离碱量计)的剂量给予式I化合物苹果酸盐A晶型水溶液、式I化合物水溶液。给药前0min,给药后15min、30min、45min、1.0h、1.5h、2h、3h、4h、6h、8h、和10h各采集血样至K
表10.不同化合物的药代动力学参数
/>
通过表10中数据可以看出:对于大鼠给予式I化合物L-苹果酸盐A晶型后,T
以上,对本发明的实施方式进行了说明。但是,本发明不限定于上述实施方式。凡在本发明的精神和原则之内,所做的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。