生产甲基吡咯烷酮的方法
文献发布时间:2023-06-19 19:30:30
技术领域
本发明涉及精细化工产品制备的技术领域,具体涉及一种生产甲基吡咯烷酮的方法。
背景技术
N-甲基吡咯烷酮(NMP)是一种极性非质子传递溶剂,具有高沸点、强极性、低粘度、强溶解能力、无腐蚀、毒性小、化学及热稳定性好等优点,主要用于芳烃萃取,乙炔、烯烃、二烯烃的纯化分离,聚合物溶剂以及聚合反应溶剂、半导体制备过程中的溶剂等。N-甲基吡咯烷酮的生产技术主要有4种:γ-丁内酯与单甲基胺无催化合成工艺、γ-丁内酯与混合甲基胺连续无催化合成工艺、γ-丁内酯与单甲基胺催化合成工艺以及1,4-丁二醇催化脱氢-胺化工艺。
专利申请CN 1263523A中公开了γ-丁内酯与单甲基胺无催化反应合成N-甲基吡咯烷酮,由于没有催化剂反应则需要较高的温度和压力,如250-310℃的反应温度和3-9MPa的压力,且反应时间长,总流程需要2-6h。专利申请CN 1384820A中公开了一种通过γ-丁内酯和混合甲基胺反应生产N-甲基吡咯烷酮的方法,巴斯夫对上述工艺进行了改进,包含3个串联反应工序,3个工序温度逐级升高,工序3的反应温度为280℃,总反应时间4h以上,压力在4-10MPa。韩国SK公司探索研究了分子筛催化合成工艺,该工艺采用纯度为99.0%以上的γ-丁内酯和40%的单甲胺溶液作原料,复合稀土铈/ZSM催化剂的加入量为单甲胺溶液的0.01%-0.5%,其中稀土铈的含量为1%-10%,反应温度为180-250℃,反应压力为4.0-6.0MPa,停留时间为0.5-2.5h(Y S Yoon,H K Shin,B S Kwak.Ring conversion of γ-butyrolactoneintoN-methyl-2-pyrrolidone over modified zeolites.CatalysisCommunications,2002,3:349-355.)。采用1,4-丁二醇的直接脱氢反应后胺化反应,虽然会省去中间步骤,减少工艺操作和能耗,但副反应多,分离提纯有难度,目前不是主流方法。
目前,市场上主流的N-甲基吡咯烷酮的工艺主要是戊内酯为原料与甲胺进行反应制备,但主要存在流程长,反应压力和反应温度较高的问题。
发明内容
本发明的目的是为了克服现有技术中生产甲基吡咯烷酮过程中流程长,反应压力和反应温度较高的问题,提供一种生产甲基吡咯烷酮的方法,该方法具有反应时间短,能耗低和合成效率高的优点。
为了实现上述目的,本发明提供一种生产甲基吡咯烷酮的方法,该方法包括如下步骤:
(1)将丁内酯与甲胺进行开环反应得到物流1;
(2)在催化剂存在条件下,将物流1、氢气以及任选地水进行成环反应;
其中,步骤(1)所述开环反应中,丁内酯的转化率不低于80%。
优选地,步骤(1)所述开环反应中,丁内酯的转化率为80-100%,进一步优选为85-100%。
本发明通过将反应分为两阶段连续进行反应,同时第二步在催化剂的作用下,提高合成甲基吡咯烷酮的效率。该反应过程分为两个合成阶段,第一阶段为开环胺化阶段,第二阶段为脱水成环阶段。控制第一阶段的反应深度,消除反应热点,充分发挥催化剂的作用,达到降低副产物,进一步提高反应效率的目的,优选情况下,通过微通道反应器将丁内酯和甲胺进行反应,可以实现连续操作。
附图说明
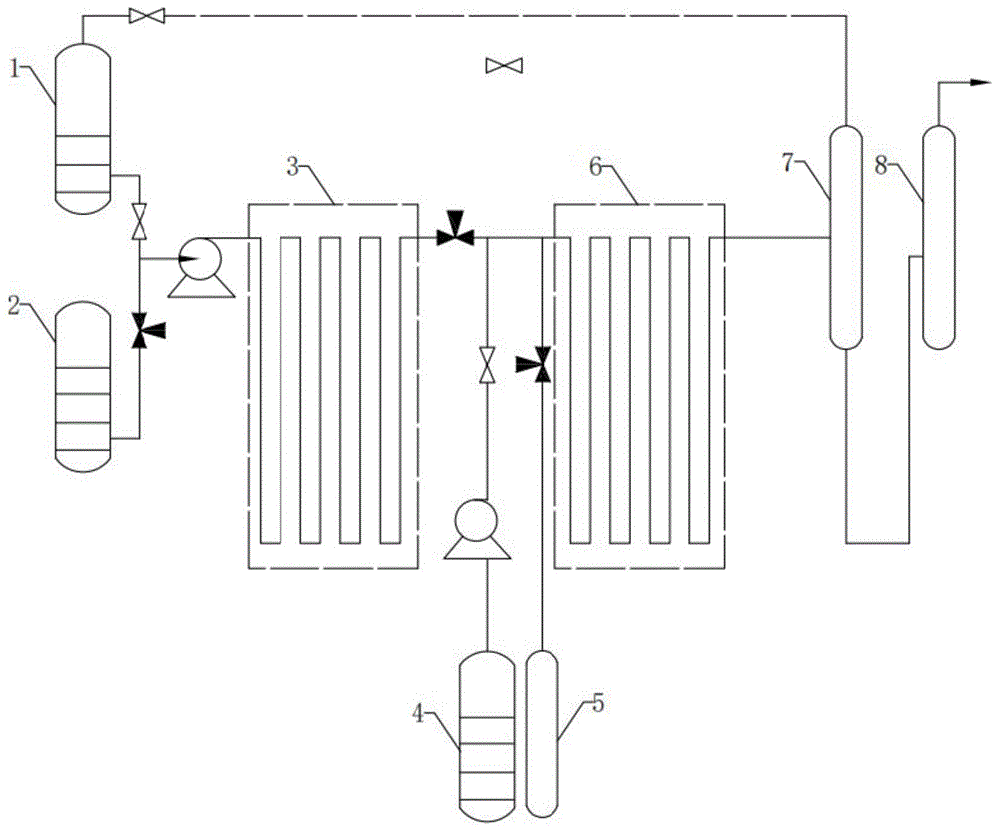
图1是本发明的反应流程图。
附图标记说明
1-缓冲罐 2-丁内酯+甲胺原料罐
3-第一微通道反应器 4-催化剂+内酯原料罐
5-氢气 6-第二微通道反应器
7-闪蒸罐 8-精馏塔
具体实施方式
在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视为在本文中具体公开。
本发明提供一种生产甲基吡咯烷酮的方法,该方法包括如下步骤:
(1)将丁内酯与甲胺进行开环反应得到物流1;
(2)在催化剂存在条件下,将物流1、氢气以及任选地水进行成环反应;
其中,步骤(1)所述开环反应中,丁内酯的转化率不低于80%。
根据本发明的方法,优选地,步骤(1)所述开环反应中,丁内酯的转化率为80-100%,优选为85-100%。
通过采用上述技术方案,将反应过程分为两个合成阶段,第一阶段为开环胺化阶段,第二阶段为脱水成环阶段。第一阶段控制反应深度,因第二阶段的脱水成环阶段为可逆反应,第一阶段开环反应不足,则需第二阶段采用更加苛刻的反应条件促使丁内酯开环,此过程不仅发生复杂的副反应,为后续的分离增加困难,而且增加能耗。本发明控制第一反应阶段的反应深度,使得丁内酯的转化率>80%,开环反应得到酰胺中间体。第二阶段,通过加入催化剂和任选地水,并通入氢气,加速脱水环化形成N-甲基吡咯烷酮,在本阶段优选加入水,加入水将进一步促进成环反应的发生。两个阶段独立连续完成,提高了反应效率,使整个反应在相对温和的条件下完成。
本发明中,对步骤(1)中丁内酯和甲胺的用量没有特别限定,只要能够使得开环反应顺利完成即可。优选地,丁内酯和甲胺的质量比例为1:0.9-4,进一步优选为1:1-1.5。
根据本发明的方法,开环反应的条件只要使得满足丁内酯的上述转化率即可,优选地,步骤(1)中所述开环反应的条件包括:反应温度为180-260℃,压力为0.2-1.5MPa,反应停留时间为5-30min;进一步优选地,步骤(1)所述开环反应的条件包括:反应温度为185-240℃,压力为0.2-1.0MPa,反应停留时间为5-20min。采用此种优选实施方式的目的是使得开环反应可以独立反应,尽量不受第二步反应影响,提高后续反应效率。
根据本发明的方法,步骤(1)中开环反应得到的物流1中包含丁酰胺及未反应的丁内酯和甲胺。
根据本发明的方法,优选地,步骤(2)中所述成环反应的条件包括:反应温度为120-180℃,压力为0.2-1.5MPa,反应停留时间为10-60min;进一步优选地,反应温度为130-160℃,压力为0.2-1.0MPa,反应停留时间为15-45min。采用此种优选实施方式的优点为提高第二步的效率,避免第一步和第二步相互间的干扰,使催化剂更为有效。
本发明中,对步骤(2)的所述的催化剂的用量没有具体限制,只要能够使得成环反应顺利进行即可。优选地,所述催化剂的加入量为丁内酯和甲胺总质量的0.1-10%,进一步优选为0.1-5%。
在一种优选情况下,水的加入量为丁内酯和甲胺总质量的1-30%。在成环反应阶段加入特定量的水能够进一步促进成环反应的发生,加速脱水形成甲基吡咯烷酮。为了进一步促进成环反应,提高反应效率,水的加入量进一步优选为3-20%。
在一种优选情况下,在成环反应阶段中通入氢气促进成环反应的发生,提高反应效率,本发明中对氢气的用量选自范围较宽。优选地,所述氢气的用量为0.1-10ml/min。
本发明中,对步骤(2)中物流1、水、催化剂以及氢气的混合顺序没有具体限制。优选地,步骤(2)所述开环反应包括:先将水与催化剂进行混合,然后与氢气进行混合,最后将得到的物流与所述物流1进行接触反应。
本发明中,步骤(2)中成环反应得到的物流包括N-甲基吡咯烷酮以及未反应的物料组分。
根据本发明的方法,在催化剂的作用下,能够提高甲基吡咯烷酮的合成效率。本发明中对催化剂的种类没有具体限制,本领域技术人员可根据具体需求进行常规选择。优选地,所述催化剂包括载体和金属活性组分。
根据本发明的方法,本发明中对载体的种类没有具体限制。优选地,所述载体选自ZSM-5、β分子筛、Y型分子筛、TiO
根据本发明的方法,本发明中对金属活性组分的种类没有具体限制,本领域技术人员可根据具体需求进行选择。优选地,所述金属活性组分选自贵金属中的至少一种。
在一种优选情况下,所述贵金属选自Pt、Ru、Pd和Ir中的至少一种,进一步优选为Pt、Ir。采用此种优选实施方式的优点为采用催化剂可提高反应效率,相同反应时间内可更快达到平衡,增加反应完成度。
本发明中,对金属活性组分的用量没有特别限制。优选地,以催化剂的总重量为基准,金属活性组分的含量为0.1-5%,进一步优选为0.1-1%。
本发明中,对所述催化剂的制备方法没有特别限制,只要能够制备上述催化剂即可。在一种优选情况下,例如,可以采用浸渍法,将贵金属前驱体引入载体上,然后采用还原剂(优选为NBH
根据本发明,所述贵金属前驱体溶液的种类没有具体限制,可以选自贵金属活性组分的水溶性化合物,例如为氯铂酸。
根据本发明,将金属活性组分前驱体溶于溶剂(例如水)中制得含有金属活性组分前驱体的溶液。
根据本发明,还原的具体条件选择范围较宽。优选地,还原温度为-10-10℃,时间为1-4h。
根据本发明,干燥的具体条件选择范围较宽。优选地,干燥温度为60-90℃,时间为1-4h。
本发明对载体的制备方法也没有特别的限定,本领域现有技术的任何制备方法均可用于本发明,优选地,以TiO
根据本发明,对S1中球磨方式没有具体限制,本领域中常规球磨方式均适用于本发明,例如干法球磨。
根据本发明,对S1和S2中干燥和焙烧的条件选择范围较宽。优选地,所述干燥温度为80-200℃,时间为1-10h;所述焙烧温度为300-400℃,时间为1-4h。
根据本发明,S2中搅拌的具体条件选择范围较宽。优选地,所述温度为60-100℃,时间为1-12h。
根据本发明,优选地,S2中搅拌过后还进行固液分离,本发明中对固液分离的方式没有具体限制,只要实现固液分离的目的即可,例如过滤。
根据本发明的方法,优选地,步骤(1)所述开环反应和步骤(2)所述成环反应均在微通道反应器中进行。采用此种优选实施方式的优点为通过微反应器将内酯和甲胺进行反应,实现精确控制反应温度,消除反应热点,充分发挥催化剂的作用,达到降低副产物和提高反应效率的目的。
在一种优选情况下,所述微通道反应器的管道内径为0.1-2mm。
在一种优选情况下,所述微通道反应器的材质为石英玻璃、高硼硅玻璃、碳化硅或聚醚醚酮管或者哈氏合金。
根据本发明的方法,优选地,该方法还包括步骤(3):从步骤(2)得到的物流中分离出甲胺。采用此种优选实施方式的目的在于去除目标产物中的甲胺,减少其对后续目标产物提纯过程造成不利影响,提高目标产物的收率。
在一种优选情况下,步骤(3)所述分离在闪蒸罐7中进行。对闪蒸罐7中的反应条件选择范围较宽,只要能分离出甲胺即可,在一种优选情况下,所述闪蒸罐中温度为10-50℃,压力0.02-0.08MPa。
根据本发明的方法,还包括缓冲阶段。优选地,该方法还包括将步骤(3)分离得到甲胺进行循环返回到缓冲罐1中,使得甲胺在缓冲罐1中暂存后汇入丁内酯+甲胺原料罐2中循环利用,提高甲胺的利用率。
根据本发明的方法,优选地,该方法包括将步骤(3)得的物料(分离出甲胺后得到的物料)进行减压蒸馏。
在一种优选情况下,所述减压蒸馏的条件包括:塔釜温度10-60℃,压力0.01-0.05MPa。采用此种优选实施方式的优点为对目标产物甲基吡咯烷酮进行提纯,获得纯度较高的甲基吡咯烷酮。
根据本发明的方法,优选地,该方法为连续生产。采用此种优选实施方式的情况下,开环反应阶段和成环反应阶段各自独立连续进行,使得整个反应过程连续不断发生,大大缩短了反应时间,减少能耗,提高了甲基吡咯烷酮的收率。
以下将通过实施例对本发明进行详细描述。以下实施例中,所用原料若无特殊说明均来自市售品。
本发明中丁内酯转化率和甲基吡咯烷酮的收率通过以下公式计算得到:
实施例1
制备Pt/TiO
S1、称取2.04g(6mol)的钛酸四丁酯与15g的硝酸钠混合球磨,将得到的固体产物在80℃下干燥12小时。
S2、取出干燥产物在350℃的马弗炉中焙烧2小时,得到的焙烧产物在80℃的去离子水中搅拌12h随后过滤干燥得到TiO
S3、称取2g TiO
甲基吡咯烷酮的生产步骤如下,反应过程如图1所示:
(1)将40g丁内酯与50g甲胺置于丁内酯+甲胺原料罐2中充分混合,用计量泵将该混合物泵入第一微通道反应器3中,投入量为4g/min;
(2)在第一微通道反应器3内反应,反应温度为160℃,压力为0.5MPa,反应停留时间为20min,检测丁内酯转化率为91%;
(3)将15g水与1g的0.2%的Pt/TiO
(4)反应后将物料转至闪蒸罐7进行闪蒸(温度为40℃,压力为0.05MPa),分离出甲胺后得到的物料进入减压蒸馏装置(精馏塔8)中进行提纯(塔釜温度60℃,压力0.05MPa),得到甲基吡咯烷酮。在第二微通道反应器6出口取样分析物料,经色谱分析,甲基吡咯烷酮收率为98.9%。
实施例2
制备Pt/ZrO
S1、称取2.30g(6mol)的锆酸四丁酯与15g的硝酸钠混合球磨,将得到的固体产物在80℃下干燥12小时。
S2、取出干燥产物在330℃的马弗炉中焙烧2小时,得到的焙烧产物在70℃的去离子水中搅拌12h随后过滤干燥得到ZrO
S3、称取2g ZrO
甲基吡咯烷酮的生产步骤如下,反应过程如图1所示:
(1)将50g丁内酯与56g甲胺置于丁内酯+甲胺原料罐2中充分混合,用计量泵将该混合物泵入第一微通道反应器3中,投入量为5g/min;
(2)在第一微通道反应器3内反应,反应温度为240℃,压力为0.4MPa,反应停留时间为10min,检测丁内酯转化率为95%;
(3)将20g水与1g的0.9%的Pt/ZrO
(4)反应后将物料转至闪蒸罐7进行闪蒸(温度为40℃,压力为0.05MPa),分离出甲胺后得到的物料进入减压蒸馏装置(精馏塔8)中进行提纯(塔釜温度40℃,压力0.03MPa),得到甲基吡咯烷酮。在第二微通道反应器6出口取样分析物料,经色谱分析,甲基吡咯烷酮收率为98.8%。
实施例3
制备Ir/ZrO
S1、称取2.30g(6mol)的锆酸四丁酯与15g的硝酸钠混合球磨,将得到的固体产物在80℃下干燥12小时。
S2、取出干燥产物在330℃的马弗炉中焙烧2小时,得到的焙烧产物在70℃的去离子水中搅拌12h随后过滤干燥得到ZrO
S3、称取2gZrO
甲基吡咯烷酮的生产步骤如下,反应过程如图1所示:
(1)将40g丁内酯与45g甲胺置于丁内酯+甲胺原料罐2中充分混合,用计量泵将该混合物泵入第一微通道反应器3中,投入量为4g/min;
(2)在第一微通道反应器3内反应,反应温度为220℃,压力为0.4MPa,反应停留时间为10min,检测丁内酯转化率为90%;
(3)将10g水与3g的0.2%的Pt/ZrO
(4)反应后将物料转至闪蒸罐7进行闪蒸(温度为40℃,压力为0.05MPa),分离出甲胺后得到的物料进入减压蒸馏装置(精馏塔8)中进行提纯(塔釜温度40℃,压力0.03MPa),得到甲基吡咯烷酮。在第二微通道反应器6出口取样分析物料,经色谱分析,甲基吡咯烷酮收率为99.1%。
实施例4
制备Pt/ZSM-5催化剂:
S1、称取10g ZSM-5粉体(购自南开催化剂厂,硅铝比为25)球磨,将得到的固体产物在80℃下干燥12小时。
S2、取出干燥产物在330℃的马弗炉中焙烧2小时,得到的焙烧产物在70℃的去离子水中搅拌12h随后过滤干燥得到ZSM-5前驱体固体产物。
S3、称取2g ZSM-5固体产物分散到15mL的0.1wt%的氯铂酸水溶液中搅拌8h,然后在5℃冰水浴中采用1M的NBH
甲基吡咯烷酮的生产步骤如下,反应过程如图1所示:
(1)将40g丁内酯与50g甲胺置于丁内酯+甲胺原料罐2中充分混合,用计量泵将该混合物泵入第一微通道反应器3中,投入量为4g/min;
(2)在第一微通道反应器3内反应,反应温度为200℃,压力为1.0MPa,反应停留时间为20min,检测丁内酯转化率为81%;
(3)将15g水与1g的0.2%的Pt/ZSM-5催化剂放入催化剂+内酯原料罐4中进行混合,流量0.8g/min,用计量泵泵入该物料,同时控制氢气5流入量为0.5ml/min,用泵将该物流和步骤(2)得到的物流共同泵入第二微通道反应器6中,反应温度为160℃,压力为1.0MPa,反应停留时间为30min;(4)反应后将物料转至闪蒸罐7进行闪蒸(温度为40℃,压力为0.05MPa),分离出甲胺后得到的物料进入减压蒸馏装置(精馏塔8)中进行提纯(塔釜温度60℃,压力0.05MPa),得到甲基吡咯烷酮。在第二微通道反应器6出口取样分析物料,经色谱分析,甲基吡咯烷酮收率为92.3%。
实施例5
制备Pd/TiO
S1、称取2.04g(6mol)的钛酸四丁酯与15g的硝酸钠混合球磨,将得到的固体产物在80℃下干燥12小时。
S2、取出干燥产物在350℃的马弗炉中焙烧2小时,得到的焙烧产物在80℃的去离子水中搅拌12h随后过滤干燥得到TiO
S3、称取2g TiO
甲基吡咯烷酮的生产步骤如下,反应过程如图1所示:
(1)将40g丁内酯与60g甲胺置于丁内酯+甲胺原料罐2中充分混合,用计量泵将该混合物泵入第一微通道反应器3中,投入量为3g/min;
(2)在第一微通道反应器3内反应,反应温度为220℃,压力为0.4MPa,反应停留时间为10min,检测丁内酯转化率为83%;
(3)将15g水与1g的0.2%的Pd/TiO
(4)反应后将物料转至闪蒸罐7进行闪蒸(温度为30℃,压力为0.05MPa),分离出甲胺后得到的物料进入减压蒸馏装置(精馏塔8)中进行提纯(塔釜温度40℃,压力0.01MPa),得到甲基吡咯烷酮。在第二微通道反应器6出口取样分析物料,经色谱分析,甲基吡咯烷酮收率为89.6%。
实施例6
制备Ru/ZrO
S1、称取2.30g(6mol)的锆酸四丁酯与15g的硝酸钠混合球磨,将得到的固体产物在80℃下干燥12小时。
S2、取出干燥产物在330℃的马弗炉中焙烧2小时,得到的焙烧产物在70℃的去离子水中搅拌12h随后过滤干燥得到ZrO
S3、称取2g ZrO
甲基吡咯烷酮的生产步骤如下,反应过程如图1所示:
(1)将40g丁内酯与50g甲胺置于丁内酯+甲胺原料罐2中充分混合,用计量泵将该混合物泵入第一微通道反应器3中,投入量为4g/min;
(2)在第一微通道反应器3内反应,反应温度为200℃,压力为1.0MPa,反应停留时间为3min,检测丁内酯转化率为75%;
(3)将17g水与3.5g的0.2%的Ru/ZrO
(4)反应后将物料转至闪蒸罐7进行闪蒸(温度为40℃,压力为0.05MPa),分离出甲胺后得到的物料进入减压蒸馏装置(精馏塔8)中进行提纯(塔釜温度60℃,压力0.05MPa),得到甲基吡咯烷酮。在第二微通道反应器6出口取样分析物料,经色谱分析,甲基吡咯烷酮收率为86.6%。
对比例1
按照实施例1的方法,不同的是,步骤(3)不加入催化剂。经色谱分析,甲基吡咯烷酮收率为55.9%。
对比例2
对比例2为控制第一阶段丁内酯的转化率为50.8%。其他反应条件同实施例1。
(1)将40g丁内酯与50g甲胺置于丁内酯+甲胺原料罐2中充分混合,用计量泵将该混合物泵入第一微通道反应器3中,投入量为4ml/min;
(2)在第一微通道反应器3内反应,反应温度为160℃,压力为0.5MPa反应停留时间为4min,检测丁内酯转化率为50.8%;
(3)将20g水与1g的0.2%的Pt/TiO
(4)反应后将物料转至闪蒸罐7进行闪蒸(温度为40℃,压力为0.05MPa),分离出甲胺后得到的物料进入减压蒸馏装置(精馏塔8)中进行提纯(塔釜温度60℃,压力0.05MPa),得到甲基吡咯烷酮。在第二微通道反应器6出口取样分析物料,经色谱分析,甲基吡咯烷酮收率为76.8%。
以上详细描述了本发明的优选实施方式,但是,本发明并不限于此。在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,包括各个技术特征以任何其它的合适方式进行组合,这些简单变型和组合同样应当视为本发明所公开的内容,均属于本发明的保护范围。