6-溴-2-甲基噻吩并2,3-d嘧啶-4-甲腈的合成方法
文献发布时间:2024-04-18 19:58:53
技术领域
本发明属于有机化学合成领域,具体涉及6-溴-2-甲基噻吩并[2,3-d]嘧啶-4-甲腈的合成方法。
背景技术
6-溴-2-甲基噻吩并[2,3-d]嘧啶-4-甲腈是制备GABAB受体正像变构调节剂ASP8062以及Ex60的关键砌块,GABAB的正变构调节作用(PAM作用),用于预防和/或治疗精神分裂症。Ex60及ASP8062化学结构式如下:
目前这一中间体主要有1条合成路线,如下式所示。该路线使用2-氨基噻吩-3-甲酸乙酯为起始原料,在酸性条件下加成、消去关环、溴代然后氯代得到目标化合物,合成路线如下:
该路线第一步收率较低,文献报道40%左右,导致整体收率30%左右。除此之外,工艺中使用了氯化氢的二氧六环溶液,对设备腐蚀性较高,存在较大的安全隐患,产生大量废水,不利于产业化。
综上所述,6-溴-2-甲基噻吩并[2,3-d]嘧啶-4-甲腈作为GABAB受体正像变构调节剂的关键砌块,具有较高的市场开发价值,其目前的合成方法还有较大改进空间。
发明内容
本发明针对现有的合成6-溴-2-甲基噻吩并[2,3-d]嘧啶-4-甲腈工艺中的安全性低,收率低,产业化难的问题,提供一种高纯度6-溴-2-甲基噻吩并[2,3-d]嘧啶-4-甲腈合成的新方法。
具体包括以下反应步骤:
1)式I化合物与酰基类物质发生反应,生成式II化合物;
2)式II化合物在碱性条件下经过缩合,得到式Ⅲ化合物;
3)式Ⅲ化合物在溴源的作用下发生取代反应,得到式IV化合物;
4)式IV化合物在氯源的作用下发生取代反应,得到式V化合物。
具体技术方案如下:
1)向反应釜中加入乙酸酐(酰基类物质)、式I化合物反应1~3小时,降至室温过滤,水洗过滤,干燥,制得式II化合物。
2)向反应釜中加入乙醇、式II化合物、5~40%的氢氧化钠溶液(碱性物质),降温至0~10℃搅拌,滴加30%双氧水搅拌0.5小时,升温至30℃搅拌0.5小时,继续升温至70~100℃反应3小时;
降至30~45℃,减压浓缩除去乙醇,加水在室温搅拌,使用6N盐酸调节PH=7~8,析出大量固体,搅拌0.5小时,过滤,干燥,制得式III化合物。
3)向反应釜中加入DMF(N,N-二甲基甲酰胺)、式III化合物,在20-30℃搅拌,加入NBS(溴源),反应1~4小时;
然后,加入水搅拌析出大量固体,搅拌1~2小时过滤,干燥,制得到式IV化合物。
4)向氢化釜中加入式IV化合物,三氯氧磷,加热到100~130℃反应1~3小时;
然后,降温至30℃左右,减压浓缩除去三氯氧磷,使用正庚烷套蒸,套蒸结束加入乙酸乙酯溶解,有机相用水洗涤,再用饱和食盐水洗涤,有机相浓缩至干得土黄色固体,加入乙腈在室温打浆0.5~2小时,过滤,干燥,得到式V化合物。
本发明优选实施方案:
步骤(1)所需的酰基类物质可以为乙酸酐、乙酰氯等,优选乙酸酐;
步骤(2)所述的所需的碱为氢氧化钠、氢氧化钾、碳酸钾、碳酸钠等,优选氢氧化钠、碳酸钾;
步骤(3)所述的所需的溴源为NBS、溴素等,优选NBS;
步骤(4)所述的所需的氯源为三氯氧磷、五氯化磷等,优选三氯氧磷。
本发明的有益效果在于:采用本发明一种合成6-溴-2-甲基噻吩并[2,3-d]嘧啶-4-甲腈的方法,工艺路线新颖,总摩尔收率大于42%,具有路线简短、化学纯度高、易于生产等特点。
附图说明
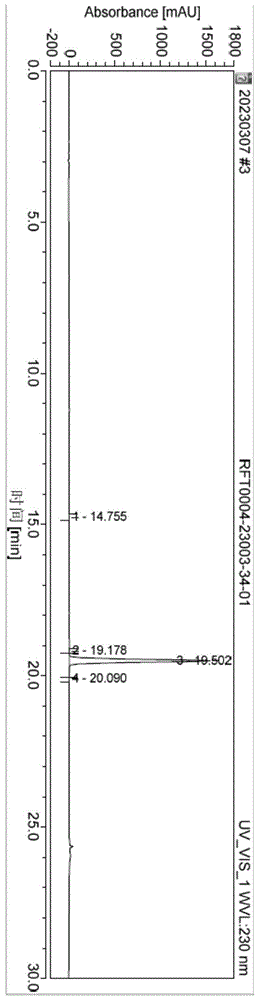
图1为实施例1制备的化合物IV的HPLC图谱;
图2为实施例1制备的化合物IV的H-NMR图谱;
图3为实施例2制备的化合物IV的HPLC图谱;
图4为实施例2制备的化合物IV的H-NMR图谱。
具体实施方式
实施例1
S1:化合物II的制备
在反应瓶中加入化合物I(40.00g.),加入乙酸酐(120ml.),升温至120℃搅拌,反应1h,反应完毕;
反应结束后,体系降温至25℃保温搅拌30min,过滤,100ml.水洗2次,得到湿品,干燥,得44.46g化合物II,纯度98.73%,收率83%。
S2:化合物III的制备
往反应瓶中加入乙醇(880ml.),加入化合物II(40.00g.),加入11%氢氧化钠溶液(146ml.)搅拌降温至0-10℃,缓慢滴加30%双氧水(220ml.),控制内温不超15℃,滴加完毕,保温搅拌30min,升温至30℃搅拌30min,升温至90℃反应3小时,反应完毕;
将反应液在45℃浓缩除去乙醇,浓缩完毕,加入水(200ml.)在室温搅拌,使用6N盐酸溶液调节PH=7-8,析出大量固体,保温搅拌30min,过滤水洗,干燥,得26.07g化合物III,纯度99.86%,收率70%。
S3:化合物IV的制备
往反应瓶中加入DMF(340ml.),加入化合物III(17.00g),30℃搅拌溶清,分批加入NBS(20.03g)反应2小时,有固体产品析出;
反应完毕,加入水(1020ml.)搅拌1小时,过滤,水洗,得湿品,干燥,得21.06g化合物IV,纯度98.82%,收率85%。
S4:化合物V的制备
在反应瓶中加入化合物IV(19.00g)、三氯氧磷(228ml.),升温至120℃,反应1h;
反应完毕,将反应液降至室温,在30℃浓缩除去三氯氧磷,使用正庚烷(190ml.)套蒸2次;浓缩完毕,得土黄色固体,加入EA(380ml.)搅拌溶解,水洗2次(190ml.*2),饱和食盐水洗1次(190ml.),得到EA相,40℃浓缩至干得土黄色固体,加入乙腈(57ml.)在室温打浆0.5小时,过滤,干燥,得18.98g化合物V,纯度99.87%,收率93%。化合物的V的HPLC图谱和H-NMR图谱如图2所示。
实施例2
S1:化合物II的制备
在反应瓶中加入二氯甲烷(400ml),化合物I(40.00g.),加入三乙胺(67.2ml.),冰水浴,滴加乙酰氯(30.3g),滴加完毕,缓慢升至20-30℃反应1-3h,反应完毕;
反应结束后,加入冰水(150ml),静置分层,饱和食盐水洗1次(190ml.),得到二氯甲烷相,40℃浓缩至干,用乙醇(100ml)和水(100ml)打浆,过滤,100ml.水洗2次,得到湿品,干燥,得44.02g化合物II,纯度98.46%,收率82%
S2:化合物III的制备
往反应瓶中加入乙醇(880ml.),加入化合物II(40.00g.),加入10%碳酸钾溶液(146ml.)搅拌降温至0-10℃,缓慢滴加30%双氧水(220ml.),控制内温不超15℃,滴加完毕,保温搅拌30min,升温至30℃搅拌30min,升温至90℃反应3小时,反应完毕;
将反应液在45℃浓缩除去乙醇,浓缩完毕,加入水(200ml.)在室温搅拌,使用6N盐酸溶液调节PH=7-8,析出大量固体,保温搅拌30min,过滤水洗,干燥,得26.61g化合物III,纯度97.79%,收率71.4%。
S3:化合物IV的制备
往反应瓶中加入DMF(340ml.),加入化合物III(17.00g),30℃搅拌溶清,分批加入NBS(20.03g)反应2小时,有固体产品析出;
反应完毕,加入水(1020ml.)搅拌1小时,过滤,水洗,得湿品,干燥,得20.56g化合物IV,纯度99.29%,收率83%。
S4:化合物V的制备
在反应瓶中加入化合物IV(19.00g)、三氯氧磷(228ml.),升温至120℃,反应1h;
反应完毕,将反应液降至室温,在30℃浓缩除去三氯氧磷,使用正庚烷(190ml.)套蒸2次;浓缩完毕,得土黄色固体,加入EA(380ml.)搅拌溶解,水洗2次(190ml.*2),饱和食盐水洗1次(190ml.),得到EA相,40℃浓缩至干得土黄色固体,加入乙腈(57ml.)在室温打浆0.5小时,过滤,干燥,得18.75g化合物V,纯度99.24%,收率92%。