一种pH响应性阳离子纳米粒子及其制备方法与应用
文献发布时间:2024-04-18 20:02:40
技术领域
本发明属于医用高分子材料领域,更具体地,涉及一种pH响应性阳离子纳米粒子及其制备方法与应用。
背景技术
炎症过程源于先天免疫系统检测到相关分子模式,进而激活蛋白相互作用、基因表达调控以及细胞内外信号通路,其中以游离核酸为代表的内源性DAMP,可被模式识别受体(pattern recognition receptors,PRR)识别,激活免疫反应。游离核酸于细胞内主要经三种通路起作用:TLR9通路、STING通路、AIM2通路。炎症失调时过量游离核酸可激活TLR9通路,启动下游各种促炎细胞因子表达,经正反馈加剧炎症反应而恶化症状。
炎症失调时过量游离核酸可激活TLR9通路,启动下游各种促炎细胞因子表达,经正反馈加剧炎症反应而恶化症状。对此常用聚氨基酸阳离子纳米颗粒(cationicnanoparticles,cNP)吸附游离核酸,以抑制其细胞内化激活TLR9。然而目前所报道cNP在体内易受蛋白黏附形成蛋白冠,失去核酸结合能力,易沉积于肝脏,受网状内皮系统(reticulo-endothelial system,RES)及肾脏识别清除。cNP通过阳离子基团结合阴离子核酸,缺乏游离核酸特异性,易无差别结合其它阴离子生物分子。局部炎症中,cNP无法靶向至炎症部位,易分布于全身,缺乏炎症部位靶向性,降低效果的同时也对健康细胞产生毒性。而阳离子聚合物因其易与细胞膜形成电荷相互作用而面临单核吞噬细胞系统快速清除,起效前便沉积于肝脾,循环时间短。因此,亟需提供一种靶向性强、不易被清除、细胞摄取能力好、在体内循环时间长的cNP。
发明内容
本发明的目的在于克服现有技术中存在的上述缺陷和不足,提供一种pH响应性阳离子聚合物。
本发明的第二个目的在于提供所述pH响应性阳离子聚合物的制备方法。
本发明的第三个目的在于提供所述pH响应性阳离子聚合物及pH响应性阳离子纳米粒子的应用。
本发明的上述目的是通过以下技术方案给予实现的:
本发明首先提供一种pH响应性阳离子聚合物,所述阳离子聚合物结构如式Ⅰ所示:
本发明的pH响应性阳离子聚合物中的PCEC作为聚合物骨架,其生物相容性优良、应用范围广泛,其中PEG外壳可屏障内部阳离子基团免于蛋白吸附及机体识别,实现长效性,较短的PEG链可暴露出内部阳离子基团,提高其ζ-电位,有利于游离核酸吸附,粒径尺度位于200nm以下,有利于cNP体内长循环。在各种胺基中,叔胺与核酸结合适中,可保证核酸稳定结合,强缓冲能力可促进内体破裂,良好生物相容性减少免疫反应及细胞毒性,因此可通过对PCEC进行叔胺化来优化cNP游离核酸清除功能,逃避RES清除,延长血液循环时间、强化抗蛋白黏附能力,同时通过其酸响应质子化能力,有效靶向至炎症部位,优化给药效率。亚细胞层面,pH响应性cNP可摄取进入内体,于酸性条件下质子化而强化其核酸结合能力,避免于胞外无差别结合生物分子。
进一步地,所述R为二乙氨基硫醚侧基。
进一步地,所述五嵌段聚合物PCL-b-P(CL-Br)-b-PEG-b-P(CL-Br)-b-PCL中PEG分子量为2000~4000。
优选地,PEG分子量为2000。
本发明还提供所述pH响应性阳离子聚合物的制备方法,包括如下步骤:
S1.环己酮经过溴化以及扩环制备得到α-溴-ε-己内酯(CL-Br);
S2.PEG引发CL-Br开环聚合(ROP)得到P(CL-Br)-b-PEG-b-P(CL-Br);
S3.以P(CL-Br)-b-PEG-b-P(CL-Br)引发ε-己内酯开环聚合制备得到可点击PCEC:PCL-b-P(CL-Br)-b-PEG-b-P(CL-Br)-b-PCL;
S4.可点击PCEC与含有二乙氨基的硫醇化合物经过S
进一步地,所述S1中扩环为拜耳-维立格氧化扩环。步骤S1具体为先使用N-溴代丁二酰亚胺与一水合对甲苯磺酸对环己酮溴化得到2-溴环己酮,再使用间氯过氧苯甲酸对2-溴环己酮进行拜耳-维立格氧化,得到CL-Br。
进一步地,所述S2中开环聚合反应以异辛酸亚锡(tin(II)octoate,Sn(Oct)
优选地,所述S2步骤中以无水甲苯溶解PEG。
优选地,所述S2步骤中所述开环聚合反应在微波反应器中于110℃下反应4h。
进一步地,所述含有二乙氨基的硫醇化合物为2-二乙氨基乙硫醇。
进一步地,所述2-二乙氨基乙硫醇的合成步骤为:将二乙胺与环硫乙烷置于微波反应管进行微波反应,反应结束后除去过量二乙胺,得到2-二乙氨基乙硫醇产物。
进一步地,所述2-二乙氨基乙硫醇的合成步骤具体为:将微波反应管烘干后转移进入手套箱中,冷却至常温后加入二乙胺,移取环硫乙烷加入二乙胺中后迅速封口,将微波反应管置于微波反应器中,60℃下反应3h,反应结束后旋蒸除去过量二乙胺得到2-二乙氨基乙硫醇产物。
本发明还提供一种pH响应性阳离子纳米粒子(pH响应性cNP),所述阳离子纳米粒子由pH响应性阳离子聚合物自组装后制得。
进一步地,所述阳离子纳米粒子的制备方法包括如下步骤:
S1.所述pH响应性阳离子聚合物用四氢呋喃震荡溶解,获得阳离子聚合物溶液;
S2.在反应瓶中加入醋酸钠缓冲液,剧烈搅拌;
S3.向S2中的反应瓶中缓慢滴入阳离子聚合物溶液,搅拌30min稳定胶束,制得自组装胶束;
S4.将S3制得的自组装胶束转移进入透析袋中,置于去离子水中透析,透析结束后,使用滤膜过滤后制得pH响应性阳离子纳米粒子。
进一步地,S2所述醋酸钠溶液为pH为5,离子浓度0.2M。
进一步地,S4所述透析袋为3500分子量;所述滤膜为220μm滤膜。
进一步地,S4所述透析为透析四次,每次透析时长大于1h。
本发明分别对所述pH响应性cNP中的PEG链长以及叔胺修饰的化合物进行比较筛选,PEG链段较长的cNP因屏蔽作用而降低了CpG结合能力,较短的PEG链可暴露出内部阳离子基团,提高其ζ-电位,有利于游离核酸吸附,而不同化合物的叔胺修饰对cNP的质子化过程以及核酸结合能力影响较大,仲胺修饰核酸吸附效果差,而硫醇修饰中又以二乙氨基硫醇化合物修饰得到的cNP核酸吸附效果好。进一步对pH响应性cNP进行核酸结合能力表征及生物活性功能测定,结果表明2kSE在pH7.72降至6.73时结合常数由6.18kM
因此,本发明提供所述pH响应性阳离子聚合物以及pH响应性阳离子纳米粒子在制备游离核酸清除剂中的应用。
本发明还提供所述pH响应性阳离子聚合物以及pH响应性阳离子纳米粒子在制备炎症调节制剂中的应用。
与现有技术相比,本发明具有以下有益效果:
本发明提供一种pH响应性阳离子聚合物,为五嵌段聚合物PCL-b-P(CL-Br)-b-PEG-b-P(CL-Br)-b-PCL叔胺化修饰后获得,本发明还提供了所述阳离子聚合物的制备方法,进一步制备获得pH响应性阳离子纳米粒子(pH响应性cNP),并对所述cNP进行核酸结合能力表征及生物活性功能测定,结果表明所述cNP可作为核酸吸附剂,具有核酸结合能力强、胞内酸性环境游离核酸吸附能力增强、靶向性提高、抗蛋白粘附性强、细胞摄取能力强、不易被清除、体内循环时间长,也能有效抑制细胞动物炎症反应中促炎细胞因子表达的特点。
附图说明
图1为合成产物的核磁氢谱图;(a)为2-溴代环己酮;(b)为CL-Br;(c)为2-二乙氨基乙硫醇。
图2为嵌段聚合物的核磁氢谱图;(a)为P(CL-Br)
图3为修饰后阳离子纳米粒子结构图及命名。
图4为阳离子纳米粒子核磁氢谱图。
图5为阳离子纳米粒子与CpG1826复合物琼脂糖凝胶电泳图。
图6为阳离子纳米粒子通过核酸染料荧光淬灭滴定表征核酸结合能力;(a)为ctDNA溶液中溴化乙锭荧光滴定;箭头方向指示彩色线条由下至上溴化乙锭浓度增加。(b)为荧光发射强度峰值关于EtBr浓度曲线。
图7为加入小牛胸腺后荧光猝灭滴定曲线;(a)为小牛胸腺DNA浓度的增加使溴化乙锭荧光强度产生变化;(b)为荧光发射强度峰值对应小牛胸腺DNA浓度(蓝色),及其一阶导数曲线(绿色)。
图8为BRB中2kSM与2kSE荧光淬灭滴定发射光谱。(a)为2kSM于pH6.73;(b)为2kSE于pH 6.73;(c)为2kSM于pH 7.72;(d)为2kSE于pH7.72。
图9为不同pH值下BRB中cNP对溴化乙锭与小牛胸腺DNA荧光淬灭,(a)为通过经典Stern-Volmer公式作图;(b)为通过修正Stern-Volmer公式作图。pH值为7.72时图中以虚线表示,溶液酸化至6.73时以实线表示,加上“+”表示。
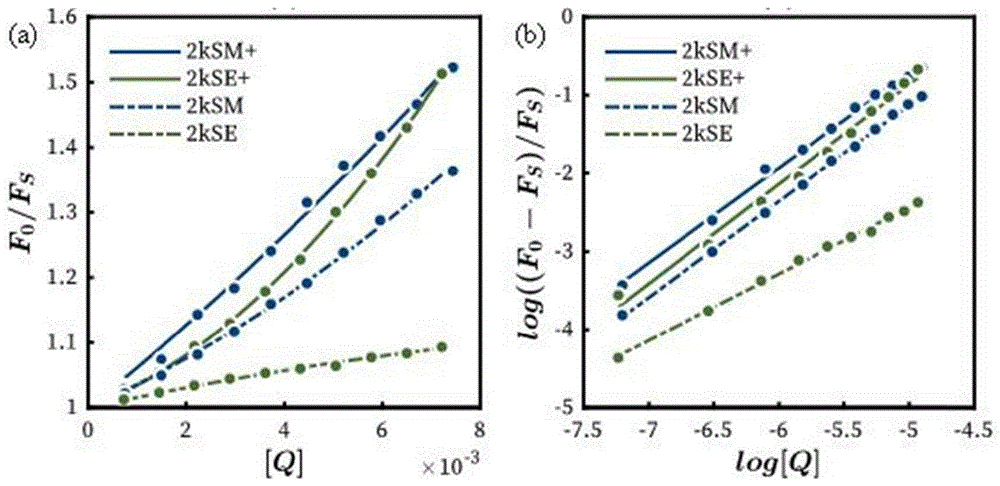
图10为在37℃下孵育1h后cNP对牛血清白蛋白吸附;各组与PAMAM显著性:*0.01 图11为流式细胞术分析;(a)、(c)为FITC-2kSM;(b)、(d)为FITC-2kSE在RAW264.7中的细胞摄取,普通细胞作为对照。 图12为细胞摄取能力表征;(a)为平均荧光强度;(b)为FITC阳性率,与阴性对照组显著性差异:*0.01 图13为腹腔注射2kSM或2kSE后小鼠器官离体荧光成像;1为脑,2为心,3为肝,4为脾,5为肺,6为肾。 图14为cNP通过结合细胞外激动剂进行炎症抑制,,同组浅色柱代表cNP浓度为12.5μg/mL,深色柱表示cNP孵育浓度为25μg/mL;各组与阳性对照组显著差异性:*0.01 图15为尾静脉注射2kSE对CpG1826所引起体温升高抑制;(a)为CpG1826;(b)为以2kSE与CpG混合溶液,(n=3)。 图16为腹腔注射2kSE对CpG1826所引起体温升高抑制表征;(a)为CpG1826或(b)为以2kSE与CpG混合溶液,(n=3)。 图17为给药cNP于TBI小鼠的治疗方案;CCI造模完成后,经颈部皮下注射cNP给药,6h后取同侧脑组织及血清检测促炎细胞因子浓度。 图18为创伤性脑损伤6h后小鼠;(a)为脑裂解液TNF-α含量,(b)为血清TNF-α浓度;与造模组差异:*0.01 图19为TBI 6h后小鼠;(a)为损伤同侧脑组织裂解液中IL-6相对总蛋白含量;(b)为血清中IL-6浓度;相对造模组:*0.01 具体实施方式 以下结合说明书附图和具体实施例来进一步说明本发明,但实施例并不对本发明做任何形式的限定。除非特别说明,本发明采用的试剂、方法和设备为本技术领域常规试剂、方法和设备。 除非特别说明,以下实施例所用试剂和材料均为市购。 实施例1可点击PCL-b-PEG-b-PCL(PCEC)制备 (1)α-溴-ε-己内酯(CL-Br)制备:CL-Br通过环己酮溴化以及拜耳-维立格(Baeyer-Villiger)氧化扩环制备获得;先使用N-溴代丁二酰亚胺与一水合对甲苯磺酸对环己酮溴化得到2-溴环己酮,再使用间氯过氧苯甲酸对2-溴环己酮进行拜耳-维立格氧化,得到CL-Br。其合成路线如反应(1-1)所示: 称取N-溴代丁二酰亚胺(NBS,57.12g,320.93mmol)与一水合对甲苯磺酸(TsOH,5.81g,30.57mmol)于500mL圆底烧瓶中并加入200mL二氯甲烷(dichloromethane,DCM)于冰水浴中搅拌。称取环己酮(30.00g,305.67mmol)于30mL DCM逐渐滴入圆底烧瓶,待冰水浴恢复室温后,升温至45℃回流反应过夜得到橙红色溶液,于-20℃冰箱中冷冻数小时后趁冷抽滤除盐,并用1:1体积饱和碳酸氢钠溶液及去离子水分别萃取洗涤3~4次,再经饱和氯化钠溶液萃取、洗涤后转移至100mL锥形瓶中以无水硫酸钠干燥过夜。干燥完成后旋蒸除去溶剂,75℃加热下减压蒸馏,收集50℃馏分于500mL圆底烧瓶中得到38.52g中间产物——2-溴代环己酮,为无色透明油状液体,产率71.2%。 将2-溴代环己酮(38.52g,217.58mmol)用250mL的DCM溶解,加入间氯过氧苯甲酸(mCPBA,55.07g,239.35mmol)于50℃下回流反应过夜得到橙红色溶液,于-20℃冰箱中冷冻后趁冷抽滤除盐,经饱和硫代硫酸钠溶液、饱和碳酸氢钠溶液及去离子水萃取,再用饱和氯化钠溶液萃取除水后以无水硫酸钠干燥过夜。旋蒸除去溶剂并使用5:1正己烷与乙酸乙酯作为洗脱剂经柱层析得到产物。用DCM/正己烷混合溶液于-20℃下重结晶得到23.48g白色晶体,抽滤得到固体后正己烷洗涤3~4次,置于双排管干燥后储存于-10℃惰性气体环境中备用,最终产率55.90%。中间产物以及最终产物通过 合成过程中间产物2-溴代环己酮粗产物为黄色油状液体,经减压蒸馏后得到透明油状液体,其核磁氢谱如图1(a)所示,峰面积及位移与产物氢相对应,证明其成功制备,CL-Br核磁氢谱图如图1(b)所示,峰面积及位移亦与预期结果相对应,证明聚合单体成功合成。 (2)可点击PCL-b-PEG-b-PCL合成:P(CL-Br)-b-PEG-b-P(CL-Br)三嵌段通过CL-Br开环聚合制备,其合成路线如反应(1-2)所示: 异辛酸亚锡(tin(II)octoate,Sn(Oct) 三嵌段聚合物P(CL-Br)-b-PEG-b-P(CL-Br)引发ε-己内酯进行ROP合成五嵌段聚合物PCL-b-P(CL-Br)-b-PEG-b-P(CL-Br)-b-PCL,其中磷酸二苯酯(diphenyl phosphate,DPP)作双作用有机催化剂,同时活化单体及可逆休眠活性链端。如反应(1-3)所示: 将P(CL-Br)-b-PEG(4000)-b-P(CL-Br)三嵌段聚合物(8.84g,1.13mmol)溶解于甲苯共沸除水三次,并在双排管中抽干后转移入手套箱备用。称量DPP(282.0mg,1.13mmol)于50mL圆底烧瓶,加入10mL无水甲苯搅拌溶解。加入ε-己内酯(CL,2.57g,22.5mmol)反应3h,反应体系单体浓度为0.25g/mL。另一批反应加入P(CL-Br)-b-PEG(2000)-b-P(CL-Br)三嵌段聚合物(8.84g,1.50mmol),磷酸二苯酯(374.53mg,1.50mmol)与己内酯(3.42g,29.9mmol)。室温搅拌三小时后转移出手套箱,将溶液于冰乙醚中沉淀,离心收集固体后沉淀溶解三次并旋蒸除去溶剂,在双排管中抽真空过夜得到白色固体。产物通过 如图2(a)所示,P(CL-Br) 实施例2阳离子纳米粒子(cNP)制备 (1)2-二乙氨基乙硫醇制备:2~5mL微波反应管烘干4h后转移进入手套箱中,冷却至常温后加入二乙胺(4.00mL,2.83g,38.7mmol),移取环硫乙烷(1.00mL,1.02g,17.0mmol)加入二乙胺中后迅速封口。将微波反应管置于微波反应器中,60℃下反应3h。反应结束后旋蒸除去过量二乙胺得到产物。反应过程通过1H NMR进行表征,速率常数通过公式(2-1)计算: 其中[R] PCEC修饰:叔胺硫醇修饰通过S 各小分子修饰完成后通过嵌段组成及修饰基团结构及命名如图3所示,各产物核磁氢谱如图4所示,因不同长度PEG所修饰产物核磁氢谱图类似,不重复展示。由PEG2000引发所形成叔胺化PCEC冠以“2k”,而引发自PEG4000则冠以“4k”;硫醇点击修饰产物命名为“S”,仲胺修饰命名为“N”;末端为二甲氨基以“M”为后缀,二乙氨基则缀上“E”。 阳离子纳米粒子(cNP)的制备:称量上述PCEC(100mg)于2.5mL离心管中,加入1mL四氢呋喃震荡溶解。另外在10mL反应瓶中加入10mL醋酸钠缓冲液(pH为5,离子浓度0.2M)。缓慢滴入阳离子PCEC溶液进于上述剧烈搅拌醋酸钠缓冲液中,搅拌30min稳定胶束,转移进入3500分子量透析袋中,置于去离子水透析四次,每次透析时长大于一小时,透析结束后,使用220μm滤膜过滤后置于无菌、4℃、避光环境储存备用。取1mL溶液于1.5mL离心管(记录空管质量)中,冻干后通过减重法计算浓度,并稀释至1mg/mL母液。 实施例3核酸结合能力表征 (1)试验方法 琼脂糖凝胶电泳:通过琼脂糖凝胶电泳法定性表征cNP核酸结合能力。通过二倍稀释法配制不同浓度cNP溶液,分别与0.4μL CpG1826溶液(1mg/mL)混合得到不同氮磷比cNP-CpG1826复合物溶液,CpG1826水溶液作为空白对照,溶液于室温孵育30min。称量琼脂粉于TAE缓冲液中配成1%w/v溶液,于微波炉低火短暂加热多次使颗粒溶解。静置降温至50~60℃后加入1%v/v核酸染料,摇匀后倒入模具静置20min成型。转移凝胶进电泳槽溶液,将上述复合物溶液与2μL甘油混合后转移至琼脂糖凝胶小孔中,80V电压及60mA电流下电泳30min后置凝胶于成像系统成像。 荧光猝灭滴定:通过溴化乙锭(ethidium bromide,EtBr)竞争法进行荧光淬灭滴定,以定量检测cNP核酸竞争能力。加EtBr溶液(1mg/mL,2.17μL)入3mL PBS或醋酸钠缓冲液溶液(sodium acetate buffer solution,SABS)中,以模拟不同pH值,测得该溶液荧光发射光谱,再加入10μL小牛胸腺DNA(calf thymus DNA,ctDNA)溶液(1mg/mL)混匀,使溶液荧光强度升高。之后依次加入不同体积cNP溶液(1mg/mL)后用移液枪吹匀。通过荧光光谱仪测定荧光发射曲线(激发波长为280nm,检测发射波长为500~800nm)。另外cNP在不同pH值伯瑞坦-罗比森缓冲溶液(Britton-Robinson buffer,BRB)荧光淬灭则在溴化乙锭浓度1μg/mL,ctDNA浓度4μg/mL下进行。淬灭能力计算如公式(3-1): 其中F K 伯瑞坦-罗比森缓冲溶液(Britton-Robinson buffer,BRB)用0.04mol/L磷酸、硼酸和醋酸配制而成,使用时加入0.2mol/L NaOH溶液调整至所需pH值。 (2)结果分析 通过琼脂糖凝胶电泳法表征cNP核酸结合能力,结果如图5所示。2kSM于氮磷比(N/P ratio)为4时即可基本结合所有CpG,而4kSM在该比时却几乎没有结合CpG,需提高氮磷比至8时才可完全结合。PEG 2000外壳cNP与对应PEG 4000外壳cNP相比可见,较短PEG链段有助于cNP结合CpG。虽然PEG链段本身不参与DNA结合,但阳离子段落与PEG嵌段通过共价连接,PEG因其亲水性而溶剂化,延长PEG链长会增大其运动范围,拖拽阳离子段落而解开氨基与DNA的相互结合,再加上PEG屏蔽阳离子与核酸相互作用,共同抑制带有长链PEG的cNP结合CpG。中性条件下2kSM因其部分质子化特点,在各氮磷比下都部分吸附CpG,4kSE在高浓度下呈现出条带拖尾,与其动态结合核酸过程相关。2kNM以及4kNM核酸吸附效果最差,实际应用价值较低。2kNE相比2kNM,中性条件下质子化程度稍强,因而表现出较强核酸结合能力,2kNE与4kNE在氮磷比为4下便可以完全吸附CpG1826,此过程中PEG链段长度影响较小。 凝胶电泳可定性表征cNP核酸结合能力,定量对比则通过核酸染料荧光淬灭滴定进行表征,其中EtBr荧光猝灭方法十分成熟,可用以淬灭常数测定及结合机理分析。EtBr通过静电相互作用及芳香环π堆积,插菲啶环于DNA双螺旋相邻碱基间形成偶联,产生强烈荧光发射;而当加入其他竞争分子取代EtBr结合DNA时,EtBr重新转变为游离状态而产生荧光淬灭。 逐渐加ctDNA入EtBr,如图6(a)所示,刚开始加入EtBr与ctDNA复合之后,在600~650nm位置产生荧光增强,继续增大EtBr浓度后,ctDNA结合趋于饱和,导致荧光增强程度逐渐减小。由图6(b)可见,滴定过程开始时蓝色曲线呈现较陡线性增长,荧光迅速增强,而随着EtBr逐渐增加,荧光增强量逐渐降低,在较高浓度下维持较平稳荧光增加,因此分别于初始、最终EtBr浓度位置(0、3.33μg/mL)对曲线作切线,其交点为理论饱和结合EtBr浓度(0.722μg/mL,1.83μM),以此作为cNP竞争结合实验浓度。ctDNA与EtBr混合于PBS中,逐渐加入cNP可使荧光发射强度逐渐降低。n表示可结合位点数。 如表1所示,荧光淬灭滴定结果线性程度较好,决定系数R 表1不同cNP与溴化乙锭与小牛胸腺DNA的荧光淬灭反应参数 如图7所示,(a)图中EtBr浓度为1μg/mL(2.54μM),逐渐加入ctDNA,箭头方向指示彩色线条由下至上代表小牛胸腺DNA浓度增加。ctDNA加入过程中荧光发射强度逐渐增加,在游离EtBr在630nm荧光逐渐降低及EtBr与ctDNA结合物610nm荧光逐渐增加共同作用下,发射波长峰值由630nm蓝移向610nm。图7(b)中,ctDNA加入2~4μg/mL时具有较高灵敏度,分别作为EtBr和ctDNA浓度,于BRB溶液体系进行荧光淬灭滴定测试。 如图8所示,黑色实线表示溶液中仅存在EtBr,加入ctDNA之后谱线升高至图中最高线条,彩色线条自上而下分别表示cNP浓度为0.33、0.67、1、1.33、1.67、2.33、2.67、3、3.33μg/mL。图8(a)、(b)、(c)中,加入cNP可将EtBr竞争下DNA,成为游离而明显降低溶液荧光发射谱线,说明2kSM在pH 6.63以及7.72下,2kSE在pH 6.63下都具有较好核酸结合能力;唯独图8(d)中2kSE在pH为7.72时,没能很好降低发射谱线,这种不同表现说明在pH由7.72酸化至6.73过程中,2kSE显著增强核酸结合能力,而2kSM则较不敏感。 对荧光淬灭滴定曲线拟合以公式(3-1)进行对比,结果如图9所示。图9(a)中2kSM在不同pH值下淬灭能力接近,并呈现线性分布,核酸结合能力具有浓度依赖性;2kSE在pH值7.72下淬灭能力较弱,而酸化至6.73则明显增强,在更高cNP浓度下斜率增大,向纵轴偏转,且可超越2kSM曲线,与其动态结合能力相关。荧光淬灭两种可能机理中,一般静态淬灭产生于荧光团与淬灭剂结合形成非荧光复合物;动态淬灭则源于荧光团激发态寿命期间与淬灭剂相遇,密切相关于淬灭剂浓度。图9(b)通过修正Stern-Volmer公式拟合,可见酸化后2kSE图线产生明显升高,结合能力明显增加,同时斜率提高表明其核酸结合协同能力亦得到提升;而与此相比,在酸化过程中2kSM改变不明显。 2kSM与2kSE在不同pH下荧光淬灭EtBr与ctDNA复合物,该过程详细参数如表2所示。2kSM结合常数由pH为7.72时155kM 表2cNP与溴化乙锭与小牛胸腺DNA荧光淬灭参数 其中a为静态淬灭常数(K 实施例4pH响应性阳离子纳米粒子对炎症调节能力的影响 (1)抗蛋白粘附 cNP抗蛋白粘附性通过BCA蛋白定量检测试剂盒检测。首先将50μL cNP溶液(1mg/mL)置于0.6mL离心管中,加入50μL牛血清白蛋白(bovine serum albumin,BSA)溶液(2mg/mL)混匀,对照组通过50μL去离子水与50μL BSA溶液混合得到。另外通过二倍稀释配制不同浓度BSA标准溶液(0.03~2mg/mL)。复合物及标准溶液在37℃摇床中孵育1h后以8000rpm转速离心5min后,取20μL上清液加入96孔板中,加入200μL现配BCA working reagent,37℃孵育30min之后使用酶标仪检测562nm吸光度,得到每个孔OD值。通过BSA标准溶液OD与已知浓度进行拟合得到标准曲线,据此转换样品孔OD值为对应BSA浓度。cNP蛋白吸附含量通过公式(C 如图10所示,37℃孵育1h后,2kSM与2kNE的BSA吸附量明显高于2kSE与2kNM,与其中性条件下较高质子化程度相关。质子化程度较高,增强了cNP与蛋白间相互作用,因而对BSA吸附量较高。相比一般阳离子树枝状高分子聚合物PAMAM,2kSM显著较高,而2kNE与其水平近似。pH响应性2kSE生理条件下蛋白吸附较低,实际应用可质子化于弱酸性条件,增强表面电荷发挥作用。 (2)细胞摄取能力 细胞摄取能力通过流式细胞术进行检测。RAW264.7铺板于12孔板中,过夜使细胞贴壁,使用温暖PBS洗涤三次后更换培养基为无血清DMEM培养基,其中含FITC-cNP(25μg/mL),置于细胞培养箱中分别避光孵育2、4、6h后用PBS洗涤三次。于12孔板中加入1mL含5%BSA的PBS后用移液枪将细胞小心从培养皿中吹下,收集于1.5mL离心管中,避光保存,门控筛选后进行荧光强度及数量测量。 一般cNP可与细胞膜上带负电荷的蛋白聚糖相互作用,通过非特异性内吞途径进入细胞。如图11所示,(a)、(b)两图中,首先圈定阳性BL1-A阈值以确保阴性对照组FITC荧光标记占比小于0.5%,再以此分析cNP不同孵育时间下的阳性率变化,可见不同时间点2kSE右移程度更大,阳性框中细胞个数均比2kSM高。(c)、(d)中,2kSM质子化程度更高,却相较低质子化2kSE表现出较少细胞摄取,原因在于其高质子化不仅增强细胞摄取能力,也更易与细胞外各种细胞因子等蛋白相互结合形成“蛋白冠”,降低表面电位而抑制其细胞摄取。2kSM孵育4h后就已饱和,无法摄取更多,6h孵育过程中只有不到10%得到细胞摄取;与之相反,2kSE却可持续摄取入细胞,孵育2、4、6h过程中,摄取率逐渐升高至40%。pH响应性2kSE细胞摄取能力更胜一筹,有助其进入内体进行游离核酸吸附,调节炎症反应。 通过平均荧光强度(mean fluorescence intensity,MFI)以及FITC阳性率,对不同孵育时长下两种cNP的细胞摄取状况进行对比,无cNP孵育的普通细胞则作为阴性对照组。图12(a)中,2kSM与2kSE在孵育后相比阴性对照组MFI都显著增强,其中2kSE相比2kSM,MFI强度增加量较大。另外图12(b)中,虽然2kSM与2kSE皆随摄取时间延长而增加细胞摄取阳性率,但2kSE细胞摄取阳性率在孵育过程中都显著高于2kSM。 两组cNP阳性率数据如表3所示,通过阴性对照组确保测得阳性率不大于0.5%,孵育2h时,FITC-2kSM仅测得2.5%摄取进入细胞,而FITC-2kSE达到了10.33%,是前者四倍有余,而在摄取4h以及6h时,FITC-2kSE摄取阳性率均能保持FITC-2kSM阳性率三倍左右,表现出优异细胞摄取能力。2kSE可通过pH响应性促进细胞内吞,基于其优异细胞摄取能力进入内体,助力其胞内游离核酸吸附;2kSM细胞摄取效率低,只能无差别吸附胞外阴离子物质,缺乏游离核酸靶向性。 表3细胞RAW264.7摄取FITC-2kSM和FITC-2kSE的阳性率 (3)生物分布定位 动物实验:动物实验均于中山大学实验动物中心SPF环境中进行,经中山大学生物伦理委员会批准,实验前后自由采食(ad libitum)。18~20g雌性昆明小鼠(KM Mouse)通过腹腔注射生理盐水溶液,其中含0.2mg/mL IR-808所标记2kSM或2kSE,cNP注射剂量为10mg/kg。分别在3、6、9、15h后处死小鼠,分别取出脑、心、肝、脾、肺、肾进行离体成像。激发及发射波长分别设置为720以及790nm,成像视野为10cm。 结果发现相比于一般cNP,pH响应性cNP 2KSE可抵抗血浆蛋白吸附及巨噬细胞清除,也因此具有更长血液循环时间。如图13所示,腹腔注射2kSM,其荧光在3h内便已达器官累积峰值,并随时间增加而迅速下降,被机体代谢。2kSM表面正电荷较强,易受血浆蛋白包裹形成“蛋白冠”,虽增强其亲水性和生物相容性,但也易受MPS识别清除而大量累积于肝脏,降低作用效果,且可能引起肝毒性。2kSE利用pH响应性增强其血液循环时间,在腹腔注射3~6h后都可维持较强荧光信号,并且可有效逃避机体识别清除,随血液分布于更多组织器官,除大脑外,图中其他器官均可观察到强弱不一的2kSE荧光信号。 (4)细胞炎症抑制 cNP炎症抑制能力通过TNF-α表达浓度进行表征。将CpG1826与cNP加入培养基使CpG1826浓度为0.5μM,而cNP浓度分别为12.5与25μg/mL。将cNP溶液替换成去离子水得到阳性对照组,培养基不含cNP与CpG1826则作为阴性对照组。将样品配制好后加入100μL于96孔板,培养24h后检测促炎细胞因子TNF-α表达,每组实验重复三次。cNP对于细胞内激动剂抑制效果通过相似步骤进行。细胞贴壁后加入0.5μM CpG1826分别培养1、2、3、4、5、6h后PBS洗涤三次以除去细胞外CpG1826,之后加入含cNP培养基培养至累计24h,通过ELISA检测促炎细胞因子TNF-α表达,实验重复三次。 TNF-α测定前一天将Coating Buffer溶液加入Capture Antibody并移取100μL于ELISA酶标板,保鲜膜封口后置于4℃冰箱包被过夜。用含0.05%Tween-20的PBS(PBST)洗板三次后,拍干ELISA酶标板剩余水分于厚纸巾,加入200μL Assay Diluent A封口并置于摇床封闭1h,洗板拍干。TNF-α标准溶液稀释于Assay Diluent A得到3.9~500ng/L的标准溶液。另外收集细胞培养96孔板各个孔中80μL样品上清液于5000rpm下离心3min后,吸取20μL上清液稀释五倍于Assay Diluent A得到样品溶液。移取100μL样品及标准溶液加入ELISA酶标板,封口并置于摇床孵育2h,洗板拍干。后面依次加入100μL Detection Antibody、Avidin-HRP,分别封口孵育1h及30min,洗板拍干,避光加入TMB Substrate并避光静置孵育15min,加入2N硫酸终止,酶标仪测定450以及570nm的OD值,做差可得TNF-α浓度依赖性OD值。通过标准溶液曲线拟合TNF-α浓度与吸光度后,将样品吸光度换算成TNF-α浓度进行对比,实验重复三次。 选用小鼠RAW264.7细胞,通过CpG刺激其TLR9通路产生炎症反应,释放TNF-α,并检测cNP干预下的促炎细胞因子表达抑制效果。如图14所示,CpG1826(0.5μM)和cNP共孵育RAW264.7持续24h。与阴性对照(negative control,NC)组仅培养DMEM相比,阳性对照(positive control,PC)组孵育CpG1826则产生显著TNF-α表达。2kNM与4kNM各浓度下均无法抑制TNF-α分泌,表明NM侧基核酸结合能力较弱。另外PEG链段较长也一定程度阻碍了cNP的炎症因子表达抑制能力,唯4kSE,及高浓度4kSM可显著抑制促炎细胞因子表达。2kSM、2kSE以及2kNE在不同浓度下都可显著抑制TNF-α释放,并显示出浓度依赖性。在所有组中,25μg/mL 2kSE炎症抑制能力最强,可将TNF-α表达降至阴性对照组水平。 (5)体温调节 将CpG1826(1mg/mL于DEPC水中)溶解在生理盐水中稀释至百分之一浓度,并以100μg/kg剂量通过尾静脉或者腹腔注射到25~30g昆明雌性小鼠体内,注射完成后将小鼠放回其饲养笼中,连续监测体温。治疗组则同时注射CpG1826及2kSE,2kSE注射量为10mg/kg。注射进行于上午9:00以避免昼夜节律影响小鼠体温,通过体温探针检测小鼠肛温,实验重复三次。 发烧过程依靠致热及退热两种细胞因子相互作用进行动态调节。其中IL-1以及IL-6为产热细胞因子;IL-10、糖皮质激素等则是退热细胞因子;TNF-α则兼具两种功能。CpG刺激TLR9,介导NF-κB上调会激活炎症反应,并提高PGE2浓度而引起发烧。 CpG ODN可通过盐水给药,并被免疫细胞自发摄取,无需要体外或体内递送系统参与,使小鼠持续数小时发热。如图15所示,以100μg/kg尾静脉注射CpG1826生理盐水溶液或者同时以1mg/kg剂量注射2kSE,并测量小鼠注射前后肛温变化。图15(a)表示通过尾静脉注射150μL CpG后2h内,小鼠显示出约0.5℃体温升高。尾静脉注射CpG与2kSE混合液则不产生明显体温升高,如图15(b)所示。说明2kSE可通过吸附CpG而抑制其激活体内炎症反应,抑制下游细胞因子激活PGE2产生而达到退烧作用。 CpG ODN以20nmol/g(约127μg/kg)剂量下腹腔注射于小鼠体内可以达到细胞因子诱导饱和,小于该浓度时发热程度则呈现与注射浓度相关依赖性。如图16(a)所示,以100μg/kg通过腹腔注射CpG1826至小鼠体内可使小鼠体温升高,由注射前37~37.5℃升高至37.5~38℃。图16(b)显示2kSE介入可使小鼠腹腔注射CpG后维持体温稳定。 (6)创伤性脑损伤(TBI)炎症调控 通过CCI建立创伤性脑损伤模型。由2~4%异氟醚诱导25~30g昆明雌鼠短暂麻醉,固定于脑定位仪,并持续维持1.5%异氟醚的空气供给。沿头皮中线切口剪开头皮,在左侧颅骨顶骨位置通过开颅手术敞开一个4.0mm直径骨窗,骨窗中点位于前囟(bregma)与人字缝尖(lambda)中点往左约3mm,移除骨瓣暴露硬脑膜并保持其完整。通过重力下落装置从25cm高度释放20.6g砝码,以2.21m/s撞击质量为6.3g,尖端直径为3mm的铝块,施加动量为45.57kg·m/s的皮质撞击损伤于骨窗暴露位置(变形深度4mm,冲击速度1.69m/s),缝合头皮,置于恒温下直至小鼠苏醒。假手术组以相同操作进行,但不进行皮质撞击。 给药组分别使用2kSM与2kSE造模后以10mg/kg通过颈部皮下注射给药。造模及给药6h后,剪去小鼠胡须,固定小鼠并压迫突出其眼球后用镊子摘除使血液流出,收集于离心管中,血液在冰浴中静置30min后,4℃下4000rpm离心10min,收集上清液,重复离心两次得到小鼠血清,保存于-80℃冰箱直至检测。小鼠颅骨中取出大脑,沿中轴线将大脑快速对半剪开,置于离心管中。左半边(损伤同侧)脑组织置于冰浴下,用研磨棒研磨使组织破碎后加入500μL RIPA裂解液,通过细胞破碎仪(150W,40kHz,50%振幅,30s)破碎后4℃下以20000rpm离心20min,收集上清液,重复离心两次得到澄清脑组织裂解液,保存于-80℃直至检测。小鼠血清以及脑组织裂解液中的TNF-α及IL-6含量通过Biolegend ELISAMAX TBI造成6h后,各种促炎细胞因子包括IL-6、TNF-α在损伤后4~6h达到浓度峰值;损伤12h后,TNF-α下降至与假手术组相当水平,但血清及损伤同侧海马体、皮质中IL-6浓度仍显著较高。因此本实施例选定损伤后6h通过脑匀浆液及血清中IL-6及TNF-α浓度对cNP炎症抑制能力进行表征。cNP若经皮下注射,可通过淋巴循环更好跨越血脑屏障,促进其脑部累积,本工作在TBI造模后,颈部皮下注射cNP给药,治疗方案如图17所示。 TBI 6h后TNF-α含量在损伤位置会增加6~10倍,损伤附近区域也会显著增加至3~5倍。本实验中,假手术组进行开颅手术但不施加皮层撞击,给药组在造模后以剂量10mg/kg通过颈部皮下分别注射2kSM与2kSE,6h后检测促炎细胞因子含量。如图18(a)所示,损伤6h后小鼠同侧半球TNF-α含量相比假手术组显著增加了1~2倍。2kSE给药可显著抑制脑损伤所造成TNF-α过多表达,使其维持在假手术组水平,2kSM抑制效果稍逊一筹,且没有显著性差异。图18(b)中,TBI 6h后血清TNF-α含量并无观察到显著变化。 TBI造成6h后,IL-6浓度于损伤区域可增加30~36倍,损伤附近位置可增加16~20倍,而损伤同侧皮层及海马体中也观察到显著升高。而在如图19(a)所示,统观TBI后损伤同侧整个半球,IL-6含量相比假手术组显著升高6~7倍,注射2kSM可降低TBI所致IL-6过度表达,但没有显著性差异(P=0.10),2kSE给药可显著降低IL-6含量至假手术组相同的水平。据报道,血清IL-6含量会在CCI造模2h后显著升高,但如图19(b)所示,6h后小鼠血清IL-6含量与假手术组并无明显差别,cNP给药也会对血清IL-6含量产生影响。 综上所述,本发明制备的2kSE为pH响应性cNP,核酸结合能力强,能够在炎症酸性部位或内体酸化环境结合游离核酸,提高cNP的靶向性;同时抗蛋白粘附能力、细胞摄取能力强,解决了cNP易受机体清除等问题,同时具有良好炎症调节能力。
- 一种具有pH响应性的席夫碱共聚物包覆的介孔二氧化硅载药纳米粒子及其制备方法和应用
- 一种具有pH响应性的两亲性白及多糖衍生物纳米颗粒及其制备方法和应用
- 一种具有pH响应性的木质素磁性纳米颗粒及其制备方法和在纤维素酶回收中的应用
- 具有pH和磁响应性杂化纳米粒子及其制备方法和在分离纳米油水乳液中的应用
- 具有pH和磁响应性杂化纳米粒子及其制备方法和在分离纳米油水乳液中的应用