小鼠人工染色体载体及其用途
文献发布时间:2023-06-19 09:49:27
技术领域
本发明涉及一种能稳定地携带在啮齿类细胞、组织或个体内,且能够传递给后代的新型小鼠人工染色体载体,具体地涉及分别来源于小鼠10号染色体和小鼠16号染色体的小鼠人工染色体。
本发明还涉及一种哺乳动物来源的细胞,其含有上述小鼠人工染色体载体。
本发明进一步涉及一种啮齿类等非人动物,其含有上述小鼠人工染色体载体。
本发明进一步涉及一种有用蛋白质或人抗体的制造方法,其利用上述细胞或上述非人动物。
背景技术
人工染色体载体由于能够导入大于约200kb的大尺寸DNA(例如,兆碱基大小的染色体片段),因此例如一直在能够用于产生人抗体、药物代谢试验、疾病模型等的非人动物制作等中使用。对于这样的载体,已知有人的人工染色体(HAC)载体、小鼠人工染色体(MAC)载体等。具体地,HAC载体在关于来源于人14号染色体和人21号染色体的载体的专利文献1、2和3,非专利文献1、2和3中有记载,此外MAC载体在关于来源于小鼠11号染色体的载体的专利文献4中有记载。
然而,在HAC载体导入小鼠中,存在着HAC载体的携带率下降、组织间或个体间产生差异、后代传递的频率不稳定等问题,因此需要经常考虑HAC载体的携带率等,此外在对特定的基因区域所具有的功能及与疾病的关系进行研究时,有时难以在组织细胞水平对目的基因的表达动态和表达产物进行详细且准确地分析,成为高重现性的均匀性分析的阻碍。此外,在小鼠细胞与人细胞进行细胞融合时,已知在小鼠细胞内人染色体不稳定。这样,包含HAC载体的人染色体在小鼠细胞内携带率不恒定,因此在将HAC载体导入到小鼠细胞时,以及在制作转基因小鼠时,无法充分发挥作为人工染色体载体的优点。
另一方面,在MAC载体导入小鼠中,基本上解决了HAC载体的携带率下降等问题。但是,仅已知MAC载体是来源于小鼠11号染色体的载体,作为在使用其他染色体时的问题点,由于关于染色体结构的信息很少,因此在制作MAC载体时需要反复试验,此外除公知的MAC载体之外的载体具有怎么样的特性也是未知的。
在这种情况下,本发明人本次尝试了来源于小鼠10号染色体和小鼠16号染色体载体这2种MAC载体的制作。
现有技术文献
专利文献
专利文献1:国际公布2009/063722号
专利文献2:国际公布2004/031385号
专利文献3:日本特开2007-295860号公报
专利文献4:日本专利第5557217号公报
非专利文献
非专利文献1:Kuroiwa et al,Nat Biotech,18:1086-1090,2000
非专利文献2:Katoh et al,BBRC,321:280-290,2000
非专利文献3:Hoshiya et al,Mol Ther,17:309-17,2009
发明内容
发明所要解决的问题
在制作其他的MAC载体而非现有公知的MAC载体时,作为所要解决的问题,例如是小鼠染色体的序列与结构相关的信息很少,且在某些情况下是未知的,或者由于这样的信息是特定的小鼠品系/个体的序列信息,因此如果品系不同则通常序列不匹配,从而存在以下问题:为了不将额外的小鼠基因引入到载体内而进行的基因操作需要大量的反复试验,包括天然着丝粒在内的染色体结构存在差异,因染色体的来源导致MAC载体的核内配置不同而有可能对宿主基因的表达控制产生影响,来源于小鼠11号染色体的MAC导入精巢来源干细胞在干细胞培养条件下显示出异常增殖等,因此需要解决这些问题。而且,由染色体编号不同的小鼠染色体制作的MAC载体与残留在载体上的小鼠基因所造成的影响同样地对稳定性、后代传递能力等性质产生如何的影响,现在还知之甚少。
理想地,MAC载体应该是MAC载体导入本身不会引起不希望的宿主基因表达波动,在啮齿类细胞或者组织内或啮齿类个体内稳定地保持,同时是能够进行后代传递的载体,如上所述,在制作这样的载体时需要反复试验。
本发明的目的在于提供一种解决上述问题的新型MAC载体。
解决问题的技术方案
本发明概括而言,包括以下特征。
(1)一种小鼠人工染色体载体,其特征在于,包含来源于小鼠染色体的天然着丝粒,所述小鼠染色体选自小鼠10号染色体和小鼠16号染色体,从靠近着丝粒的小鼠10号染色体长臂的染色体位点即基因Gm8155删除了长臂远端的来源于小鼠10号染色体的长臂片段或从靠近着丝粒的小鼠16号染色体长臂的染色体位点即基因Gm35974删除了长臂远端的来源于小鼠16号染色体的长臂片段,以及端粒序列;以及,在啮齿类的细胞、组织或个体中稳定地携带且能够传递给后代。
(2)根据上述(1)所述的小鼠人工染色体载体,其包含保藏细胞株DT40(10MAC)T5-26(NITE BP-02656)中所含的小鼠人工染色体。
(3)根据上述(1)所述的小鼠人工染色体载体,其包含保藏细胞株DT40(16MAC)T1-14(NITE BP-02657)中所含的小鼠人工染色体。
(4)根据上述(1)~(3)中任一项所述的小鼠人工染色体载体,其中,啮齿类为小鼠或大鼠。
(5)根据上述(1)~(4)中任一项所述的小鼠人工染色体载体,其还包含一个或更多个DNA序列插入位点。
(6)根据上述(5)所述的小鼠人工染色体载体,其中,DNA序列插入位点包含选自loxP序列、FRT序列、
(7)根据上述(1)~(6)中任一项所述的小鼠人工染色体载体,其还包含报告基因、选择标记基因或它们两者。
(8)根据上述(1)~(7)中任一项所述的小鼠人工染色体载体,其还包含外源DNA序列。
(9)根据上述(8)所述的小鼠人工染色体载体,其中,外源DNA序列为人DNA序列。
(10)根据上述(9)所述的小鼠人工染色体载体,其中,外源DNA序列是人染色体长臂或短臂的基因或基因座的DNA序列。
(11)根据上述(9)或(10)所述的小鼠人工染色体载体,其中,外源DNA序列是人免疫球蛋白重链基因或基因座、人免疫球蛋白轻链基因或基因座,或者人免疫球蛋白重链和轻链基因或基因座这两者的DNA序列。
(12)根据上述(8)~(10)中任一项所述的小鼠人工染色体载体,其中,外源DNA序列是选自如下的基因或DNA的序列:编码细胞因子类、激素类、生长因子类、营养因子类、造血因子类、凝血/溶血因子类、G蛋白偶联受体类、酶类等多肽类的基因或DNA的序列,或者肿瘤、肌肉营养不良、血友病、神经变性疾病、自身免疫性疾病、过敏性疾病、遗传性疾病等疾病相关的治疗用基因或DNA的序列,以及T细胞受体(TCR)、人白细胞抗原(HLA)等免疫系统基因或DNA的序列。
(13)一种哺乳动物来源的细胞,其含有上述(1)~(12)中任一项所述的小鼠人工染色体载体。
(14)根据上述(13)所述的细胞,其中,哺乳动物来源的细胞选自体细胞、干细胞和前体细胞。
(15)根据上述(13)或(14)所述的细胞,其中,哺乳动物来源的细胞是啮齿类来源的细胞。
(16)一种非人动物,其含有上述(1)~(12)中任一项所述的小鼠人工染色体载体。
(17)根据上述(16)所述的非人动物,其中,非人动物为啮齿类动物。
(18)根据上述(17)所述的非人动物,其中,啮齿类动物为小鼠或大鼠。
(19)根据上述(16)~(18)中任一项所述的非人动物,其是能够产生人抗体的动物。
(20)根据上述(16)~(19)中任一项所述的非人动物,其中,对应于小鼠人工染色体载体所含的外源DNA的内源基因被破坏或所述内源基因的表达下降。
(21)一种蛋白质的制造方法,其包括:培养含有小鼠人工染色体载体的上述(13)~(15)中任一项所述的细胞,所述小鼠人工染色体载体包含外源DNA序列,并回收所产生的由该DNA编码的蛋白质。
(22)一种人抗体的制造方法,其包括:利用含有小鼠人工染色体载体的上述(19)所述的非人动物产生人抗体,所述小鼠人工染色体载体包含人抗体重链和轻链基因或基因座,并回收该人抗体。
(23)根据上述(22)所述的方法,其中,人抗体轻链基因或基因座是人抗体λ和κ轻链基因或基因座。
本说明书包含了作为本申请的优先权基础的日本专利申请号2016-213844号的公开内容。
发明的效果
根据本发明,能够提供分别来源于小鼠10号染色体和16号染色体的小鼠染色体的、在啮齿类组织内稳定的新型小鼠人工染色体载体,从而可以制作利用这些载体的能产生人抗体的小鼠、大鼠等啮齿类动物,以及利用该动物制造人抗体。
附图说明
图1是表示通过将携带耐药基因标记的小鼠10号染色体或16号染色体的小鼠胚胎成纤维细胞与小鼠A9细胞进行细胞融合,制作携带各小鼠染色体的A9样细胞的图。
图2是表示通过微核细胞融合法将各小鼠染色体从含有耐药基因标记的小鼠10号染色体或16号染色体的A9样细胞转入鸡DT40细胞的图。
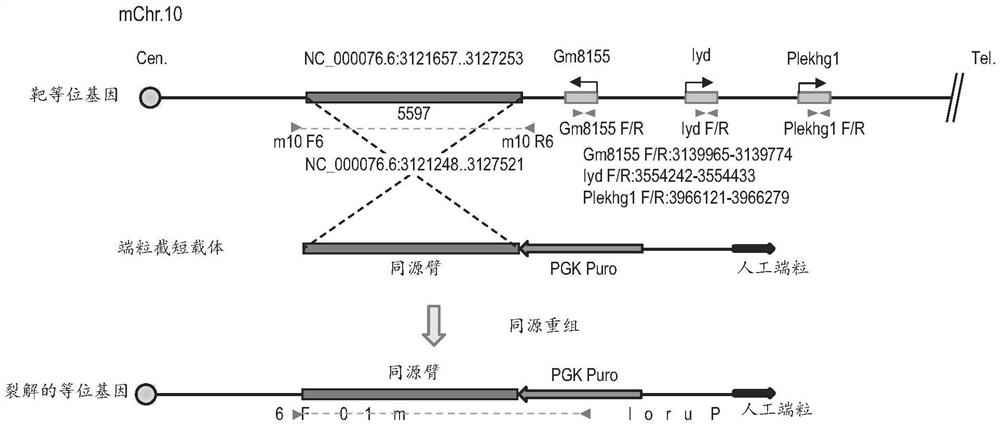
图3是表示通过端粒截短分别从天然的小鼠10号和16号染色体构建小鼠人工染色体(MAC)的步骤,以及在该MAC插入含loxP、EGFP基因等的盒的步骤的示意图。图中,Cen.表示着丝粒,Tel.表示端粒。此外,HS4表示绝缘子,CAG和PGK分别表示启动子,EGFP表示编码荧光蛋白的基因,NeoR、Puro、Neo分别表示耐药基因,HPRT表示次黄嘌呤鸟嘌呤磷酸核糖转移酶。
图4是表示小鼠10号染色体的端粒截短的示意图。
图5是表示长臂部分被删除前的小鼠10号染色体和长臂部分被删除的小鼠10号染色体(10MAC;T5-26、T6-37、T7-34)的FISH分析图像。
图6是表示向小鼠10号染色体(10MAC)搭载loxP和EGFP表达单元的示意图。
图7是表示在小鼠10号染色体(10MAC)上搭载loxP和EGFP表达单元之前与之后(10MAC1)的FISH分析图像。图中,搭载前是T5-26和T6-37,搭载后是T5-26L1-2、T5-26L2-3、T6-37L1-5。箭头指的是10MAC、10MAC1。
图8是表示携带10MAC1的CHO细胞的FISH分析图像。图中,箭头指的是10MAC1。
图9是表示小鼠16号染色体的端粒截短的示意图。
图10是表示小鼠16号染色体通过端粒截短而得到的长臂大部分被删除的2个模式的等位基因的图。图中,其中一个模式是T1-14,另外一个模式是T2-64、T2-65。
图11是表示长臂部分被删除前的小鼠16号染色体和长臂部分被删除的小鼠16号染色体(16MAC;T1-14、T2-64、T2-65)的FISH分析图像。箭头指的是小鼠16号染色体各自的16MAC。
图12是表示向删除长臂部分后的小鼠16号染色体搭载loxP和EGFP表达单元的示意图(模式1)。
图13是表示向删除长臂部分后的小鼠16号染色体搭载loxP和EGFP表达单元的示意图(模式2)。
图14是表示在小鼠16号染色体(16MAC)上搭载loxP和EGFP表达单元之前(16MAC;T1-14、T2-64、T2-65)与之后(16MAC1HA;T1-14 L1-4、16MAC1Gm;T2-64 L1-4、T2-65 L1-4)的各FISH分析图像。箭头指的是16MAC、16MAC1HA、16MAC1Gm。
图15是表示携带10MAC1的小鼠ES细胞TT2F的FISH分析图像。箭头指的是10MAC1。
图16是表示10MAC1传递给后代的小鼠的明视野(上)与GFP荧光(下)的图。由于在10MAC1上搭载有GFP表达盒,因此携带10MAC1的细胞为GFP荧光阳性。
图17是表示IGHK-NAC构建与含IGHK-NAC的产生人抗体的小鼠或大鼠的制作示意图。
图18是表示携带修饰的人2号染色体和NAC(本发明的新型人工染色体载体)的CHO细胞的FISH分析图像。左侧箭头指的是NAC,右侧箭头指的是修饰的人2号染色体。
图19是表示由Cre/loxP系统产生的相互易位的IGK-NAC构建的示意图。图中,关于HAT抗性,在loxP序列发生重组时,5’HPRT和3’HPRT连接,发生HPRT基因的重建,由此获得HAT抗性,因此能够用药剂HAT进行筛选。图中,Igκ表示免疫球蛋白轻链κ基因(座)。图中,Cen.表示着丝粒,Tel.表示端粒。此外,HS4表示绝缘子,CAG、CMV、PGK分别表示启动子,EGFP表示编码荧光蛋白的基因,Bsd、hyg、Puro、Neo分别表示耐药基因,HPRT表示次黄嘌呤鸟嘌呤磷酸核糖转移酶。
图20是表示由Flp/FRT系统产生的相互易位的IGHK-NAC构建的示意图。图中,关于HAT抗性,在FRT序列发生重组时,5’HPRT和3’HPRT连接,发生HPRT基因的重建,由此获得HAT抗性,因此能够用药剂HAT进行筛选。图中,Igκ表示免疫球蛋白轻链κ基因(座)。图中,IgH表示免疫球蛋白重链基因(座)。图中,Cen.表示着丝粒,Tel.表示端粒。此外,HS4表示绝缘子,CAG、CMV、PGK分别表示启动子,EGFP表示编码荧光蛋白的基因,Bsd、hyg、Neo分别表示耐药基因,HPRT表示次黄嘌呤鸟嘌呤磷酸核糖转移酶。
图21是表示IGHL-NAC构建与含IGHL-NAC的产生人抗体的小鼠或大鼠的制作示意图。
图22是表示由Cre/loxP系统产生的相互易位的IGL-NAC构建的示意图。图中,关于HAT抗性,在loxP序列发生重组时,5’HPRT和3’HPRT连接,发生HPRT基因的重建,由此获得HAT抗性,因此能够用药剂HAT进行筛选。图中,Igλ表示免疫球蛋白轻链λ基因(座)。图中,Cen.表示着丝粒,Tel.表示端粒。此外,HS4表示绝缘子,CAG、CMV、PGK分别表示启动子,EGFP表示编码荧光蛋白的基因,Bsd、hyg、Puro、Neo分别表示耐药基因,HPRT表示次黄嘌呤鸟嘌呤磷酸核糖转移酶。
图23是表示由Flp/FRT系统产生的相互易位的IGHL-NAC构建的示意图。图中,关于HAT抗性,在FRT序列发生重组时,5’HPRT和3’HPRT连接,发生HPRT基因的重建,由此获得HAT抗性,因此能够用药剂HAT进行筛选。图中,Cen.表示着丝粒,Tel.表示端粒。此外,HS4表示绝缘子,CAG、CMV、PGK分别表示启动子,EGFP表示编码荧光蛋白的基因,Bsd、hyg、Neo分别表示耐药基因,HPRT表示次黄嘌呤鸟嘌呤磷酸核糖转移酶。
图24是表示向小鼠10号染色体(10MAC)搭载用于插入环状DNA的loxP序列和新霉素抗性基因后的10MAC2构建的示意图。
图25是表示向小鼠10号染色体(10MAC)搭载GFP荧光表达单元和用于插入环状DNA的loxP序列和新霉素抗性基因后的10MAC3构建的示意图。
图26是表示携带10MAC2的DT40细胞的FISH分析图像。箭头指的是经构建的10MAC2。
图27是表示携带10MAC3的DT40细胞的FISH分析图像。箭头指的是经构建的10MAC3。
图28是表示携带10MAC2的CHO细胞的FISH分析图像。箭头指的是10MAC2。
图29是表示携带10MAC3的CHO细胞的FISH分析图像。箭头指的是10MAC3。
具体实施方式
将更详细地描述本发明。
如上所述,本发明提供一种小鼠人工染色体载体,其特征在于,包含来源于小鼠染色体的天然着丝粒,所述小鼠染色体选自小鼠10号染色体和小鼠16号染色体,从靠近着丝粒的小鼠10号染色体长臂的染色体位点即基因Gm8155(NCBI;NC_000076.6)删除了长臂远端的来源于小鼠10号染色体的长臂片段或从靠近着丝粒的小鼠16号染色体长臂的染色体位点即基因Gm35974(NCBI;NC_000082.6)删除了长臂远端的来源于小鼠16号染色体的长臂片段,以及端粒序列;以及,在啮齿类的细胞、组织或个体中稳定地携带且能够传递给后代。
本说明书中使用的术语“来源于小鼠染色体的天然着丝粒”是指小鼠10号染色体或小鼠16号染色体的整个着丝粒(完整的着丝粒)。因此,在这样的着丝粒中,不包括使用小鼠染色体的着丝粒序列的一部分偶然或人工得到的具有着丝粒功能的结构体,以及其他动物种类的染色体的着丝粒。
本说明书中使用的“小鼠人工染色体”或“小鼠人工染色体载体”是通过自上而下法构建的人工染色体,不是通过自下而上法构建的人工染色体。自上而下的方法是指通过染色体修饰将基因区域从自然染色体中删除,将天然着丝粒作为人工染色体载体的一部分进行构建的方法。另一方面,自下而上法是指将着丝粒序列的一部分作为克隆DNA而获得,再通过转染至哺乳类细胞来构建具有着丝粒功能的人工染色体的方法,本发明中不使用该方法。
本说明书中使用的“从靠近着丝粒的小鼠10号(或16号)染色体长臂的染色体位点即基因Gm8155(或基因Gm35974)删除了长臂远端的来源于小鼠10号(或16号)染色体的长臂片段”是指为了使本发明的载体优选在小鼠、大鼠等啮齿类的细胞或个体组织中稳定地携带,并为了不妨碍啮齿类的个体发生和后代传递而尽可能地排除内源基因的影响,因此,以基本上除去上述小鼠染色体的长臂中的内源基因的方式在靠近着丝粒的长臂位点的基因Gm8155(或基因Gm35974)的上游区域删除长臂远端而得到的着丝粒侧的长臂片段。这里,“基本上除去”是指小鼠10号染色体或小鼠16号染色体的全部内源基因(数)的至少99.5%,优选为至少99.7%,更优选为至少99.8%,最优选为99.9%~100%被除去。此外,“上游区域”是指上述基因的5’末端区域,优选为从转录起始位点到5’非翻译区末端的区域。
本说明书中使用的小鼠人工染色体在小鼠、大鼠等啮齿类的细胞、组织或个体中稳定地携带时的“携带率”是指在培养细胞或在啮齿类的组织及细胞中,存在人工染色体的细胞的比例。
本发明的染色体载体的“稳定地携带”是指在细胞分裂时该染色体载体不易发生脱落,即,即使在分裂后也能细胞内稳定地携带,从而该染色体载体有效地后代传递给子细胞或后代小鼠。
在来源于小鼠10号染色体片段的人工染色体载体的情况下,上述长臂片段非限定地例如由比该10号染色体的长臂的标记基因Gm8155更远端的区域被删除的长臂片段组成。此外,在来源于小鼠16号染色体片段的人工染色体载体的情况下,上述长臂片段非限定地例如由比该16号染色体的长臂的标记基因Gm35974更远端的区域被删除的长臂片段组成。或者,上述长臂片段包含保藏细胞株DT40(10MAC)T5-26(保藏编号:NITE BP-02656)或保藏细胞株DT40(16MAC)T1-14(保藏编号:NITE BP-02657)中所含的小鼠人工染色体作为基本结构。这些基本结构中,还可包含用于插入外源DNA、基因或基因座的loxP、FRT等DNA序列插入位点。
本发明的载体可包含用于插入外源DNA或基因序列的位点,因此通过在该位点插入目的外源DNA、基因或基因座,在该载体被导入到任何细胞时能够表达该目的外源DNA、基因或基因座,因此可应用于蛋白质生产、治疗用药剂的筛选、药物代谢试验、DNA的功能分析、基因治疗、有用的非人动物的制作等。
本说明书中的“DNA”,如无特别说明,则是指适用于包括基因或基因座、cDNA、化学修饰的DNA在内的所有种类的DNA核酸。
另外,本说明书中的“外源基因”或“外源DNA”是指插入到载体的基因插入位点的搭载于载体的目的基因或目的DNA,是应在作为目标的细胞内表达的、本来不存在于该细胞内的基因或DNA,或者它们的序列。
本说明书中的“DNA序列插入位点”是指人工染色体中可以插入目的DNA(例如基因、基因座等)序列的位点,例如,位点特异性重组酶的识别位点等。这样的识别位点非限定地包括例如loxP(Cre重组酶识别位点)、FRT(Flp重组酶识别位点)、
本说明书中的“位点特异性重组酶”是指用于在这些酶的识别位点特异性地与目的DNA序列发生重组的酶。其举例有:Cre整合酶(也称为Cre重组酶)、Flp重组酶、
本发明的载体还用于修饰小鼠染色体,将来源于小鼠的天然着丝粒直接用于载体的制作。
作为本发明的载体的有用的特性,在小鼠、大鼠、仓鼠等啮齿类的细胞或个体组织中携带率提高,由此在细胞内稳定地携带,因此能够长期稳定地含有目的基因(组),在啮齿类的个体间或组织间,能够使导入基因在量上无差异地长期表达,此外能够使经由分化为多能性细胞(例如ES细胞、iPS细胞等)的啮齿类的个体发生和后代传递的效率提高。携带率在试验的组织(例如,来源于肝脏、肠、肾脏、脾脏、肺、心脏、骨骼肌、脑或骨髓的组织)中约为90%以上,此外本发明的小鼠人工染色体能够有效地增殖,能够在细胞内携带复数(多个)拷贝。
小鼠的10号和16号染色体的序列信息可以从DDBJ/EMBL/GenBank、Santa CruzBiotechnology,Inc.等染色体数据库处获得。
本说明书中的染色体的“长臂”是指从小鼠染色体的着丝粒侧起包括基因区域的染色体区域。另一方面,小鼠染色体中基本不存在短臂。
本说明书中的“远端”是指远离着丝粒的区域(即,端粒侧)。相反地,靠近着丝粒的区域(即,着丝粒侧)称为“近端”。长臂远端是指位于比长臂的特定裂解位点更靠近端粒侧的区域,长臂近端是指位于比长臂的特定裂解位点更靠近着丝粒侧的区域。该特定裂解位点是存在于小鼠10号染色体或小鼠16号染色体长臂的全部内源基因(数)的至少99.5%,优选为至少99.7%,更优选为至少99.8%,最优选为99.9%~100%被删除的位点(位置)。
本说明书中的“端粒序列”是同种或异种的天然端粒序列,或者人工端粒序列。这里,同种是指与人工染色体载体的染色体片段所来源的小鼠为同种的动物,而异种是指除该小鼠之外的哺乳动物(其中,包括人)。此外,人工端粒序列是指(TTAGGG)n序列(n是指重复)等人工制作的具有端粒功能的序列。向人工染色体导入端粒序列例如可通过国际公布WO00/10383号中记载的端粒截短(端粒序列的替换)来进行。端粒截短可以在本发明的人工染色体的制作中用于缩短染色体。
本说明书中的术语“非人动物”包括除人之外的哺乳动物,如猴、黑猩猩等灵长类;小鼠、大鼠、仓鼠、豚鼠等啮齿类、牛、猪、绵羊、山羊等有蹄类等,但并不限于这些。
本说明书中的“胚胎干细胞”或“ES细胞”是从来源于哺乳动物的受精卵胚囊的内细胞团建立的具有分化多能性和半永久性增殖能力的干细胞(M.J.Evans andM.H.Kaufman(1981)Nature 292:154-156;J.A.Thomson et al.(1999)Science 282:1145-1147;J.A.Thomson et al.(1995)Proc.Natl.Acad.Sci.USA 92:7844-7848;J.A.Thomsonet al.(1996)Biol.Reprod.55:254-259;J.A.Thomson and V.S.Marshall(1998)Curr.Top.Dev.Biol.38:133-165)。具有与该细胞同等的性质,且通过体细胞的重编程进行人工诱导的细胞是“人工多能干细胞”或“iPS细胞”(K.Takahashi and S.Yamanaka(2006)Cell 126:663-676;K.Takahashi et al.(2007)Cell 131:861-872;J.Yu et al.(2007)Science 318:1917-1920)。
<小鼠人工染色体载体的制作和用途>
下面,对本发明的小鼠人工染色体载体的制作及其用途进行说明。具体地,在后述的实施例和附图中记载有其操作步骤。
(1)小鼠人工染色体载体的制作
本发明的人工染色体载体可通过包括以下步骤(a)~(c)的方法来制作:
(a)获得含小鼠染色体的细胞的步骤;
(b)删除小鼠染色体的长臂远端以不含大部分(99.5%以上且100%以下)内源基因(数)的步骤;以及
(c)在长臂近端插入1个以上的DNA序列插入位点的步骤。
这里,步骤(b)和(c)的顺序也可以反过来。
步骤(a):
在制作本发明的人工染色体载体时,首先制作含小鼠染色体的细胞。例如,可通过以下方式制作:将含耐药基因(例如,G418抗性基因即neo基因)标记的小鼠染色体的小鼠成纤维细胞即小鼠胚胎成纤维细胞(mChr11-neo)与导入杀稻瘟素S(blasticidin S)抗性基因即BSr基因的小鼠A9细胞(ATCC VA20110-2209)即小鼠A9(BSr)进行细胞融合,从含耐药基因标记的小鼠染色体的小鼠A9杂种细胞即小鼠A9x小鼠胚胎成纤维细胞(BSr;mChr-neo)将其染色体转移到同源重组率高的细胞来制作。小鼠成纤维细胞可以根据文献所述的方法获得,例如,小鼠成纤维细胞可通过能从日本CLEA株式会社得到的ICR品系或C57B6品系的小鼠建立。作为同源重组率高的细胞,例如可利用鸡DT40细胞(Dieken et al.,NatureGenetics,12:174-182,1996)。再者,上述转移可通过公知的染色体转移法,例如微核细胞融合法(Koi et al.,Jpn.J.Cancer Res.,80:413-418,1973)来进行。
步骤(b):
在含来源于小鼠的单一染色体的细胞中,删除该小鼠染色体的长臂远端。此时,重要的是删除(或除去或缺失)存在于长臂上的大部分内源基因而构建含小鼠着丝粒的人工染色体。这是以删除(或除去或缺失)存在于长臂上的全部内源基因的至少99.5%,优选为至少99.7%,更优选为至少99.8%,最优选为99.9%~100%地确定裂解位置。由此,人工染色体可以稳定并以高携带率在导入有人工染色体的来源于啮齿类的细胞、组织或个体中携带,所述啮齿类优选为小鼠或者大鼠,可用于目的基因(组)的准确分析、物质生产等。上述内源基因的删除例如可通过WO00/10383号所述的端粒截短来进行。具体地,在含小鼠染色体的细胞中,构建包含人工端粒序列的打靶载体,通过同源重组获得在染色体上所希望的位置插入有(人工)端粒序列的克隆,由此通过端粒截短得到缺失突变体。即,所希望的位置(或位点)是应删除的长臂远端的裂解位置,在该位置,人工端粒序列可通过同源重组被替换、插入而删除长臂远端。该位置可通过在构建打靶载体时的靶序列设计来适当设定。例如,在后述的实施例中,根据小鼠10号染色体长臂的NC_000076.6(GenBank登录号)的DNA序列和小鼠16号染色体长臂的NC_000082.6(GenBank登录号)的DNA序列设计靶序列,以在比该靶序列更靠近端粒侧引起端粒截短的方式进行设定。由此得到内源基因的大部分被删除的小鼠10号染色体片段或小鼠16号染色体片段。
步骤(c):
作为DNA序列插入位点,优选为可以插入位点特异性重组酶的识别位点。即,已知有某种酶识别特定的识别位点并特异性地在该识别位点引起DNA的重组的现象,在本发明的小鼠人工染色体载体中,可以利用由这样的酶与该酶的识别位点组成的系统,插入并搭载作为目标的基因或DNA序列。作为这样的系统,例如可举出:来源于噬菌体P1的Cre酶和其识别位点即loxP序列的系统(Cre/loxP系统;B.Sauer in Methods of Enzymology;1993,225:890-900)、来源于出芽酵母的Flp酶和其识别位点即FRT(Flp Recombination Target)序列的系统(Flp/FRT系)、来源于链霉菌噬菌体的
为了插入这样的位点特异性重组酶的识别位点,可利用公知的方法,例如同源重组法,插入位置和数量可以在长臂近端和短臂近端内适当设定。
在本发明中,可以插入一个单一种类识别位点或不同种类的识别位点。通过设定识别位点,可以确定外源基因或外源DNA的插入位置,因此插入位置固定,不会受到预想外的位置效应(position effect)。在后述的实施例中所例示的小鼠人工染色体的情况下,可以组织特异性地表达在小鼠染色体上的位点特异性重组酶的识别位点loxP序列中所插入的基因。
在本发明的具有DNA序列插入位点的小鼠人工染色体载体中,优选可以保留目标基因或DNA序列的插入位点,并预先插入报告基因。作为报告基因,没有特别限定,例如可举出:荧光蛋白(例如,绿色荧光蛋白(GFP、EGFP等)基因、黄色荧光蛋白(YFP)等)、标签蛋白质编码DNA、β-半乳糖苷酶基因、发光基因(萤光素酶基因等)等,优选GFP或EGFP。
本发明的小鼠人工染色体载体中还可包含选择标记基因。选择标记在筛选利用该载体进行转化的细胞时有效。作为选择标记基因,可例示有正选择标记基因和负选择标记基因中的任一种或它们两者。正选择标记基因包括耐药基因,例如新霉素抗性基因(Neo或者NeoR)、氨苄青霉素抗性基因、杀稻瘟菌素S(BS)抗性基因、嘌呤霉素抗性基因(Puro)、遗传霉素(G418)抗性基因、潮霉素抗性基因(Hyg)等。此外,负选择标记基因包含例如单纯疱疹病毒胸苷激酶(HSV-TK)基因、白喉毒素A片段(DT-A)基因等。通常HSV-TK与更昔洛韦或阿昔洛韦组合使用。
作为在本发明的小鼠人工染色体载体中插入报告基因或目的外源基因或者目的DNA的方法,可优选使用同源重组法。同源重组可使用打靶载体来进行,所述打靶载体是在与小鼠染色体上的插入位置的5’侧区域和3’侧区域的碱基序列(各约1~6kb,优选为约2~4kb)一致的两个序列(5’臂和3’臂)之间连接应插入的DNA盒而得到。作为以此目的而使用的载体,例如可举出质粒、噬菌体、粘粒、病毒等,优选为质粒。用于构建打靶载体的基本质粒的示例为V907或V913(Lexicon Genetics)等,但并不限于这些。基本载体中也可包含启动子、增强子、选择标记基因、复制起点等在构建载体时通常插入的1个或2个以上的序列或元件。启动子的示例可包含磷酸甘油酸激酶(PGK)启动子、鸡β-肌动蛋白(CAG)启动子、巨细胞病毒(CMV)启动子、延伸因子1α(EF1α)启动子等。
利用上述方法制作的小鼠人工染色体载体包含来源于小鼠的染色体片段(其中包含天然着丝粒的至少99%、优选为至少99.5%的内源基因被删除的长臂片段和(当存在时)短臂)、人工端粒序列。此外,上述着丝粒是用于制作人工染色体而被利用的小鼠的染色体的整个着丝粒结构。
本发明的小鼠人工染色体载体的示例是在后述的实施例中制作的小鼠人工染色体载体,该人工染色体是在小鼠10号染色体中将长臂远端在基因Gm8155的上游区域中删除而得到的载体在和小鼠16号染色体中将长臂远端在基因Gm35974的上游区域中删除而得到的载体。这些载体包含保藏细胞株DT40(10MAC)T5-26(保藏编号:NITE BP-02656)或保藏细胞株DT40(16MAC)T1-14(保藏编号:NITE BP-02657)中所含的小鼠人工染色体作为基本结构。由于是基本结构,因此可以在该DNA结构中插入如下所述的DNA序列插入位点、选择标记基因、外源基因(或DNA)等。
上述小鼠人工染色体载体优选包含一个或更多个DNA序列插入位点,例如位点特异性重组酶的识别位点(例如Cre酶识别位点即loxP序列)。这里,位点特异性重组酶的识别位点例如是GFP-PGKneo-loxP-3’HPRT型的loxP序列,或者是5’HPRT-loxP-hyg型,或者是PGKneo-loxP-3’HPRT型的loxP序列或者是GFP-5’HPRT-loxP-PGKhyg型的loxP序列,但并不限于这些。这里,GFP是绿色荧光蛋白基因,PGKneo是磷酸甘油酸激酶启动子/新霉素抗性基因盒,HPRT是次黄嘌呤-鸟嘌呤磷酸核糖基转移酶基因,hyg是潮霉素抗性基因。
上述小鼠人工染色体载体中还可包含报告基因、选择标记基因(正选择标记基因、负选择标记基因等)。
上述小鼠人工染色体载体中还可包含外源基因或DNA序列。
外源基因或DNA没有限定,包括人基因或DNA等,其中例如包括人染色体长臂或短臂的基因或基因座的DNA等(参照下述(2))。
作为本发明的小鼠人工染色体载体的优点,可举出现有的人工染色体载体的优点:1)由于未插入到宿主染色体中而独立地保持,因此不会破坏宿主基因;2)由于稳定地携带一定的拷贝数(可为复数(多个)拷贝),且受到宿主细胞的生理的表达控制,因此不会发生所插入的基因的过量表达和表达消失;3)由于对可导入的DNA大小没有限制,因此除了可导入含表达调控区的基因、多个基因/异构体(isoform)之外,还在啮齿类细胞或者啮齿类个体中的携带率提高,实现导入基因在长期中的稳定表达,且后代传递率提高,由此转基因小鼠的制作效率提高;4)载体导入后的组织间差异小,即携带率约为90%以上等优点。
(2)外源基因或DNA的导入
在本发明的小鼠人工染色体载体中也可以导入外源基因或DNA。
外源基因或DNA序列的大小没有特别限制,可以是20kb以下,或者可以大于20kb,例如50kb以上、100kb以上、200kb以上、500kb以上、700kb以上、1Mb以上、10Mb以上、20Mb以上、30Mb以上、40Mb以上、或50Mb以上。本发明的载体可以用BAC、PAC、YAC等人工染色体载体搭载不易搭载的尺寸、即1Mb以上的外源DNA(染色体片段)。而且,本发明的载体如上所述可以将200kb以上、例如1Mb以上的大尺寸外源基因或DNA以稳定地且高携带率地包含在啮齿类的细胞内、组织内或个体内。
作为本发明的实施方式之一,本发明提供一种可使200kb以上的巨大尺寸的外源基因或DNA以90%以上的携带率稳定地保持在啮齿类的细胞、组织或个体中的载体,以及制作该载体的方法。
外源基因或DNA是来源于除小鼠、大鼠等啮齿类之外的生物物种的核酸,并没有特别限定,是来源于任何生物物种或者来源于任何组织或细胞的基因或DNA,优选为来源于哺乳动物的基因或DNA,更优选为来源于人的基因或DNA。这样的基因或DNA中包括对细胞因子类、激素类、生长因子类、营养因子类、造血因子类、免疫球蛋白类、G蛋白偶联受体类、酶类等多肽类进行编码的基因或DNA,其他与包括肿瘤、肌肉营养不良、血友病、神经变性疾病(例如阿尔茨海默病、亨廷顿舞蹈病、帕金森病等)、自身免疫性疾病、过敏性疾病、遗传性疾病、传染病、闭塞性动脉硬化症、囊性纤维化、ADA (腺苷脱氨酶)缺少症等各种疾病相关的治疗用基因或DNA,T细胞受体(TCR)、人白细胞抗原(HLA)等免疫系统基因或DNA,(人)药物代谢酶的基因(组)或DNA、(人)药物代谢相关基因或DNA、人染色体的长臂或短臂的DNA、(人)基因组文库等,但并不限于这些。
细胞因子类中例如包括干扰素类(例如IF-α,IF-β,IF-γ等)、白介素类(例如IL-1,IL-2,IL-4,IL-6,IL-11,IL-12等)、肿瘤坏死因子(例如TNF-α,TNF-β)、TGF-β家族蛋白(例如骨形态发生蛋白(BMP)等)等。
激素类中例如包括生长激素、人绒毛膜促性腺激素(hCG)、人胎盘催乳素(hPL)、人垂体促性腺激素、促甲状腺激素(TSH)、促黄体生成激素释放因子、胰岛素、胰高血糖素、生长激素抑制素、催乳激素等。
生长因子类或营养因子类中例如包括胰岛素样生长因子、脑源性神经营养因子(BDNF)、白蛋白融合睫状神经营养因子、血小板源性神经营养因子(PDNF)、转化生长因子、神经生长因子(NGF)、TNF生长因子等。
凝血/溶血因子类中例如包括因子VII、因子VIII、因子X、t-PA等。
造血因子类中例如包括红细胞生成素、(粒细胞)集落刺激因子、血小板生成素等。
G蛋白偶联受体类中包括肾上腺素受体、毒蕈碱型乙酰胆碱受体、腺苷受体、GABA受体(B型)、血管紧张素受体、胆囊收缩素受体、多巴胺受体、胰高血糖素受体、组胺受体、嗅觉受体、阿片受体、分泌素受体、生长激素抑制素受体、胃泌素的受体、P2Y受体等。
酶类中例如包括天冬酰胺酶、超氧化物歧化酶、尿酸酶、链激酶、多巴胺合成酶、腺苷脱氨酶等。
与包括肿瘤、肌肉营养不良、神经变性疾病(例如阿尔茨海默病、亨廷顿舞蹈病、帕金森病等)、自身免疫性疾病、过敏性疾病、遗传性疾病等各种疾病相关的治疗用基因中例如包括抗肌萎缩蛋白基因、IL-12基因、TNF-α基因、肿瘤抑制基因、多巴胺合成相关酶基因、遗传性酶缺陷的基因类(例如肾上腺酶(例如细胞色素酶(P450)、3β-羟基类固醇脱氢酶、21羟化酶(P450 c21)等)的基因等)等。
免疫系统基因中包括对T细胞受体(TCR)、人白细胞抗原(HLA)、Fcγ受体(FCGR)、杀伤细胞Ig样受体(killer cell Ig-like receptor(KIR))、白细胞Ig样受体(leukocyteIg-like receptor(LILR))等与免疫系统相关的蛋白进行编码的基因或DNA等。
药物代谢酶是与用于分解/排泄药和毒物等异物的代谢反应相关的酶,包括与第一相反应(氧化、还原、水解)相关的酶和与第二相反应(结合)相关的酶。与第一相反应相关的酶中例如包括细胞色素P450(“CYP”)等公知的酶,具体地是CYP1A、CYP1B、CYP2A、CYP2B、CYP2C、CYP2D、CYP2E、CYP2J、CYP3A、CYP4A、CYP4B及它们的亚家族,以及CES;等。作为CYP亚家族,例如CYP3A的亚家族中包括CYP3A4、CYP3A43、CYP3A5、CYP3A7等,此外CYP2C的亚家族中包括CYP2C8、CYP2C9、CYP2C18、CYP2C19等。另一方面,与第二相反应(结合)相关的酶中例如包括UGT1和UGT2等。
药物代谢相关基因中例如包括编码转运蛋白的基因、编码核内受体的基因等。编码转运蛋白的基因的示例是MDR1、MDR2、MRP2、OAT、OATP、OCT、BCRP等,编码核内受体的基因的示例是PXR、AhR、CAR、PPARα等。
这样,可导入本发明的载体中的药物代谢相关的外源DNA序列可包含选自由编码与第一相反应相关的酶的基因、编码与第二相相关的酶的基因、编码转运蛋白的基因和编码核内受体的基因组成的组中的至少1个基因的序列或者至少2个基因的序列。
编码免疫球蛋白类的基因或DNA优选为人抗体基因或基因座,具体地是人免疫球蛋白重链基因或基因座、人免疫球蛋白轻链基因或基因座,或者人免疫球蛋白重链和轻链基因或基因座这两者的DNA。该轻链基因或基因座是λ轻链基因或基因座和/或κ轻链基因或基因座。
本说明书中使用的“人抗体基因或基因座”,如无特别说明,则是指来源于人14号染色体的人抗体重链基因或基因座、来源于人2号染色体的人抗体轻链κ基因或基因座,以及/或者来源于人22号染色体的人抗体轻链λ基因或基因座。具体地,人抗体基因或基因座例如由人14号染色体的免疫球蛋白重链基因座(人)NC_000014.9((碱基编号105586437..106879844)或(碱基编号105264221..107043718))、人2号染色体的免疫球蛋白κ基因座(人)NC_000002.12((碱基编号88857361..90235368)或(碱基编号88560086..90265666)),以及人22号染色体的免疫球蛋白λ基因座(人)NC_000022.11((碱基编号22026076..22922913)或(碱基编号21620362..23823654))中记载的碱基序列来表示。人抗体重链基因或基因座约为1.3Mb碱基长度,人抗体轻链κ基因或基因座约为1.4Mb碱基长度,人抗体轻链λ基因或基因座约为0.9Mb碱基长度。
附带说明,小鼠的抗体重链基因或基因座位于小鼠第12号染色体上,小鼠的抗体轻链κ基因或基因座位于小鼠第6号染色体上,小鼠抗体轻链λ基因或基因座位于小鼠第16号染色体上。具体地,小鼠抗体重链基因或基因座例如由染色体12,NC_000078.6(113258768..116009954,互补序列)记载的碱基序列表示,小鼠抗体轻链κ基因或基因座由染色体6,NC_000072.6(67555636..70726754)记载的碱基序列表示,小鼠抗体轻链λ基因或基因座由染色体16,NC_000082.6(19026858..19260844,互补序列)中记载的碱基序列表示。
另外,大鼠的抗体重链基因或基因座位于大鼠第6号染色体上,大鼠的抗体轻链κ基因或基因座位于大鼠第4号染色体上,大鼠抗体轻链λ基因或基因座位于大鼠第11号染色体上。同样,这些基因或基因座的碱基序列可以从美国NCBI(GenBank等)、公知文献等处获得。
本发明中,包含上述人抗体基因的上述小鼠人工染色体载体是包含来源于人14号染色体的人抗体重链基因或基因座和来源于人2号染色体的人抗体轻链κ基因或基因座的小鼠人工染色体载体,或者是包含来源于人14号染色体的人抗体重链基因或基因座和来源于人22号染色体的人抗体轻链λ基因或基因座的小鼠人工染色体载体,或者是包含来源于人14号染色体的人抗体重链基因或基因座、来源于人2号染色体的人抗体轻链κ基因或基因座和来源于人22号染色体的人抗体轻链λ基因或基因座这三者的小鼠人工染色体载体。这些载体可通过使用本说明书记载的染色体步骤技术来制作。
本发明的非人动物是含有如下小鼠人工染色体载体的动物,所述小鼠人工染色体载体是包含上述的来源于人14号染色体的人抗体重链基因或基因座和来源于人2号染色体的人抗体轻链κ基因或基因座的小鼠人工染色体载体,和包含来源于人14号染色体的人抗体重链基因或基因座和来源于人22号染色体的人抗体轻链λ基因或基因座的小鼠人工染色体载体,或者是含有如下小鼠人工染色体载体的动物,所述小鼠人工染色体载体是包含来源于人14号染色体的人抗体重链基因或基因座、来源于人2号染色体的人抗体轻链κ基因或基因座和来源于人22号染色体的人抗体轻链λ基因或基因座的小鼠人工染色体载体。由此,上述的非人动物在被给予抗原物质时,可以产生针对该物质的人抗体。
本说明书中的人抗体可以是人免疫球蛋白(Ig)的任一类和亚类。这样的类中包括IgG、IgA、IgM、IgD和IgE,亚类中包括IgG1、IgG2、IgG3、IgG4、IgA1和IgA2。这些类和亚类可以根据重链的不同而分类,IgG链称为γ链,IgG1~IgG4相应地称为γ1、γ2、γ3和γ4链,IgA、IgM、IgD、IgE分别称为α链(α1和α2)、μ链、δ链、ε链。已知任何抗体的轻链中都有κ链和λ链,在免疫球蛋白基因的重排的过程中,如果κ链基因的重构以无效结束,则会发生λ链基因的重构。此外,人抗体重链基因座从5’起至3’依次含有:包含VH1,VH2,....,VHm(其中,m例如为38~46)的V(variable,可变)区基因;包含DH1,DH2..DHn(其中,n例如为23)的D(diversity,多样性)区基因;包含JH1,JH2..JHr(其中,r为6)的J(joining,连接)区基因;包含Cμ,Cδ,Cγ3,Cγ1,Cα1,Cγ2,Cγ4,Cε,Cα2的C(constant,恒定)区基因。免疫系统中,经由上述的人免疫球蛋白基因的重排而产生的抗体是人抗体。
人抗体分子由2条人抗体重链和2条人抗体轻链组成,各重链和各轻链通过2个二硫键结合,以及2条重链具有通过在恒定(C)区的2个二硫键结合而成的结构。此外,抗体分子的可变(V)区有3个高变区,被称为互补决定区(complementarity-determining region(CDR)),从N末端侧起,叫做CDR1、CDR2和CDR3。根据该CDR区的序列的不同,抗体对抗原的结合特性发生变化。已知免疫球蛋白基因的重构会产生抗体的多样性。
可以使至少1个绝缘子序列存在于本发明的小鼠人工染色体载体中的外源基因或外源DNA的插入位点的附近或插入位点的两侧。绝缘子序列具有增强子阻断效应(即,相邻的基因互相不受影响)或染色体边界效应(将保证基因表达的区域和抑制基因表达的区域隔开进行区别)。这样的序列包含例如人β珠蛋白HS1~HS5、鸡β珠蛋白HS4等。
外源基因或DNA的导入可利用作为上述的DNA序列插入位点而被插入的上面所例示那样的位点特异性重组酶的体系来进行。例如,构建包含Cre酶的识别位点即loxP序列和外源基因或DNA的打靶载体,或者包含插入了Cre酶的识别位点即loxP序列的外源基因或DNA的染色体片段,在含有本发明的小鼠人工染色体载体的细胞内使Cre酶表达,由此通过在loxP序列中与该打靶载体或者该染色体片段的位点特异性重组,可以导入外源基因或DNA。
在本发明的小鼠人工染色体载体中也可以插入含位点特异性重组酶的识别位点(例如loxP序列、FRT序列等)的环状DNA,也可利用以大肠杆菌为宿主的质粒或以酵母为宿主的环状YAC等现有的载体插入经克隆的DNA。优选的loxP序列是来源于P1噬菌体的野生序列,向利用Cre酶的人工染色体载体上的loxP序列插入环状插入体(インサート)的反应是可逆的。一旦插入环状插入体时,在人工染色体载体上残留有2个loxP序列。因此,再次使Cre酶表达时,也有可能引起切出环状插入体的逆反应,而难以二次插入插入体来进一步进行人工染色体载体的修饰。但是,利用碱基替换的突变loxP序列、
(3)小鼠人工染色体载体向细胞的转移和非人动物的制作
本发明的小鼠人工染色体载体,或者包含外源基因或DNA的本发明的小鼠人工染色体载体可以转移或导入到任何细胞。为此的方法例如包括微核细胞融合法、脂质体转染法、磷酸钙法、微注射法、电穿孔等,优选的方法为微核细胞融合法。
微核细胞融合法是通过含有本发明的小鼠人工染色体载体且具有微核形成能力的细胞(例如小鼠A9细胞)与所希望的其他细胞的微核融合,将该载体转移到该其他细胞的方法。具有微核形成能力的细胞用多倍体诱导剂(例如秋水仙酰胺、秋水仙素等)处理,形成微核多核细胞,通过细胞松弛素处理形成微核体,在进行上述处理后,与所希望的细胞进行细胞融合。
可导入上述的载体的细胞包括:动物细胞,优选为包括人细胞在内的哺乳动物细胞,例如卵母细胞、精子细胞等生殖种系细胞;胚胎干(ES)细胞、精子干(GS)细胞、成体干细胞等干细胞;体细胞;胚胎细胞;成体细胞;正常细胞;疾病细胞;原代培养细胞;继代细胞或建立的细胞株等。干细胞中例如包括:ES细胞、胚胎生殖(EG)细胞、胚胎癌(EC)细胞、mGS细胞、人间充质干细胞等多能干细胞;人工多能性干(iPS)细胞;来源于核移植克隆胚胎的胚胎干(ntES)细胞等。优选的细胞选自来源于哺乳动物(优选包括小鼠在内的啮齿类)的体细胞、非人生殖种系细胞、干细胞和前体细胞。当细胞是来源于啮齿类等哺乳类的细胞时,在导入有本发明载体的哺乳类(例如小鼠等啮齿类)的细胞或组织中,更能稳定地携带载体,即载体从细胞的脱落明显降低,或不会发生脱落。
细胞例如为:肝细胞、肠细胞、肾细胞、脾细胞、肺细胞、心脏细胞、骨骼肌细胞、脑细胞、骨髓细胞、淋巴细胞、巨核细胞、精子、卵子等。
组织例如为:肝脏、肠、肾脏、胸腺、脾脏、肺、心脏、肌肉(例如骨骼肌)、脑、骨髓、精巢、卵巢等组织。
ES细胞可以从对象动物的受精卵的胚囊提取内细胞团,将丝裂霉素C处理小鼠胚胎成纤维细胞作为饲养层来建立并保持(M.J.Evans和M.H.Kaufman(1981)Nature 292:154-156)。
iPS细胞通过向体细胞(含成体干细胞)导入某种特定的重编程化因子(DNA或蛋白质),用适当的培养基进行培养、继代培养,从而在约3~5周时间内生成集落。重编程化因子例如已知有:由Oct3/4、Sox2、Klf4和c-Myc组成的组合;由Oct3/4、Sox2和Klf4组成的组合;由Oct4、Sox2、Nanog和Lin28组成的组合;或者由Oct3/4、Sox2、Klf4、c-Myc、Nanog和Lin28组成的组合等(K.Takahashi and S.Yamanaka,Cell 126:663-676(2006);WO 2007/069666;M.Nakagawa et al.,Nat.Biotechnol.26:101-106(2008);K.Takahashi et al.,Cell 131:861-872(2007);J.Yu et al.,Science 318:1917-1920(2007);J.Liao et al.,Cell Res.18:600-603(2008))。培养例包括将经丝裂霉素C处理的小鼠胚胎成纤维细胞株(例如STO)作为饲养层细胞,在该饲养层细胞层上使用ES细胞用培养基,在约37℃的温度培养载体导入体细胞(约10
已知ES细胞和iPS细胞有助于生殖种系,因此可以通过包括以下内容的方法来制作非人动物(或转基因动物(人除外)):将导入有含目的基因或目的DNA的本发明的小鼠人工染色体载体的这些细胞注入到该细胞所来源的同种的哺乳动物的胚胎胚囊中,将该胚胎移植到寄养母体的子宫中,使其生产。再者,通过使得到的雌雄转基因动物进行交配,可以制作纯合性动物,进而制作其后代动物。
经由本发明的小鼠人工染色体载体,向ES细胞和iPS细胞等分化多能性细胞、其他的上述细胞类中导入人抗体基因、疾病治疗用基因、药物代谢相关基因等外源的基因或DNA,由此可以制作能够产生人抗体的细胞或非人动物,此外可以制作能够产生治疗用蛋白的细胞,再者可以制作药物代谢相关疾病等疾病模型非人动物。
在这样的非人动物中,有时也优选对应于小鼠人工染色体载体所含的外源基因的内源基因被破坏或所述内源基因的表达下降。破坏的方法可使用基因打靶法。内源基因的表达的降低法可使用RNAi法、miRNA法等。例如,对于这样的外源基因的例子,可举出:药物代谢相关基因、人抗体基因等。内源基因被破坏的非人动物可通过以下方式制作:使含有如下小鼠人工染色体载体的嵌合体非人动物或其后代与对应的内源基因整簇缺失的嵌合体动物或后代进行交配,再使得到的该内源基因杂合性缺失的动物间相互交配,其中,所述小鼠人工染色体载体包含外源基因。
通过上述方法,可以制作含有小鼠人工染色体载体的细胞或转基因非人动物。具体的非人动物的示例是含有小鼠人工染色体载体的小鼠或大鼠等啮齿类。
即,本发明提供一种细胞或非人动物,其特征在于,含有小鼠人工染色体载体。
再者,从本发明的非人动物得到的细胞、组织或器官,也可以用于从这些制作产生由外源基因表达的蛋白质的细胞株。
(4)产生人抗体的非人动物
本发明的非人动物如上所述,是含有如下小鼠人工染色体载体的动物,所述小鼠人工染色体载体是包含来源于人14号染色体的人抗体重链基因或基因座和来源于人2号染色体的人抗体轻链κ基因或基因座的小鼠人工染色体载体、包含来源于人14号染色体的人抗体重链基因或基因座和来源于人22号染色体的人抗体轻链λ基因或基因座的小鼠人工染色体载体,或者是含有如下小鼠人工染色体载体的动物,所述小鼠人工染色体载体是包含来源于人14号染色体的人抗体重链基因或基因座、来源于人2号染色体的人抗体轻链κ基因或基因座和来源于人22号染色体的人抗体轻链λ基因或基因座的小鼠人工染色体载体。
具体地,例如通过图17、图21所示的操作步骤,可以制作能够产生人抗体的本发明的非人动物(小鼠和大鼠)。
下面,对于利用小鼠人工染色体的非人动物的制作例进行说明。
将导入位点特异性重组酶的识别位点(例如loxP和FRT)而修饰的包含来源于人2号染色体的人抗体轻链κ基因或基因座的动物细胞(例如DT40),以及包含来源于人22号染色体的人抗体轻链λ基因或基因座的动物细胞(例如DT40),利用细胞融合法分别转移到含有小鼠人工染色体(MAC)的啮齿类细胞(例如CHO)中,然后通过诱导位点特异性重组酶(例如Cre)表达,制作含有包含人抗体轻链κ基因或基因座的MAC的啮齿类细胞,以及制作含有包含人抗体轻链λ基因或基因座的MAC的啮齿类细胞
向动物细胞(例如DT40)中携带的人14号染色体上的人抗体重链基因或基因座的附近导入位点特异性重组酶的识别位点(例如FRT),然后将经修饰的包含人抗体重链基因或基因座的动物细胞,利用细胞融合法转移到含有MAC的啮齿类细胞(例如CHO)中,制作含有包含人抗体重链基因或基因座的MAC的啮齿类细胞。
使含有包含上述的人抗体轻链κ基因或基因座的MAC的啮齿类细胞,以及使含有包含人抗体轻链λ基因或基因座的MAC的啮齿类细胞,分别与包含上述的人抗体重链基因或基因座的啮齿类细胞进行融合,由此向包含人抗体重链基因或基因座的啮齿类细胞转移包含人抗体轻链κ基因或基因座的MAC或包含人抗体轻链λ基因或基因座的MAC,然后通过诱导位点特异性重组酶(例如FLP)表达,从而分别制作含有包含来源于人14号染色体的人抗体重链基因或基因座和来源于人2号染色体的人抗体轻链κ基因或基因座的MAC的啮齿类细胞,以及制作含有包含来源于人14号染色体的人抗体重链基因或基因座和来源于人22号染色体的人抗体轻链λ基因或基因座的MAC的啮齿类细胞。
利用微核细胞融合法,使含有包含上述的来源于人14号染色体的人抗体重链基因或基因座和来源于人2号染色体的人抗体轻链κ基因或基因座的MAC的啮齿类细胞,以及使含有包含来源于人14号染色体的人抗体重链基因或基因座和来源于人22号染色体的人抗体轻链λ基因或基因座的MAC的啮齿类细胞,分别与非人动物(例如小鼠或大鼠)分化多能干细胞(例如ES细胞或iPS细胞)进行融合,制作含有包含来源于人14号染色体的人抗体重链基因或基因座和来源于人2号染色体的人抗体轻链κ基因或基因座的MAC的非人动物分化多能干细胞,以及制作含有包含来源于人14号染色体的人抗体重链基因或基因座和来源于人22号染色体的人抗体轻链λ基因或基因座的MAC的非人动物分化多能干细胞。
使含有包含上述的来源于人14号染色体的人抗体重链基因或基因座和来源于人2号染色体的人抗体轻链κ基因或基因座的MAC的非人动物分化多能干细胞,以及使含有包含来源于人14号染色体的人抗体重链基因或基因座和来源于人22号染色体的人抗体轻链λ基因或基因座的MAC的非人动物分化多能干细胞,分别移植到非人动物的早期胚胎(例如8细胞期胚胎或胚囊期胚胎)中,制作含有上述各MAC的嵌合体动物,再制作后代动物。进一步通过后代动物间相互交配,制作含有上述各MAC的后代动物。
通过使用相同的方法,可以制作含有如下小鼠人工染色体载体的非人动物,所述小鼠人工染色体载体是包含上述的人抗体重链基因或基因座、人抗体轻链κ基因或基因座的小鼠人工染色体载体,以及是包含人抗体轻链λ基因或基因座的小鼠人工染色体载体。
使含有包含上述的来源于人14号染色体的人抗体重链基因或基因座和来源于人2号染色体的人抗体轻链κ基因或基因座的MAC的非人动物,或者使含有包含包含来源于人14号染色体的人抗体重链基因或基因座和来源于人22号染色体的人抗体轻链λ基因或基因座的MAC的非人动物,与对应于人抗体重链基因或基因座和人抗体轻链κ和λ基因或基因座的内源抗体基因或基因座被敲除的同种的非人动物之间进行交配,制作含有包含来源于人14号染色体的人抗体重链基因或基因座和来源于人2号染色体的人抗体轻链κ基因或基因座的MAC且对应于人抗体重链基因或基因座和人抗体轻链κ和λ基因或基因座的内源抗体基因或基因座被敲除的非人动物,或者制作含有包含来源于人14号染色体的人抗体重链基因或基因座和来源于人22号染色体的人抗体轻链λ基因或基因座的MAC且对应于人抗体重链基因或基因座和人抗体轻链κ和λ基因或基因座的内源抗体基因或基因座被敲除的非人动物。
或者,使含有包含上述的来源于人14号染色体的人抗体重链基因或基因座和来源于人2号染色体的人抗体轻链κ基因或基因座的MAC的非人动物,以及使含有包含来源于人14号染色体的人抗体重链基因或基因座和来源于人22号染色体的人抗体轻链λ基因或基因座的MAC的非人动物,与对应于人抗体重链基因或基因座和人抗体轻链κ和λ基因或基因座的内源抗体基因或基因座被敲除的同种的非人动物之间进行交配,制作含有包含来源于人14号染色体的人抗体重链基因或基因座和来源于人2号染色体的人抗体轻链κ基因或基因座的MAC且对应于人抗体重链基因或基因座和人抗体轻链κ和λ基因或基因座的内源抗体基因或基因座被敲除的非人动物,以及制作含有包含来源于人14号染色体的人抗体重链基因或基因座和来源于人22号染色体的人抗体轻链λ基因或基因座的MAC且对应于人抗体重链基因或基因座和人抗体轻链κ和λ基因或基因座的内源抗体基因或基因座被敲除的非人动物。
或者,使含有包含上述的人抗体重链基因或基因座、人抗体轻链κ基因或基因座和人抗体轻链λ基因或基因座的小鼠人工染色体载体的非人动物,与对应于人抗体重链基因或基因座和人抗体轻链κ和λ基因或基因座的内源抗体基因或基因座被敲除的同种的非人动物进行交配,制作含有MAC且该动物的该内源抗体基因或基因座被敲除的非人动物。
5)有用的蛋白质的生产方法
本发明还提供一种蛋白质的生产方法,其包括:培养含有小鼠人工染色体载体的细胞,所述小鼠人工染色体载体以可表达方式包含外源DNA序列,并回收所产生的由该DNA编码的蛋白质。
作为蛋白质,例如可举出在上述的医疗方面、诊断方面、农业方面等产业方面有用的蛋白质或多肽类。在启动子(以及根据需要,增强子)的存在下,使编码这些蛋白质或多肽类的DNA以可表达方式插入到小鼠人工染色体载体中,转化或转染适当的细胞。培养所得到的细胞,使该DNA表达而产生该蛋白质或多肽,将其从细胞或培养基中回收。
细胞除了哺乳动物细胞之外,也可以使用Sf细胞等昆虫细胞;鸟类细胞、酵母细胞、植物细胞等真核细胞。
包括培养基在内的培养条件可根据细胞的种类进行选择,培养条件可使用公知的条件。例如,动物细胞的培养基包括:MEM培养基、DMEM培养基、Ham’s F12培养基、Eagle’sMEM培养基、Iscove’s EME培养基、RPMI1640培养基、这些的混合培养基等。
蛋白质或多肽类的回收(或分离)可将凝胶过滤色谱、离子交换色谱、亲和色谱、HPLC、FPLC等色谱法、盐析法、硫酸铵沉淀法、有机溶剂沉淀法、超滤法、结晶等公知的手段以单独或组合地实施。
(6)人抗体的制造法
本发明还提供一种人类抗体的制造方法,其包括:使用含有小鼠人工染色体载体的上述(4)的非人动物产生人抗体,所述小鼠人工染色体载体包含人抗体基因,并回收该人抗体。
人抗体基因是对人IgG、IgM、IgA、IgD、IgE中的任一类或者对人IgG1、IgG2、IgG3、IgG4、IgA1、IgA2中的任一亚类进行编码的基因。优选的人抗体基因为IgG类及其亚类。
人抗体由2条相同序列的重链(H)和2条相同序列的轻链(L)构成,H链和L链均由可变区和恒定区构成。人H链和L链的可变区均由3个超可变区(从N末端侧至C末端侧以CDR1、CDR2、CDR3的顺序)和4个构架区(从N末端侧至C末端侧以FR1、FR2、FR3、FR4的顺序)组成,由人H链和L链的各3个CDR序列决定抗体的特异性。
人IgG抗体时,由重链即μ链和轻链即λ链或者κ链构成,这些抗体链基因分别存在于人14号染色体、22号染色体、2号染色体上。作为本发明中使用的人抗体基因,使用包含各抗体基因座的人染色体片段,将这些整合到不同或相同的小鼠人工染色体中。抗体基因序列可以从NCBI(美国)的数据库等处获得。该一系列的方法例如是日本特开2005-230020号公报中所记载的技术的改良。
能够生产完全人抗体的非人动物通过使含有如下小鼠人工染色体载体的非人动物,所述小鼠人工染色体载体包含人μ链基因座,与含有如下小鼠人工染色体载体的同种的非人动物进行交配,所述小鼠人工染色体载体包含人λ链基因座和/或人κ链基因座,由此可以得到保有两者的H链和L链基因座的嵌合体非人动物及其后代动物。
通过包括用某种特定的抗原肽或者抗原多肽免疫如上述制作的能够产生完全人抗体的非人动物(例如小鼠、大鼠等啮齿类),并从该动物的血液分离人抗体的方法,能够生产人抗体。
或者,摘出用某种特定的抗原免疫的非人动物的脾脏,使其与骨髓瘤细胞融合,也可以得到产生单克隆抗体的杂交瘤。
(7)治疗物质的筛选法
本发明还提供一种对治疗疾病有效的物质进行筛选的方法,其包括对疾病模型动物即上述非人动物给予候选药剂,并对该药剂的治疗效果进行评价。
疾病模型非人动物是具有如下疾病的人工制作的动物,所述疾病是以因某种蛋白质的缺陷、突变等引起的生物功能异常、药物代谢异常、染色体异常等异常为病因的疾病。具有染色体异常的模型非人动物的示例并不限于以下具有人18号或21号染色体三体的动物。
这样的非人动物可通过如下方法制作:制作在基因或染色体上含有如上述的异常的基因或染色体片段,将其整合到本发明的小鼠人工染色体载体中,导入到ES细胞或iPS细胞,注入受精卵的胚囊中,将该细胞移植到非人动物的寄养母体的子宫中,使其生产。
向如上述制作的非人动物给予候选药剂,对该药剂的治疗效果进行评价,由此对治疗该疾病有效的物质进行筛选。
候选药剂非限定地,例如是低分子化合物、高分子化合物、(糖)蛋白质、肽、(磷或糖)脂质、糖类、核酸类等。
(8)药物或食品的药理作用、代谢或毒性的试验方法
根据本发明的实施方式,本发明还提供一种药物或食品的药理作用和/或代谢和/或毒性的试验方法,其包括:对含有小鼠人工染色体载体的上述非人动物或者来源于该动物的细胞、器官或组织给予药物或食品,所述小鼠人工染色体载体包含人药物代谢相关基因,并对该药物或食品的药理作用和/或代谢和/或毒性进行测定。
本发明还提供一种药物或食品的毒性的试验方法,其包括:将从含有小鼠人工染色体载体的上述非人动物得到的微粒体或微粒体级分S9与培养细胞或细菌和药物或/和食品一起培养,所述小鼠人工染色体载体包含人药物代谢相关基因,并测定药物或食品对该细胞或细菌造成的(不良)影响(例如突变等)。
人药物代谢相关基因是上述例示的基因。此外,非人动物的制作方法也与上述记载的方法相同。
在使用含有具有人药物代谢相关基因的小鼠人工染色体载体的上述的非人动物的上述方法中,例如,通过观察该动物的状态、对脏器和染色体造成的影响的试验等,可以对药物或食品的药理作用、代谢或毒性进行判定。
本发明的另一方法中,将由该非人动物得到的微粒体或者微粒体级分S9(9000g级分即含有催化水解、还原、氧化、结合等多种酶的级分)与培养细胞(特别是动物细胞,优选为哺乳类细胞)或细菌(优选为沙门氏菌)一起在药物和/或食品的存在下进行培养。药物或食品对于细胞的毒性的检测可通过艾姆斯(Ames)试验或小核试验来进行。在艾姆斯试验中,根据沙门氏菌的突变对毒性进行判定。在小核试验中,根据细胞核的染色体的异常对毒性进行判定。这些试验法是众所周知的,可在本发明的方法中使用。
实施例
下面,举出实施例进一步详细地说明本发明,但本发明的范围并不限定于这些具体例。
[实施例1]用于以小鼠人工染色体载体构建为目的的染色体修饰的携带小鼠染色体的DT40细胞的制作
为了小鼠人工染色体载体构建,经由微核形成率高的小鼠A9细胞,向同源重组频率高的鸡DT40细胞转移小鼠染色体。
[A]A9细胞与小鼠成纤维细胞(Bsd;mChr-Neo)的杂交细胞建立
将含有耐药基因(neo抗性基因)标记的小鼠10号染色体和16号染色体的小鼠成纤维细胞即小鼠胚胎成纤维细胞(mChr10-Neo、mChr16-Neo)与在公知的小鼠A9细胞中插入有杀稻瘟菌素S抗性基因即Bsd基因的小鼠A9(mouse A9)(Bsd)进行细胞融合,建立携带耐药基因标记的小鼠染色体的小鼠A9杂种细胞即小鼠A9x小鼠胚胎成纤维杂种细胞(Bsd;mChr10-Neo与Bsd;mChr16-Neo)(图1)。为了利用微核细胞融合法将耐药基因标记的小鼠染色体导入到同源重组频率高的鸡DT40细胞,通过细胞融合将耐药基因标记的小鼠染色体导入到已知微核形成率高的小鼠A9细胞。
[A.1]细胞融合和双重耐药克隆的分离
对G418抗性基因即neo基因插入到小鼠染色体上的小鼠成纤维细胞即小鼠胚胎成纤维细胞(mChr10-Neo和mChr16-Neo)与插入有杀稻瘟菌素S抗性基因即Bsd基因的小鼠A9细胞即小鼠A9(bsd)分别用PBS(-)清洗细胞表面,然后通过添加胰蛋白酶使细胞分散,悬浮于培养液(10%FBS、DMEM)中,将各细胞1×10
[B]耐药基因标记的小鼠染色体向DT40细胞的导入
从含有耐药基因标记的小鼠染色体的小鼠A9杂种细胞即小鼠A9x小鼠胚胎成纤维杂种细胞(bsd;mChr10-neo与Bsd;mChr16-neo)将耐药基因标记的小鼠染色体导入到鸡来源的DT40细胞(图2)。为了有效地进行因人工端粒(TTAGGG)n序列(尺寸:约1kb)的插入导致的染色体位点特异性裂解即端粒截短,以及通过同源重组在小鼠染色体中DNA序列插入位点即loxP序列的插入,利用微核细胞融合法将耐药基因标记的小鼠染色体导入到同源重组频率高的DT40细胞。
[B.1]微核细胞融合和耐药克隆的分离
为了有效地进行染色体修饰,将小鼠染色体从A9杂种细胞克隆即A9x小鼠胚胎成纤维杂种细胞(bsd;mChr10-neo与Bsd;mChr16-neo)转入同源重组频率高的鸡来源的细胞即DT40中。用烧瓶×24培养的供体细胞即A9x小鼠胚胎成纤维杂种细胞(bsd;mChr10-neo与Bsd;mChr16-neo)在各烧瓶中达到汇合度70%时,在37℃、5%CO
由于受体细胞即DT40细胞是浮游细胞,因此需要形成一次附着状态。为了使DT40附着在6孔板(Nunc)中的孔中,用调整为50μg/ml的聚L-赖氨酸(SIGMA)1.5ml对孔在37℃,温育过夜进行包被。回收聚L-赖氨酸,用PBS(-)清洗培养板,将约1×10
将纯化的微核细胞再次悬浮于含PHA-P(SIGMA)的无血清培养液2ml中,轻轻地接种在除去了无血清培养液(DMEM)的附着DT40上。将培养板在37℃,以1200rpm离心3分钟。除去上清,用PEG1000(Wako)[将5g的PEG1000完全溶解于无血清DMEM培养基,添加1ml二甲基亚砜并进行过滤灭菌]溶液以1ml准确融合1分钟。用无血清培养液(DMEM)以4ml清洗4次,用通常的DT40培养液3ml进行移液器吹打,使附着DT40恢复为浮游状态,在37℃,接种到2块24孔板中,温育过夜。加入G418使达到1500μg/ml,选择培养3~4周时间。将用A9x小鼠胚胎成纤维杂种细胞(bsd;mChr10-neo)#4、A9x小鼠胚胎成纤维杂种细胞(bsd;mChr16-neo)#2的微核细胞融合得到的分别合计10和2个抗性集落进行分离并使其增殖,进行下面的分析(克隆名称:DT40(mChr10-neo)和DT40(mChr16-neo))。
[B.2]耐药克隆的筛选
[B.2.1]FISH分析
对于在上述得到的DT40(mChr10-neo)和DT40(mChr16-neo)的克隆,通过Shinohara等人的报告(Human Molecular Genetics,10:1163-1175,2001)中记载的方法,进行了以小鼠cot-1DNA作为探针的FISH分析,结果分别在90%的DT40(mChr10-neo)1和93%的DT40(mChr16-neo)3中,每个正常核型(2n)的小鼠染色体数为单拷贝,因此用于下面的步骤。
[实施例2]利用小鼠10号染色体修饰的小鼠人工染色体(10MAC)的构建
在构建小鼠人工染色体载体时,需要尽可能地删除内源的基因。在同源重组效率高的鸡DT40细胞内,通过称为端粒截短的方法删除包含内源基因的小鼠10号染色体长臂的大部分。
[A]在鸡DT40细胞内利用端粒截短从小鼠10号染色体区域靠近着丝粒处位点特异性裂解远端
作为小鼠人工染色体载体,除导入目的基因之外的内源基因少时,可减轻对实验体系造成的影响,且需要尽可能不残留内源基因中印记基因那样的对小鼠个体发生产生因基因表达量的变化导致的影响的基因,因此删除小鼠长臂的大部分(图3)。
[A.1]端粒截短载体制作
在短臂近端位点特异性裂解用的基本载体中使用pBS-TEL/puro构建体(Kuroiwaet al.Nature Biotech 2002)。
在pBS-TEL/puro的EcoRI位点插入自退火(self-annealing)的合成寡核苷酸(SIGMA)。合成寡核苷酸的序列如下所示:
EcoRI-AscI-EcoRI:5’-AATTCGGCGCGCCG-3’(SEQ ID NO:1)
由从GenBank数据库得到的(NC_000076.6)小鼠10号染色体长臂近端的碱基序列设计同源重组靶序列。从DT40(mChr10-neo)1提取基因组DNA作为模板,用于PCR扩增同源重组靶序列的引物的序列如下所示:
AscI_m10T F2:5’-TCGAGGCGCGCCAGCCTTCTAGGGAACAGGAGATGTTCAA-3’(SEQ IDNO:2)
BamHI_m10T R3:5’-TCGAGGATCCGCCTTGAGTGGGGTTCTAGTCATCTTTC-3’(SEQ ID NO:3)
PCR按照推荐的条件进行,作为热循环仪使用Perkin-Elmer公司制造的GeneAmp9600,Taq聚合酶使用KOD FX(TOYOBO),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品。温度、循环条件是在98℃1分钟的热变性后,按98℃15秒、68℃6分钟进行30个循环,将得到的PCR产物用AscI和BamHI(NEB)进行消化,并利用琼脂糖凝胶进行分离纯化,然后克隆到经AscI和BamHI消化的pBS-TEL/puro(载体名称:pBS-TEL/puro_10MAC)。打靶载体、靶序列和通过同源重组产生的染色体等位基因如图4所示。
[A.2]同源重组体的筛选
在从小鼠10号染色体区域近端起的远端进行位点特异性裂解的载体使用上述的pBS-TEL/puro_10MAC进行转染,并进行嘌呤霉素抗性克隆的分离和同源重组体的筛选。
从得到的克隆提取DNA并将其作为模板进行PCR,进行位点特异性裂解的确认。引物序列如下所示:
m10 F6:5’-AACTACCCAGTTCTGCATTTGGTGTGAG-3’(SEQ ID NO:4)
m10 R6:5’-ATCAGTCATCAGTACCCCCAACCTCTCT-3’(SEQ ID NO:5)
m10 F6(同上)
PuroI:5’-GAGCTGCAAGAACTCTTCCTCACG-3’(SEQ ID NO:6)
PCR按照推荐的条件进行,作为热循环仪使用Perkin-Elmer公司制造的GeneAmp9600,Taq聚合酶使用KOD FX(TOYOBO),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品。温度、循环条件是在98℃1分钟的热变性后,按98℃15秒、68℃8分钟进行35个循环。
Plekhg1 F:5’-TGGATGGGTTTCAATGCCACT-3’(SEQ ID NO:7)
Plekhg1 R:5’-GGCATTCTCCCCTGTTGTGG-3’(SEQ ID NO:8)
Gm8155 F:5’-ACCCCTCGAACCCCTATTGC-3’(SEQ ID NO:9)
Gm8155 R:5’-CACGCCATCGGTGATGGATA-3’(SEQ ID NO:10)
Iyd F:5’-TGGGATGACCCCCACTTCTTT-3’(SEQ ID NO:11)
Iyd R:5’-TTTTGGCCTCTTGCCCCATA-3’(SEQ ID NO:12)
PCR按照推荐的条件进行,作为热循环仪使用Perkin-Elmer公司制造的GeneAmp9600,Taq聚合酶使用Ampli Taq Gold(Applied Biosystems),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品。温度、循环条件在95℃10分钟的热变性后,按94℃30秒、60℃30秒、72℃30秒进行35个循环。
其结果是确认到可裂解小鼠10号染色体区域的3个克隆(克隆名称:DT40(10MAC))。打靶载体、靶序列和通过同源重组产生的染色体等位基因如图4所示。
[A.3]利用双色FISH分析的耐药克隆的筛选
对于在上述得到的DT40(10MAC)的3个克隆,通过Shinohara等人的报告(HumanMolecular Genetics,10:1163-1175,2001)中记载的方法,进行了以小鼠cot-1DNA和PGKPuro质粒作为探针的FISH分析,结果在全部3个克隆中,确认到小鼠10号染色体的长臂部分在靠近着丝粒处被裂解(图5)。下面的步骤中使用克隆T5-26、T6-37。
[实施例3]小鼠人工染色体载体10MAC1的构建
通过在小鼠人工染色体10MAC插入作为DNA插入序列的GFP-PGKneo-loxP-3’HPRT型的loxP序列来构建小鼠人工染色体载体10MAC1,导入到以基因搭载为目的的hprt缺陷CHO细胞株。
[A]利用向小鼠人工染色体10MAC插入GFP-PGKneo-loxP-3’HPRT型的loxP序列的小鼠人工染色体载体10MAC1构建
向小鼠人工染色体10MAC搭载基因搭载位点loxP和能够监视其存在的GFP表达单元。
[A.1]GFP-PGKneo-loxP-3’HPRT型的loxP打靶载体制作
用于在DT40(10MAC)插入loxP序列的基本质粒中使用V913(Lexicon genetics)。loxP插入位点即小鼠10号染色体的DNA序列从GenBank数据库得到(NC_000076.6)。从耐药克隆提取基因组DNA作为模板,用于同源重组的二个靶序列扩增的引物的序列如下所示:
KpnI_m10 LA F:
5’-TCGAGGTACCTCTAAGTCAGGGAAAGATCCCCTTCTTG-3’(SEQ ID NO:13)
XhoI_m10 LA R:
5’-TCGACTCGAGGACCATGAAGATGGTCCAACTAAAGCAA-3’(SEQ ID NO:14)
SalI_m10 RA F:
5’-TCGAGTCGACCACTGCTCTTTCTTTAGTTACATGCAGCCC-3’(SEQ ID NO:15)
NotI_m10 RA R:
5’-TCGAGCGGCCGCATTCTTGCCAAGCTACTCTTCCGAGCTA-3’(SEQ ID NO:16)
PCR按照推荐的条件进行,作为热循环仪使用Perkin-Elmer公司制造的GeneAmp9600,Taq聚合酶使用KOD FX(TOYOBO),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品。温度、循环条件是在98℃1分钟的热变性后,按98℃15秒、68℃3分钟和5分钟进行35个循环。
将各PCR产物用KpnI(NEB)与XhoI(NEB)和SalI(NEB)与NotI(NEB)进行消化,并利用琼脂糖凝胶进行分离纯化,然后克隆到V913的KpnI/XhoI或者SalI/NotI位点(载体名称:V913-m10LARA)。3’HPRT-loxP是将经寡核苷酸合成的loxP序列克隆到V820(Lexicongenetics)的XbaI位点。将HPRT基因的第3至第9外显子即3’HPRT-loxP克隆到V907(Lexicongenetics)的EcoRI和AscI(载体名称:X3.1)。进一步将利用KpnI和NotI切出的PGKneo序列克隆到X3.1的KpnI位点和EcoRI位点(载体名称:X4.1)。将利用KpnI和AscI从X4.1切出的PGKneo-loxP-3’HPRT克隆到V913的KpnI位点和AscI位点(载体名称:pVNLH)。将在NotI和SalI消化后进行平端化的HS4-CAG-EGFP-HS4(由大阪大学的冈部博士和NIH的Felsenfeld博士赠与)克隆到pVNLH的EcoRV位点(载体名称:pVGNLH)。将利用SalI和AscI从pVGNLH切出的GFP-PGKneo-loxP-3’HPRT盒克隆到V913-m10LARA的XhoI位点和AscI位点(载体名称:p10MAC1)。打靶载体、靶序列和通过同源重组产生的染色体等位基因如图6所示。
[A.2]转染和G418抗性克隆的分离
鸡DT40细胞的培养在添加有10%胎牛血清(Gibco,以下记为“FBS”)、1%鸡血清(Gibco)、10
[A.3]同源重组体的筛选
[A.3.1]PCR分析
为了提取G418抗性株的基因组DNA作为模板来筛选重组体,使用以下引物进行PCR,确认了在小鼠10号染色体上是否发生位点特异性地重组。其引物序列如下所示:
m10 F1:5’-TGAGAAATACCGAATGGCAGAGAAACAC-3’(SEQ ID NO:17)
EGFP-F(L):5’-CCTGAAGTTCATCTGCACCA-3’(SEQ ID NO:18)
kj neo:5’-CATCGCCTTCTATCGCCTTCTTGACG-3’(SEQ ID NO:19)
m10 R2:5’-GAGAGGAGGGAAGCTTGATGAGAAAATG-3’(SEQ ID NO:20)
KpnI m10 LA F(同上)
XhoI m10 LA R(同上)
PCR按照推荐的条件进行,作为热循环仪使用Perkin-Elmer公司制造的GeneAmp9600,Taq聚合酶使用KOD FX(TOYOBO),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品。温度、循环条件是在98℃1分钟的热变性后,按98℃15秒、68℃5分钟、8分钟、2.5分钟进行35个循环。
其结果提示对于DT40(10MAC)T5-26、T6-37,进行了各8个、3个克隆的目的重组。
[A.3.2]双色FISH分析
在由上述得到的DT40(10MAC1)中,按照松原等人(FISH实验方案,秀润社,1994)进行双色FISH分析。将小鼠cot-1DNA和GFP-PGKneo-loxP-3’HPRT(pVGNLH)盒作为探针进行FISH分析,结果在打靶有loxP序列的小鼠10号染色体片段的着丝粒附近检测到探针来源的FITC信号,且检测到在阴性对照的打靶前的小鼠10号染色体片段(DT40(10MAC))中未出现的信号,因此可视觉确认发生位点特异性地重组(图7)。由这些结果可得到如下结论:得到携带小鼠人工染色体载体10MAC1的DT40细胞克隆。在下面的步骤中,采用DT40(10MAC1)T5-26 L1-2、T5-26L2-3、T6-37 L1-5这3个克隆。
[B]从含有小鼠人工染色体载体10MAC1的DT40细胞向CHO细胞导入10MAC1
为了经由CHO细胞将小鼠人工染色体载体10MAC1导入到小鼠ES细胞,或者为了在CHO细胞内经由小鼠人工染色体载体10MAC1的DNA序列插入位点即loxP稳定地插入目的基因(组)等,例如CYP3A簇、人抗体基因等,而导入到CHO细胞。
[B.1]微核细胞融合与耐药克隆的分离
使用供体细胞即DT40(10MAC1),与上述同样地对CHO hprt缺陷细胞(由日本健康科学研究资源库(HSRRB)获得,登录号JCRB0218)即CHO(HPRT
[B.2]耐药克隆的筛选
[B.2.1]PCR分析
为了提取G418抗性株的基因组DNA作为模板来筛选重组体,使用以下引物进行PCR,确认了小鼠人工号染色体载体10MAC1是否能导入到CHO细胞。其引物序列如下所示:
m10 F6(同上)
PuroI(同上)
m10 F1(同上)
EGFP-F(L)(同上)
kj neo(同上)
m10 R2(同上)
PCR按照推荐的条件进行,作为热循环仪使用Perkin-Elmer公司制造的GeneAmp9600,Taq聚合酶使用KOD FX(TOYOBO),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品。温度、循环条件是在98℃1分钟的热变性后,按98℃15秒、68℃8分钟、2.5分钟进行35个循环。其结果是对于PCR阳性的克隆进行下面的分析。
[B.2.2]单色FISH分析
对于在上述得到的CHO(HPRT
[C]向10MAC1插入重组的确认
对10MAC1的重组序列即LoxP-3’HPRT是否发挥作用进行验证。
[C.1]利用Cre/loxP系统向MAC1插入环状DNA
将CHO(HPRT-;10MAC1)在6cm培养皿培养至汇合。使用Lipofectamine2000并按照制造商的方案,一起导入Cre表达质粒(载体名称:pBS185)和5’HPRT-LoxP的质粒。转基因24小时后,将细胞在10块10cm培养皿中进行继代培养,进一步自24小时后用HAT进行药剂选择。对得到的耐药克隆24个克隆进行下面的分析。
[C.2]耐药克隆的分析
如果发生期待的位点特异性重组,插入携带5’HPRT-LoxP的环状DNA,则在10MAC1中发生HPRT基因的重构,形成HAT抗性。从耐药克隆提取DNA,进行检测该重组接头的PCR。使用的引物如下所示:
TRANS L1:5’-TGGAGGCCATAAACAAGAAGAC-3’(SEQ ID NO:21)
TRANS R1:5’-CCCCTTGACCCAGAAATTCCA-3’(SEQ ID NO:22)
关于这些引物,使用LA taq(Takara),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品,按照推荐的条件进行。温度、循环条件是在98℃1分钟的热变性后,按94℃10秒、60℃30秒、72℃3分钟进行30个循环。其结果是对于全部耐药24个克隆均为PCR阳性,因此表示对于得到的耐药克隆,高效率地发生利用位点特异性重组的环状DNA的插入。
[实施例4]利用小鼠16号染色体修饰的小鼠人工染色体(16MAC)的构建
在构建小鼠人工染色体载体时,需要尽可能地删除内源的基因。在同源重组效率高的鸡DT40细胞内,通过称为端粒截短的方法删除包含内源基因的小鼠16号染色体长臂部。
[A]在鸡DT40细胞内利用端粒截短从小鼠16号染色体区域靠近着丝粒处位点特异性裂解远端
作为小鼠人工染色体载体,除导入目的基因之外的内源基因少时,可减轻对实验体系造成的影响,且需要尽可能不残留内源基因中印记基因那样的对小鼠个体发生产生因基因表达量的变化导致的影响的基因,因此删除小鼠长臂的大部分(图3)。
[A.1]端粒截短载体制作
在短臂近端位点特异性裂解用的基本载体中使用pBS-TEL/puro构建体(Kuroiwaet al.Nature Biotech 2002)。
由从GenBank数据库得到的小鼠16号染色体长臂近端的碱基序列设计同源重组靶序列。将从DT40(mChr16-neo)3提取基因组DNA作为模板,用于PCR扩增同源重组靶序列的引物的序列如下所示:
BamHI_m16T F2:
5’-TCGAGGATCCGGGAGTAATTTTCAATCCTTGAGGCAGA-3’(SEQ ID NO:23)
BglII_m16T R2:
5’-TCGAAGATCTCATCAGTGTACACCACAATCCCATCTGT-3’(SEQ ID NO:24)
PCR按照推荐的条件进行,作为热循环仪使用Perkin-Elmer公司制造的GeneAmp9600,Taq聚合酶使用KOD FX(TOYOBO),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品。温度、循环条件是在98℃1分钟的热变性后,按98℃15秒、68℃7分钟进行35个循环,将得到的PCR产物用BamHI和BglII(NEB)进行消化,并利用琼脂糖凝胶进行分离纯化,然后克隆到经BamHI消化的pBS-TEL/puro(载体名称:pBS-TEL/puro_16MAC)。打靶载体、靶序列和通过同源重组产生的染色体等位基因如图9所示。
[A.2]同源重组体的筛选
在从小鼠16号染色体区域近端起的远端进行位点特异性裂解的载体使用上述的pBS-TEL/puro_16MAC进行转染,并进行嘌呤霉素抗性克隆的分离和同源重组体的筛选。
从得到的克隆提取DNA并将其作为模板进行PCR,进行位点特异性裂解的确认。引物序列如下所示:
m16 F5:5’-cctcttcttgacggttaccacattttgc-3’(SEQ ID NO:25)
PuroI(同上)
PCR按照推荐的条件进行,作为热循环仪使用Perkin-Elmer公司制造的GeneAmp9600,Taq聚合酶使用KOD FX(TOYOBO),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品。温度、循环条件是在98℃1分钟的热变性后,按98℃15秒、68℃9分钟进行35个循环。
Vmn1r-ps137 F:5’-TGAATTGGTCCCTCCTGCTCA-3’(SEQ ID NO:26)
Vmn1r-ps137 R:5’-CAGGCCATGAGACCCAGACA-3’(SEQ ID NO:27)
Mefv F:5’-TCCTCGGAGAATGGCTCCTG-3’(SEQ ID NO:28)
Mefv R:5’-GGCAGGTTGATGGGAACTGG-3’(SEQ ID NO:29)
Slx4 F:5’-AACCAGGGTCCCCATCCTGT-3’(SEQ ID NO:30)
Slx4 R:5’-TGGGCTGGTTTCAATGCTGA-3’(SEQ ID NO:31)
Gm4106 F:5’-GTGTGGCCATGGCTGGAGTA-3’(SEQ ID NO:32)
Gm4106 R:5’-TGTTCCTCTGCTGCCACTCG-3’(SEQ ID NO:33)
Gm35974 F:5’-ACCCAGCCACTCCCACCATA-3’(SEQ ID NO:34)
Gm35974 R:5’-AAGGGCATGGCTATCCCACA-3’(SEQ ID NO:35)
PCR按照推荐的条件进行,作为热循环仪使用Perkin-Elmer公司制造的GeneAmp9600,Taq聚合酶使用Ampli Taq Gold(Applied Biosystems),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品。温度、循环条件在95℃10分钟的热变性后,按94℃30秒、60℃30秒、72℃30秒进行35个循环。
其结果是确认到提示小鼠10号染色体区域裂解的3个克隆(克隆名称:DT40(16MAC))。对于这些克隆,确认裂解位点不同,提示存在2个模式。预想裂解后的等位基因的2个模式如图10所示。
[A.3]利用单色FISH分析的耐药克隆的筛选
对于在上述得到的DT40(16MAC)的3个克隆,通过Shinohara等人的报告(HumanMolecular Genetics,10:1163-1175,2001)中记载的方法,进行了以小鼠cot-1DNA作为探针的FISH分析时,结果在全部3个克隆,确认到小鼠16号染色体的长臂部分在靠近着丝粒处被裂解(图11)。
[实施例5]小鼠人工染色体载体16MAC1的构建
通过在小鼠人工染色体16MAC插入作为DNA插入序列的GFP-PGKneo-loxP-3’HPRT型的loxP序列来构建小鼠人工染色体载体16MAC1,导入到以基因搭载为目的的hprt缺陷CHO细胞株。
[A]GFP-PGKneo-loxP-3’HPRT型的loxP序列向小鼠人工染色体载体16MAC的插入
[A.1]GFP-PGKneo-loxP-3’HPRT型的loxP打靶载体制作
对于裂解位点不同的2种16MAC,构建以不同序列为靶标的打靶载体。
作为1型,构建以用于端粒截短的相同序列为靶标的打靶载体。
用于在DT40(16MAC)插入loxP序列的基本质粒中使用V913(Lexicon genetics)。loxP插入位点即小鼠16号染色体的DNA序列从GenBank数据库得到(NC_000082.6)。从耐药克隆提取基因组DNA作为模板,用于同源重组的二个靶序列扩增的引物的序列如下所示:
KpnI_m16 HAtLA F:
5’-TCGAGGTACCGGGAGTAATTTTCAATCCTTGAGGCAGA-3’(SEQ ID NO:36)
XhoI_m16 HAtLA R:
5’-TCGACTCGAGTGGCACTGACCCCTTAATTACGTACAGA-3’(SEQ ID NO:37)
SalI_m16 HAtRA F:
5’-TCGAGTCGACAAAGATTTGCATCCTTGGCCATGACTC-3’(SEQ ID NO:38)
NotI_m16 HAtRA R:
5’-TCGAGCGGCCGCCATCAGTGTACACCACAATCCCATCTGT-3’(SEQ ID NO:39)
PCR按照推荐的条件进行,作为热循环仪使用Perkin-Elmer公司制造的GeneAmp9600,Taq聚合酶使用KOD FX(TOYOBO),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品。温度、循环条件是在98℃1分钟的热变性后,按98℃15秒、68℃4分钟进行35个循环。
将各PCR产物用KpnI(NEB)与XhoI(NEB)和SalI(NEB)与NotI(NEB)进行消化,并利用琼脂糖凝胶进行分离纯化,然后克隆到V913的KpnI/XhoI或者SalI/NotI位点(载体名称:V913-m16HA)。3’HPRT-loxP是将经寡核苷酸合成的loxP序列克隆到V820(Lexicongenetics)的XbaI位点。将HPRT基因的第3至第9外显子即3’HPRT-loxP克隆到V907(Lexicongenetics)的EcoRI和AscI(载体名称:X3.1)。进一步将利用KpnI和NotI切出的PGKneo序列克隆到X3.1的KpnI位点和EcoRI位点(载体名称:X4.1)。将利用KpnI和AscI从X4.1切出的PGKneo-loxP-3’HPRT克隆到V913的KpnI位点和AscI位点(载体名称:pVNLH)。将在NotI和SalI消化后进行平端化的HS4-CAG-EGFP-HS4(由大阪大学的冈部博士和NIH的Felsenfeld博士赠与)克隆到pVNLH的EcoRV位点(载体名称:pVGNLH)。将利用SalI和AscI从pVGNLH切出的GFP-PGKneo-loxP-3’HPRT盒克隆到V913-m10LARA的XhoI位点和AscI位点(载体名称:p16HAMAC1)。打靶载体、靶序列和通过同源重组产生的染色体等位基因如图12所示。
作为2型,构建以残存的小鼠16号染色体近端序列为靶标的打靶载体。
用于在DT40(16MAC)插入loxP序列的基本质粒中使用V913(Lexicon genetics)。loxP插入位点即小鼠16号染色体的DNA序列从GenBank数据库得到(NC_000082.6)。从耐药克隆提取基因组DNA作为模板,用于同源重组的二个靶序列扩增的引物的序列如下所示:
KpnI_m16 GmLA F:
5’-TCGAGGTACCAAGAACAAGCTTCAGAACACAGCCAGAC-3’(SEQ ID NO:40)
XhoI_m16 GmLA R:
5’-TCGACTCGAGAACTTGTCACACAGATCCTACTGGAGGTG-3’(SEQ ID NO:41)
SalI_m16 GmRA F:
5’-TCGAGTCGACCCACAGACTGAAGCAATTGACCTCAAAAG-3’(SEQ ID NO:42)
NotI_m16 GmRA R:
5’-TCGAGCGGCCGCAAAGCAGTTATCCGCTATTTGGGACCTT-3’(SEQ ID NO:43)
PCR按照推荐的条件进行,作为热循环仪使用Perkin-Elmer公司制造的GeneAmp9600,Taq聚合酶使用KOD FX(TOYOBO),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品。温度、循环条件是在98℃1分钟的热变性后,按98℃15秒、68℃4分钟进行35个循环。
将各PCR产物用KpnI(NEB)与XhoI(NEB)和SalI(NEB)与NotI(NEB)进行消化,并利用琼脂糖凝胶进行分离纯化,然后克隆到V913的KpnI/XhoI或者SalI/NotI位点(载体名称:V913-m16Gm)。3’HPRT-loxP是将经寡核苷酸合成的loxP序列克隆到V820(Lexicongenetics)的XbaI位点。将HPRT基因的第3至第9外显子即3’HPRT-loxP克隆到V907(Lexicongenetics)的EcoRI和AscI(载体名称:X3.1)。进一步将利用KpnI和NotI切出的PGKneo序列克隆到X3.1的KpnI位点和EcoRI位点(载体名称:X4.1)。将利用KpnI和AscI从X4.1切出的PGKneo-loxP-3’HPRT克隆到V913的KpnI位点和AscI位点(载体名称:pVNLH)。将在NotI和SalI消化后进行平端化的HS4-CAG-EGFP-HS4(由大阪大学的冈部博士和NIH的Felsenfeld博士赠与)克隆到pVNLH的EcoRV位点(载体名称:pVGNLH)。将利用SalI和AscI从pVGNLH切出的GFP-PGKneo-loxP-3’HPRT盒克隆到V913-m10LARA的XhoI位点和AscI位点(载体名称:p16GmMAC1)。打靶载体、靶序列和通过同源重组产生的染色体等位基因如图13所示。
[A.2]转染和G418抗性克隆的分离
鸡DT40细胞的培养在添加有10%胎牛血清(Gibco,以下记为“FBS”)、1%鸡血清(Gibco)、10
[A.3]同源重组体的筛选
[A.3.1]PCR分析
为了提取G418抗性株的基因组DNA作为模板来筛选重组体,使用以下引物进行PCR,确认到在小鼠16号染色体上发生位点特异性地重组。其引物序列如下所示:
模式1(p16MAC1HA):
m16 F5(同上)
EGFP-F(L)(同上)
kjneo(同上)
PuroI(同上)
PCR按照推荐的条件进行,作为热循环仪使用Perkin-Elmer公司制造的GeneAmp9600,Taq聚合酶使用KOD FX(TOYOBO),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品。温度、循环条件是在98℃1分钟的热变性后,按98℃15秒、68℃7.5分钟进行35个循环。
其结果提示对于DT40(16MAC)T1-14来源的5个克隆进行了目的重组。(克隆名称:DT40(16MAC1HA))
模式2(p16GmMAC1):
m16 F6:5’-CATGCACATTTGCTTACACACAGAGGTT-3’(SEQ ID NO:44)
EGFP-F(L)(同上)
kjneo(同上)
m16 R7:5’-ATCTGGGCACTGGGGTACAACTGTTAAT-3’(SEQ ID NO:45)
PCR按照推荐的条件进行,作为热循环仪使用Perkin-Elmer公司制造的GeneAmp9600,Taq聚合酶使用KOD FX(TOYOBO),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品。温度、循环条件是在98℃1分钟的热变性后,按98℃15秒、68℃7.5分钟进行35个循环。
其结果提示对于DT40(16MAC)T2-64、T2-65来源的各11、9个克隆进行了目的重组(克隆名称:DT40(16MAC1Gm))。
[A.3.2]双色FISH分析
在由上述得到的DT40(16MAC1HA和16MAC1Gm)中,按照松原等人(FISH实验方案,秀润社,1994)进行双色FISH分析。将小鼠cot-1DNA和GFP-PGKneo-loxP-3’HPRT(pVGNLH)盒作为探针进行FISH分析,结果在打靶有loxP序列的小鼠10号染色体片段的着丝粒附近检测到探针来源的FITC信号,且检测到在阴性对照的打靶前的小鼠16号染色体片段(DT40(16MAC)T1-14、T2-64、T2-65)中未出现的信号,因此可视觉确认发生位点特异性地重组(图14)。由这些结果可得到如下结论:得到携带小鼠人工染色体载体16MAC1HA和16MAC1Gm的DT40细胞克隆。
[B]从含有小鼠人工染色体载体16MAC1HA和16MAC1Gm的DT40细胞向CHO细胞导入16MAC1HA和16MAC1Gm
为了经由CHO细胞将小鼠人工染色体载体16MAC1HA和16MAC1Gm导入到小鼠ES细胞,或者为了在CHO细胞内经由小鼠人工染色体载体16MAC1HA和16MAC1Gm的DNA序列插入位点即loxP稳定地插入目的基因(组)等,例如CYP3A簇、人抗体基因等,将16MAC1HA和16MAC1Gm导入到CHO细胞。
[B.1]微核细胞融合与耐药克隆的分离
使用供体细胞即DT40(16MAC1HA)和DT40(16MAC1Gm),与上述同样地对CHO hprt缺陷细胞(由日本健康科学研究资源库(HSRRB)获得,登录号JCRB0218)即CHO(HPRT
[B.2]耐药克隆的筛选
[B.2.1]PCR分析
为了提取G418抗性株的基因组DNA作为模板来筛选重组体,使用以下引物进行PCR,确认小鼠人工号染色体16MAC1HA和16MAC1Gm能够导入到CHO细胞。其引物序列如下所示:16MAC1HA的确认:
m16 F5(同上)
EGFP-F(L)(同上)
kjneo(同上)
PuroI(同上)
16MAC1Gm的确认
m16 F6(同上)
EGFP-F(L)(同上)
kjneo(同上)
m16 R7(同上)
PCR按照推荐的条件进行,作为热循环仪使用Perkin-Elmer公司制造的GeneAmp9600,Taq聚合酶使用KOD FX(TOYOBO),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品。温度、循环条件是在98℃1分钟的热变性后,按98℃15秒、68℃7.5分钟进行35个循环。对于PCR阳性的克隆进行下面的分析。
[B.2.2]单色FISH分析
对于在上述得到的CHO(HPRT
[实施例6]小鼠人工染色体载体在小鼠个体中的稳定性评价
通过检验10MAC1、10MAC1HA、10MAC1Gm在小鼠ES细胞中的稳定性,再制作导入有各小鼠人工染色体载体的后代传递小鼠,来检验在个体组织的稳定性。
[A]从含有各小鼠人工染色体载体的CHO细胞向小鼠ES细胞导入小鼠人工染色体载体10MAC1、16MAC1HA、16MAC1Gm
为了检验小鼠ES细胞和小鼠个体内小鼠人工染色体载体10MAC1、16MAC1HA、16MAC1Gm的稳定性,将各小鼠人工染色体10MAC1、16MAC1HA、16MAC1Gm导入到小鼠ES细胞,制作含有各小鼠人工染色体载体10MAC1、16MAC1HA、16MAC1Gm的嵌合体小鼠,以及后代传递小鼠。
[A.1]微核细胞融合与耐药克隆的分离
将受体细胞即CHO(HPRT
供体细胞使用C57B6品系小鼠的ES细胞即B6-ES,以及对B6-ES细胞进行6TG处理的HPRT缺陷株即B6(HPRT
[A.2]耐药克隆的筛选
[A.2.1]PCR分析
为了提取G418抗性株的基因组DNA作为模板来筛选重组体,使用以下引物进行PCR,确认各小鼠人工号染色体载体能否导入到小鼠ES细胞。其引物序列如下所示:
10MAC1的确认:
m10 F6(同上)
PuroI(同上)
m10 F1(同上)
EGFP-F(L)(同上)
kj neo(同上)
m10 R2(同上)
PCR按照推荐的条件进行,作为热循环仪使用Perkin-Elmer公司制造的GeneAmp9600,Taq聚合酶使用KOD FX(TOYOBO),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品。温度、循环条件是在98℃1分钟的热变性后,按98℃15秒、68℃8分钟、2.5分钟进行35个循环。其结果对于得到的PCR阳性的9个克隆进行下面的分析。
16MAC1HA的确认:
m16 F5(同上)
EGFP-F(L)(同上)
kjneo(同上)
PuroI(同上)
16MAC1Gm的确认:
m16 F6(同上)
EGFP-F(L)(同上)
kjneo(同上)
m16 R7(同上)
PCR按照推荐的条件进行,作为热循环仪使用Perkin-Elmer公司制造的GeneAmp9600,Taq聚合酶使用KOD FX(TOYOBO),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品。温度、循环条件是在98℃1分钟的热变性后,按98℃15秒、68℃7.5分钟进行35个循环。对于PCR阳性的克隆进行下面的分析。
[A.2.2]单色FISH分析
对于在上述得到的B6-ES(MAC1)和B6(HPRT
[B]各小鼠人工染色体载体10MAC1、16MAC1HA、16MAC1Gm的稳定性
[B.1]小鼠人工染色体载体10MAC1、16MAC1HA、16MAC1Gm在CHO细胞中的稳定性
对于在上述得到的CHO克隆(例如CHO(HPRT
[B.2]小鼠人工染色体载体10MAC1、16MAC1HA、16MAC1Gm在小鼠ES细胞中的稳定性
对于在上述得到的小鼠ES克隆(例如KO56(10MAC1)和TT2F(10MAC1)可在上述实施例6[A]中获得),通过在0~100PDL的非选择培养下的长期培养后的FISH分析,测量10MAC1携带细胞的比例。
[B.3]携带小鼠人工染色体载体10MAC1、16MAC1HA、16MAC1Gm的嵌合体小鼠的制作
使用在上述实施例6[A]中得到的ES细胞克隆,通过Tomizuka等人的方法(NatureGenet.16:133,1997)制作嵌合体小鼠。作为宿主,使用通过MCH(ICR)(白色,从日本CLEA株式会社购入)雌雄交配而得到的8细胞期胚胎。将注入胚移植到寄养母体,结果出生的仔小鼠可以通过毛色判定是否为嵌合体。将注入10MAC1携带ES克隆TT2F(10MAC1)(例如其他,KO56 10MAC1,可在上述实施例6[A]中获得)的胚胎移植到寄养母体。对于出生的嵌合体小鼠,通过毛色(毛色中看到深棕色的部分)判定了嵌合体率。由此确认了携带小鼠人工染色体载体10MAC1的ES细胞株(KO56和TT2F)是否保持嵌合体形成能力,即小鼠个体的正常组织中保持分化的能力。
[B.4]10MAC1、16MAC1HA、16MAC1Gm从携带各种小鼠人工染色体载体10MAC1、16MAC1HA、16MAC1Gm的嵌合体小鼠的后代传递
使上述[B.3]中制作的雌嵌合体小鼠(嵌合体率约100%)与MCH(ICR)(白色,从日本CLEA株式会社购入)雄小鼠交配。对于通过嵌合体小鼠出生的仔小鼠,通过GFP荧光研究了各种小鼠人工染色体载体的携带。将各小鼠人工染色体传递给后代的小鼠品系称为TC(10MAC1、16MAC1HA、16MAC1Gm)。对于从TT2F(MAC1)来源的嵌合体得到的后代传递个体,得到能够在全身确认GFP荧光蛋白表达的个体(图16)。
[B.5]TC(10MAC1、16MAC1HA、16MAC1Gm)小鼠品系的体细胞中10MAC1、16MAC1HA、16MAC1Gm的稳定性
[B.5.1]实体荧光显微镜观察
在上述得到的雄和雌TC(10MAC1、16MAC1HA、16MAC1Gm)小鼠中,在实体荧光显微镜观察下对脑、胸腺、心脏、肺、肝脏、肾脏、脾脏、小肠、肌肉、精巢(或卵巢)进行观察,在全部的组织中观察GFP阳性。
[B.5.2]血液系细胞的FACS分析
使用针对B细胞(CD19)、T细胞(CD4,CD8)、巨核细胞(CD41)的特异性抗体(Becton,Dickinson and Company),研究骨髓和脾脏细胞中的GFP阳性率。
[B.5.3]荧光原位杂交(FISH)分析
另外,使用由与上述相同的个体制备的尾部成纤维细胞,按照Shinohara等人的报告(Human Molecular Genetics,10:1163-1175,2001)中记载的方法,进行以小鼠小卫星(minor satellite)DNA作为探针的FISH分析,视觉上确认MAC的存在,确认与宿主染色体独立地保持。
[实施例7]使用新型小鼠人工染色体载体,制作产生人抗体的(IGHK-NAC)小鼠和大鼠
根据10MAC1、16MAC1HA、16MAC1Gm各小鼠人工染色体载体的功能性评价,减少所使用的载体(将筛选出的小鼠人工染色体称为“NAC”)、搭载人抗体基因(IGH、IGK),制作产生人抗体的小鼠和大鼠(图17)。选择10MAC1,作为NAC,进行下面的实验。
[A]修饰的人2号染色体向携带NAC的CHO细胞的转移
通过携带修饰的人2号染色体的CHO细胞,将修饰的人2号染色体导入到携带NAC的CHO细胞。
[A.1]利用微核细胞融合法的修饰的人2号染色体向携带NAC的Hprt缺陷CHO细胞的转移
将供体细胞即修饰的人2号染色体CHO细胞(CHO hChr2LF)在细胞培养皿培养,在达到汇合时,更换为添加有20%FBS、0.1μg/ml秋水仙酰胺的F12培养基,再培养48小时培养后,培养基更换为添加有20%FBS、0.1μg/ml秋水仙酰胺的F12培养基,再温育过夜以形成微细胞。除去培养液,在离心用烧瓶中装满预先在37℃保温的细胞松弛素B(10μg/ml,SIGMA)溶液,在34℃,以8000rpm离心1小时。将微核(也称为“微细胞”)悬浮于无血清DMEM培养基,用8μm、5μm、3μm过滤器进行纯化。纯化后,将微细胞悬浮于用DMEM制备的0.05mg/ml PHA-P(SIGMA)溶液2mL中,在除去培养液后添加到在6cm细胞培养皿达到汇合时的受体即携带NAC的Hprt缺陷CHO细胞株。温育15分钟,使微核贴附到CHO细胞。然后,用PEG1000(Wako)溶液[将5g的PEG1000完全溶解于6mL的无血清DMEM培养基,添加1ml二甲基亚砜并进行过滤灭菌]以1ml准确融合1分钟。用5mL的无血清DMEM进行4次清洗操作以除去PEG,然后添加CHO培养液。24小时后,将细胞接种到10块10cm细胞培养皿,添加8μg/mL杀稻瘟菌素S,选择培养10天。对于得到的耐药株,进行下面的分析。
[A.2]利用PCR分析的耐药克隆的筛选
提取杀稻瘟菌素S抗性株的基因组,作为模板进行PCR分析,确认修饰的人2号染色体的携带。其引物序列如下所示:
确认修饰的人2号染色体loxP序列的引物:
cos138 sp L:5’-CTGAGAAGAGTCATTGTTTATGGTAGACT-3’(SEQ ID NO:46)
cos138 sp R:5’-ATCCCCATGTGTATCACTGGCAAACTGT-3’(SEQ ID NO:47)
x6.1cosRa L:5’-GGGGAATAAACACCCTTTCCAAATCCTC-3’(SEQ ID NO:48)
x6.1cosRa R:5’-ACCAAGTAACCGATCAAACCAACCCTTG-3’(SEQ ID NO:49)
对于cos138 sp L、cos138 sp R的引物,使用Accuprime Taq DNA聚合酶(ThermoFisher Scientific),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品,按照推荐的条件进行。温度、循环条件在94℃2分钟的热变性后,94℃15秒、60℃15秒、68℃5分钟进行35个循环。
对于x6.1cosRa L,x6.1cosRa R的引物,使用KOD FX(TOYOBO),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品,按照推荐的条件进行。温度、循环条件是在98℃1分钟的热变性后,按98℃15秒、68℃12分钟进行30个循环。
确认修饰的人2号染色体FRT序列的引物:
kD9 tcLa L:5’-TGAGAACACAGGGGTCTCCATTCTGACT-3’(SEQ ID NO:50)
kD9 tcLa R:5’-ACAATCAACAGCATCCCCATCTCTGAAG-3’(SEQ ID NO:51)
kD9 tcRa L:5’-GACGTGCTACTTCCATTTGTCACGTCCT-3’(SEQ ID NO:52)
kD9 tcRa R:5’-TGGTCACTGAAGCTTTCCATCTGCTCTT-3’(SEQ ID NO:53)
关于这些引物,使用KOD FX(TOYOBO),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品,按照推荐的条件进行。温度、循环条件是在98℃1分钟的热变性后,按98℃15秒、68℃5分钟进行35个循环。
除此之外,确认在人2号染色体上是否携带有使用引物的区域。其引物序列如下所示:
D2S177 F:5’-AGCTCAGAGACACCTCTCCA-3’(SEQ ID NO:54)
D2S177 R:5’-CTGTATTAGGATACTTGGCTATTGA-3’(SEQ ID NO:55)
FABP1-F:5’-TATCAAGGGGGTGTCGGAAATCGTG-3’(SEQ ID NO:56)
FABP1-R:5’-ACTGGGCCTGGGAGAACCTGAGACT-3’(SEQ ID NO:57)
EIF2AK3-F:5’-AGGTGCTGCTGGGTGGTCAAGT-3’(SEQ ID NO:58)
EIF2AK3-R:5’-GCTCCTGCAAATGTCTCCTGTCA-3’(SEQ ID NO:59)
RPIA-F:5’-CTTACCCAGGCTCCAGGCTCTATT-3’(SEQ ID NO:60)
RPIA-R:5’-CTCTACCTCCCTACCCCATCATCAC-3’(SEQ ID NO:61)
IGKC-F:5’-TGGAAGGTGGATAACGCCCT-3’(SEQ ID NO:62)
IGKC-R:5’-TCATTCTCCTCCAACATTAGCA-3’(SEQ ID NO:63)
IGKV-F:5’-AGTCAGGGCATTAGCAGTGC-3’(SEQ ID NO:64)
IGKV-R:5’-GCTGCTGATGGTGAGAGTGA-3’(SEQ ID NO:65)
Vk3-2 F:5’-CTCTCCTGCAGGGCCAGTCA-3’(SEQ ID NO:66)
Vk3-2 R:5’-TGCTGATGGTGAGAGTGAACTC-3’(SEQ ID NO:67)
D2S159_1F:5’-CTCTAACTGAATCAAGGGAATGAAC-3’(SEQ ID NO:68)
D2S159_1R:5’-AGCAGTTTGAGTTTAGGATGAAGG-3’(SEQ ID NO:69)
Taq聚合酶使用Ampli Taq Gold(Applied Biosystems),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品,按照推荐的条件进行。温度、循环条件在95℃10分钟的热变性后,按95℃30秒、60℃30秒、72℃1分钟进行35个循环。
对于PCR为阳性的克隆,进行下面的分析。其结果是获得保持有修饰的人2号染色体和NAC(新型人工染色体载体)的克隆。
[A.3]双色FISH分析
根据上述的结果,对于PCR阳性克隆,按照松原等人(FISH实验方案,秀润社,1994)进行双色FISH分析。将人cot-1DNA和小鼠cot-1DNA作为探针进行FISH分析,确认到修饰的人2号染色体和NAC与宿主染色体独立地稳定地保持(图18)。
[B]利用易位克隆的人2号染色体IGK区域向小鼠人工染色体载体(NAC)的搭载
在携带NAC的CHO细胞中,将人2号染色体上IGK区域易位克隆到NAC。易位克隆中使用Cre/loxP系统,通过使人2号染色体与NAC相互易位,从而将IGK区域搭载到NAC(图19)。
[B.1]利用Cre表达的HAT抗性染色体重组体的获得
NAC中搭载loxP位点,在Cre重组酶存在下与修饰的人2号染色体的loxP位点发生重组。此外,发生重组时成为副产物的NAC中未搭载的人2号染色体区域的5’HPRT与成为副产物的NAC末端的3’HPRT相连接,发生HPRT基因的重构,CHO(hprt-/-)获得HAT抗性。
对于携带修饰的人2号染色体和NAC的Hprt缺陷CHO细胞,在10cm细胞培养皿达到汇合时,使用Lipofectamine2000(Thermo Fisher Scientific),参照制造商的操作步骤加入18μg的Cre表达质粒(载体名称:pBS185)。添加后经过6小时,更换培养液,24小时后,接种到10块10cm细胞培养皿,用1×HAT(SIGMA)、4μg/mL杀稻瘟菌素进行药剂选择。将得到的耐药株用于下面的分析。
[B.2]利用PCR分析的耐药克隆的筛选
为了将提取HAT抗性株的基因组DNA作为模板筛选相互易位克隆,使用以下引物进行PCR,确认在人2号染色体片段和NAC上发生染色体相互易位。其引物序列如下所示:
TRANS L1(同上)
TRANS R1(同上)
KJneo:5’-CATCGCCTTCTATCGCCTTCTTGACG-3’(SEQ ID NO:70)
PGKr-2:5’-ATCTGCACGAGACTAGTGAGACGTGCTA-3’(SEQ ID NO:71)
关于这些引物,使用LA taq(Takara),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品,按照推荐的条件进行。温度、循环条件是在98℃1分钟的热变性后,按94℃10秒、60℃30秒、72℃3分钟进行30个循环。
除此之外,进行PCR以确认是否保持人2号染色体区域与FRT序列。引物如下所示:
人2号染色体区域确认引物:
D2S177 F(同上)
D2S177 R(同上)
FABP1-F(同上)
FABP1-R(同上)
EIF2AK3-F(同上)
EIF2AK3-R(同上)
RPIA-F(同上)
RPIA-R(同上)
IGKC-F(同上)
IGKC-R(同上)
IGKV-F(同上)
IGKV-R(同上)
Vk3-2 F(同上)
Vk3-2 R(同上)
D2S159_1F(同上)
D2S159_1R(同上)
Taq聚合酶使用Ampli Taq Gold(Applied Biosystems),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品,按照推荐的条件进行。温度、循环条件在95℃10分钟的热变性后,按95℃30秒、60℃30秒、72℃1分钟进行35个循环。
确认人2号染色体上FRT序列的引物:
kD9 tcLa L(同上)
kD9 tcLa R(同上)
kD9 tcRa L(同上)
kD9 tcRa R(同上)
关于这些引物,使用KOD FX(TOYOBO),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品,按照推荐的条件进行。温度、循环条件是在98℃1分钟的热变性后,按98℃15秒、68℃5分钟进行35个循环。对于PCR阳性克隆,进行下面的分析。
[B.3]双色FISH分析
对于PCR阳性克隆,将人cot-1DNA和小鼠cot-1DNA作为探针进行FISH分析。确认NAC与修饰的人2号染色体发生相互易位,且IGK区域搭载在NAC上的IGK-NAC、副产物独立携带。将该克隆称为CHO IGK-NAC。
[C]IGK-NAC向修饰的人14号染色体携带CHO(hprt-/-)细胞株的转移
将制作的IGK-NAC转入到携带修饰的人14号染色体的CHO(hprt-/-)细胞株,使发生利用FRT/Flp系统的重组,将IGH区域搭载到IGK-NAC,来制作IGHK-NAC(图20)。
[C.1]微核细胞融合与耐药克隆的分离
将供体细胞即CHO IGK-NAC在细胞培养皿培养,达到汇合时,更换为添加有20%FBS、0.1μg/ml秋水仙酰胺的F12培养基,再培养48小时培养后,培养基更换为添加有20%FBS、0.1μg/ml秋水仙酰胺的F12培养基,再温育过夜以形成微细胞。除去培养液,在离心用烧瓶中装满预先在37℃保温的细胞松弛素B(10μg/ml,SIGMA)溶液,在34℃,以8000rpm离心1小时。将微核(也称为“微细胞”)悬浮于无血清DMEM培养基,用8μm、5μm、3μm过滤器进行纯化。纯化后,将微细胞悬浮于用DMEM制备的0.05mg/ml PHA-P(SIGMA)溶液2mL中,除去培养液后添加在6cm细胞培养皿达到汇合时的受体即CHO hprt-/-14FRT。温育15分钟,使微核贴附到CHO细胞。然后,用PEG1000(Wako)溶液[将5g的PEG1000完全溶解于6mL的无血清DMEM培养基,添加1ml二甲基亚砜并进行过滤灭菌]以1ml准确融合1分钟。用5mL的无血清DMEM进行4次清洗操作以除去PEG,然后添加CHO培养液。24小时后,将细胞接种到10块10cm细胞培养皿,添加600μg/mL G418和6μg/mL杀稻瘟菌素,选择培养10天。对于得到的耐药株,进行下面的分析。
[C.2]利用PCR分析的耐药克隆的筛选
为了确认IGK-NAC是否转移到保持有修饰的人14号染色体的CHO(hprt-/-)株,是否保持修饰的人14号染色体,进行PCR分析。使用的引物如下所示。
IGK-NAC的确认引物:
KJneo(同上)
PGKr-2(同上)
关于这些引物,使用LA taq(Takara),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品,按照推荐的条件进行。温度、循环条件是在98℃1分钟的热变性后,按94℃10秒、60℃30秒、72℃3分钟进行30个循环。
IGK-NAC上的FRT插入位点确认引物:
kD9 tcLa L(同上)
kD9 tcLa R(同上)
kD9 tcRa L(同上)
kD9 tcRa R(同上)
关于这些引物,使用KOD FX(TOYOBO),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品,按照推荐的条件进行。温度、循环条件是在98℃1分钟的热变性后,按98℃15秒、68℃5分钟进行35个循环。
人2号染色体区域确认引物:
D2S177 F(同上)
D2S177 R(同上)
EIF2AK3-F(同上)
EIF2AK3-R(同上)
RPIA-F(同上)
RPIA-R(同上)
IGKC-F(同上)
IGKC-R(同上)
IGKV-F(同上)
IGKV-R(同上)
Vk3-2 F(同上)
Vk3-2 R(同上)
Taq聚合酶使用Ampli Taq Gold(Applied Biosystems),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品,按照推荐的条件进行。温度、循环条件在95℃10分钟的热变性后,按95℃30秒、60℃30秒、72℃1分钟进行35个循环。
确认修饰的人14号染色体上FRT序列的引物:
14TarC_La F:5’-AGCAATTAGGGCCTGTGCATCTCACTTT-3’(SEQ ID NO:72)
14TarC_La R:5’-CCAGCTCATTCCTCCCACTCATGATCTA-3’(SEQ ID NO:73)
14TarC_Ra F:5’-CATCTGGAGTCCTATTGACATCGCCAGT-3’(SEQ ID NO:74)
14TarC_Ra R:5’-CTTATTCCTCCTTCTGCCCACCCTTCAT-3’(SEQ ID NO:75)
关于这些引物,使用KOD FX(TOYOBO),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品,按照推荐的条件进行。温度、循环条件是在98℃1分钟的热变性后,按98℃15秒、68℃6分钟进行35个循环。
14号染色体区域确认引物:
MTA1-F3:5’-AGCACTTTACGCATCCCAGCATGT-3’(SEQ ID NO:76)
MTA1-R3:5’-CCAAGAGAGTAGTCGTGCCCCTCA-3’(SEQ ID NO:77)
ELK2P2-F:5’-CCCACTTTACCGTGCTCATT-3’(SEQ ID NO:78)
ELK2P2-R:5’-ATGAAGGTCCGTGACTTTGG-3’(SEQ ID NO:79)
g1(g2)-F:5’-ACCCCAAAGGCCAAACTCTCCACTC-3’(SEQ ID NO:80)
g1(g2)-R:5’-CACTTGTACTCCTTGCCATTCAGC-3’(SEQ ID NO:81)
VH3-F:5’-AGTGAGATAAGCAGTGGATG-3’(SEQ ID NO:82)
VH3-R:5’-CTTGTGCTACTCCCATCACT-3’(SEQ ID NO:83)
CH3F3:5’-AGGCCAGCATCTGCGAGGAT-3’(SEQ ID NO:84)
CH4R2:5’-GTGGCAGCAAGTAGACATCG-3’(SEQ ID NO:85)
Taq聚合酶使用Ampli Taq Gold(Applied Biosystems),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品,按照推荐的条件进行。温度、循环条件在95℃10分钟的热变性后,按95℃30秒、60℃30秒或者56℃30秒、72℃1分钟进行35个循环。将PCR阳性的克隆用于下面的分析。
[C.3]双色FISH分析
对于筛选的克隆,将人cot-1DNA和小鼠cot-1DNA作为探针进行FISH分析,确认IGK-NAC与修饰的人14号染色体独立地保持各单拷贝,用于下面的步骤。
[D]使用FRT/Flp重组系统的IGHK-NAC的构建
通过将IGK-NAC与修饰的人14号染色体利用FRT/Flp系统进行相互易位,在IGK-NAC上将人14号染色体来源的IGH区域进行易位克隆,构建IGHK-NAC。
[D.1]利用FLP表达的HAT抗性染色体重组体的获得
使用IGK-NAC上的FRT位点与修饰的人14号染色体上的FRT位点,在FLP重组酶存在下使其发生相互易位。此外,发生重组时,在IGHK-NAC上,5’HPRT与3’HPRT相连接,发生HPRT基因的重构,获得HAT抗性。携带IGK-NAC与修饰的人14号染色体的CHO(hprt-/-)株在10cm细胞培养皿中达到汇合时,使用Lipofectamine2000(Thermo Fisher Scientific),参照制造商的操作步骤加入18μg的FLP表达质粒。添加后经过6小时,更换培养液,24小时后,接种到10块10cm细胞培养皿,用1×HAT、6μg/mL杀稻瘟菌素进行药剂选择。对于得到的HAT抗性克隆进行下面的分析。
[D.2]利用PCR分析的耐药克隆的筛选
使用FRT/FLP系统发生期待的相互易位,为了确认是否构建IGHK-NAC,提取耐药克隆的DNA,作为模板进行PCR分析。使用的引物如下所示:
相互易位连接位点的确认引物:
TRANS L1(同上)
TRANS R1(同上)
CMVr-1:5’-CCTATTGGCGTTACTATGGGAACATACG-3’(SEQ ID NO:86)
PGKr-2(同上)
KJneo(同上)
PGKr-2(同上)
关于这些引物,使用LA taq(Takara),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品,按照推荐的条件进行。温度、循环条件是在98℃1分钟的热变性后,按94℃10秒、60℃30秒、72℃3分钟进行30个循环。
人2号染色体区域确认引物:
D2S177 F(同上)
D2S177 R(同上)
EIF2AK3-F(同上)
EIF2AK3-R(同上)
RPIA-F(同上)
RPIA-R(同上)
IGKC-F(同上)
IGKC-R(同上)
IGKV-F(同上)
IGKV-R(同上)
Vk3-2 F(同上)
Vk3-2 R(同上)
Taq聚合酶使用Ampli Taq Gold(Applied Biosystems),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品,按照推荐的条件进行。温度、循环条件在95℃10分钟的热变性后,按95℃30秒、60℃30秒、72℃1分钟进行35个循环。
人14号染色体区域确认引物:
MTA1-F3(同上)
MTA1-R3(同上)
ELK2P2-F(同上)
ELK2P2-R(同上)
g1(g2)-F(同上)
g1(g2)-R(同上)
VH3-F(同上)
VH3-R(同上)
CH3F3(同上)
CH4R2(同上)
使用这些引物的PCR按照推荐的条件进行,Taq聚合酶使用Ampli Taq Gold(Applied Biosystems),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品。温度、循环条件在95℃10分钟的热变性后,按95℃30秒、60℃30秒或者56℃30秒、72℃1分钟进行35个循环。对于PCR阳性的克隆进行下面的分析。
[D.3]双色FISH分析
对于筛选的克隆,将人cot-1DNA和小鼠cot-1DNA作为探针进行FISH分析。在作为副产物的NAC中未搭载的人14号染色体中,如果额外的人2号染色体区域易位而染色体变长,则提示发生了相互易位。而且,作为探针,使用BAC克隆CH17-405H5(IGK区域:CHORI)与CH17-262H11(IGH区域:CHORI)与CH17-216K2(IGK区域:CHORI)与CH17-212P11(IGH区域:CHORI)的组合进行双色FISH分析,详细分析是否构建了实际IGHK-NAC。将能确认到认为是IGHK-NAC的染色体以单拷贝独立地存在的克隆用于下面。
[E]IGHK-NAC向CHO K1细胞株的转移
在IGHK-NAC和用于构建IGHK-NAC的相互易位时形成的副产物这两者中均携带Neo抗性基因,在通过微核细胞融合法转移到目标细胞时,当用G418进行药剂筛选时,会获得转移单独的IGHK-NAC或副产物或这两者的细胞。由于在NAC上搭载有EGFP,因此能确认IGHK-NAC是否转移到目标细胞,但为了以有效进行染色体导入的供体细胞制作仅携带IGHK-NAC的细胞,将IGHK-NAC转移到CHO K1细胞株。
[E.1]微核细胞融合与耐药克隆的分离:利用染色体转移,制作仅携带IGHK-NAC的细胞株。
将供体细胞即CHO IGHK-NAC在细胞培养皿培养,达到汇合时,更换为添加有20%FBS、0.1μg/ml秋水仙酰胺的F12培养基,再培养48小时培养后,培养基更换为添加有20%FBS、0.1μg/ml秋水仙酰胺的F12培养基,再温育过夜以形成微细胞。除去培养液,在离心用烧瓶中装满预先在37℃保温的细胞松弛素B(10μg/ml,SIGMA)溶液,在34℃,以8000rpm进行1小时的离心。将微核(也称为“微细胞”)悬浮于无血清DMEM培养基,用8μm、5μm、3μm过滤器进行纯化。纯化后,将微细胞悬浮于用DMEM制备的0.05mg/ml PHA-P(SIGMA)溶液2mL中,在除去培养液后添加到在6cm细胞培养皿达到汇合时的受体即CHO K1细胞株。温育15分钟,使微核贴附到CHO细胞。然后,用PEG1000(Wako)溶液[将5g的PEG1000完全溶解于6mL的无血清DMEM培养基,添加1ml二甲基亚砜并进行过滤灭菌]以1ml准确融合1分钟。用5mL的无血清DMEM进行4次清洗操作以除去PEG,然后添加CHO培养液。24小时后,将细胞接种到10块10cm细胞培养皿,添加800μg/mL G418,选择培养10天。对于得到的耐药株,确认IGHK-NAC上GFP基因的荧光蛋白表达,用于下面的分析。
[E.2]利用PCR分析的耐药克隆的筛选
为了确认IGHK-NAC转移到CHO K1细胞株,提取耐药克隆的DNA,并将其作为模板进行PCR分析。使用的引物如下所示:
相互易位连接位点确认引物:
TRANS L1(同上)
TRANS R1(同上)
CMVr-1(同上)
PGKr-2(同上)
KJneo(同上)
PGKr-2(同上)
关于这些引物,使用LA taq(Takara),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品,按照推荐的条件进行。温度、循环条件是在98℃1分钟的热变性后,按94℃10秒、60℃30秒、72℃3分钟进行30个循环。
人2号染色体区域确认引物:
EIF2AK3-F(同上)
EIF2AK3-R(同上)
RPIA-F(同上)
RPIA-R(同上)
IGKC-F(同上)
IGKC-R(同上)
IGKV-F(同上)
IGKV-R(同上)
Vk3-2 F(同上)
Vk3-2 R(同上)
Taq聚合酶使用Ampli Taq Gold(Applied Biosystems),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品,按照推荐的条件进行。温度、循环条件在95℃10分钟的热变性后,按95℃30秒、60℃30秒、72℃1分钟进行35个循环。
人14号染色体区域确认引物:
MTA1-F3(同上)
MTA1-R3(同上)
ELK2P2-F(同上)
ELK2P2-R(同上)
g1(g2)-F(同上)
g1(g2)-R(同上)
VH3-F(同上)
VH3-R(同上)
CH3F3(同上)
CH4R2(同上)
使用这些引物的PCR按照推荐的条件进行,Taq聚合酶使用Ampli Taq Gold(Applied Biosystems),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品。温度、循环条件在95℃10分钟的热变性后,按95℃30秒、60℃30秒或者56℃30秒、72℃1分钟进行35个循环。根据PCR的结果,对于提示只携带IGHK-NAC的克隆进行下面的分析。
[E.3]双色FISH分析
对于筛选的克隆,将人cot-1DNA和小鼠cot-1DNA作为探针进行FISH分析,确认所期待的那样只携带IGHK-NAC。除此之外,作为探针,使用BAC克隆CH17-405H5(IGK区域:CHORI)与CH17-262H11(IGH区域:CHORI)与CH17-216K2(IGK区域:CHORI)与CH17-212P11(IGH区域:CHORI)的组合进行双色FISH分析,详细分析是否构建了实际IGHK-NAC。
[F]IGHK-NAC向小鼠ES细胞的转移
为了制作产生人抗体的小鼠,需要将IGHK-NAC转移到小鼠ES细胞,在受精卵8细胞期进行注射,制作嵌合体小鼠,使IGHK-MAC传递给后代。制作携带IGHK-NAC的小鼠ES细胞。
[F.1]微核细胞融合与耐药克隆的分离
供体细胞使用CHO K1 IGHK-NAC。将供体细胞在细胞培养皿培养,达到汇合时,更换为添加有20%FBS、0.1μg/ml秋水仙酰胺的F12培养基,再培养48小时培养后,培养基更换为添加有20%FBS、0.1μg/ml秋水仙酰胺的F12培养基,再温育过夜以形成微细胞。除去培养液,在离心用烧瓶中装满预先在37℃保温的细胞松弛素B(10μg/ml,SIGMA)溶液,在34℃,以8000rpm离心1小时。将微核(也称为“微细胞”)悬浮于无血清DMEM培养基,用8μm、5μm、3μm过滤器进行纯化。纯化后,以2000rpm离心10分钟。以2000rpm离心10分钟,悬浮于5ml的无血清DMEM培养基。再以2000rpm离心10分钟。受体细胞使用小鼠ES细胞HKD31 6TG-9(小鼠的Igh和Igk基因被破坏。记载于国际公布WO98/37757号中)和XO ES9(抗体基因未被破坏)。培养是在DMEM(Dulbecco’s Modified Eagle’s Medium-high glucose(高糖培养基):SIGMA)中添加10%FCS、LIF(小鼠白血病抑制因子)、1×10
[F.2]利用PCR分析的耐药克隆的筛选
为了确认IGHK-NAC转移到各种小鼠ES细胞株,提取耐药克隆的DNA,并将其作为模板进行PCR分析。使用的引物如下所示:
相互易位连接位点确认引物:
TRANS L1(同上)
TRANS R1(同上)
KJneo(同上)
PGKr-2(同上)
关于这些引物,使用LA taq(Takara),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品,按照推荐的条件进行。温度、循环条件是在98℃1分钟的热变性后,按94℃10秒、60℃30秒、72℃3分钟进行30个循环。
人2号染色体区域确认引物:
EIF2AK3-F(同上)
EIF2AK3-R(同上)
RPIA-F(同上)
RPIA-R(同上)
IGKC-F(同上)
IGKC-R(同上)
IGKV-F(同上)
IGKV-R(同上)
Vk3-2 F(同上)
Vk3-2 R(同上)
Taq聚合酶使用Ampli Taq Gold(Applied Biosystems),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品,按照推荐的条件进行。温度、循环条件在95℃10分钟的热变性后,按95℃30秒、60℃30秒、72℃1分钟进行35个循环。
人14号染色体区域确认引物:
MTA1-F3(同上)
MTA1-R3(同上)
ELK2P2-F(同上)
ELK2P2-R(同上)
g1(g2)-F(同上)
g1(g2)-R(同上)
VH3-F(同上)
VH3-R(同上)
CH3F3(同上)
CH4R2(同上)
使用这些引物的PCR按照推荐的条件进行,Taq聚合酶使用Ampli Taq Gold(Applied Biosystems),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品。温度、循环条件在95℃10分钟的热变性后,按95℃30秒、60℃30秒或者56℃30秒、72℃1分钟进行35个循环。根据PCR的结果,对于提示携带IGHK-NAC的克隆进行下面的分析。
[F.3]双色FISH分析
将人cot-1DNA和小鼠cot-1DNA作为探针进行FISH分析,确认IGHK-NAC的独立携带和宿主的核型为正常。对于得到期待结果的克隆,用于嵌合体小鼠制作。
[G]IGHK-NAC向大鼠ES细胞的转移
为了制作产生人抗体的大鼠,需要将IGHK-NAC转移到大鼠ES细胞,在8细胞期胚胎进行注射,制作嵌合体大鼠,使IGHK-MAC传递给后代。
[G.1]微核细胞融合与耐药克隆的分离
将CHO K1 IGHK-NAC作为供体,使用与上述F.1记载的向小鼠ES细胞的微核细胞融合法为相同的方法,进行IGHK-NAC向大鼠ES细胞的导入。融合后,温育过夜,加入G418使达到150μg/mL,选择培养3~4周时间。筛选显示为耐药和GFP阳性的株,进行下面的分析。
[G.2]利用PCR分析的耐药克隆的筛选
为了确认IGHK-NAC转移到大鼠ES细胞株,提取耐药克隆的DNA,并将其作为模板进行PCR分析。使用的引物如下所示:
相互易位连接位点确认引物:
TRANS L1(同上)
TRANS R1(同上)
KJneo(同上)
PGKr-2(同上)
关于这些引物,使用LA taq(Takara),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品,按照推荐的条件进行。温度、循环条件是在98℃1分钟的热变性后,按94℃10秒、60℃30秒、72℃3分钟进行30个循环。
人2号染色体区域确认引物:
EIF2AK3-F(同上)
EIF2AK3-R(同上)
RPIA-F(同上)
RPIA-R(同上)
IGKC-F(同上)
IGKC-R(同上)
IGKV-F(同上)
IGKV-R(同上)
Vk3-2 F(同上)
Vk3-2 R(同上)
Taq聚合酶使用Ampli Taq Gold(Applied Biosystems),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品,按照推荐的条件进行。温度、循环条件在95℃10分钟的热变性后,按95℃30秒、60℃30秒、72℃1分钟进行35个循环。
人14号染色体区域确认引物:
MTA1-F3(同上)
MTA1-R3(同上)
ELK2P2-F(同上)
ELK2P2-R(同上)
g1(g2)-F(同上)
g1(g2)-R(同上)
VH3-F(同上)
VH3-R(同上)
CH3F3(同上)
CH4R2(同上)
使用这些引物的PCR按照推荐的条件进行,Taq聚合酶使用Ampli Taq Gold(Applied Biosystems),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品。温度、循环条件在95℃10分钟的热变性后,按95℃30秒、60℃30秒或者56℃30秒、72℃1分钟进行35个循环。根据PCR的结果,对于提示携带IGHK-NAC的克隆进行下面的分析。
[G.3]双色FISH分析
将人cot-1DNA和小鼠cot-1DNA作为探针进行FISH分析,对于能确认到独立携带所期待的IGHK-NAC,保持大鼠ES的正常核型(42条)的株,用于制作嵌合体大鼠。
[H]携带IGHK-NAC的小鼠的制作
进行携带IGHK-NAC的小鼠的制作和分析。对于过程中得到的嵌合体也进行分析。
[H.1]嵌合体小鼠的制作
使用得到的携带IGHK-NAC的小鼠ES细胞,并按照(基因打靶,实验医学,1995)的方法,制作嵌合体小鼠。作为宿主,使用通过MCH(ICR)(白色,从日本CLEA株式会社购入)雌雄交配而得到的桑椹胚和8细胞期胚胎。将注入胚移植到寄养母体,结果出生的仔小鼠可以通过毛色判定是否为嵌合体。
[H.2]嵌合体小鼠的携带IGHK-NAC的分析
按照(胜木元也,发育工程实验手册,讲谈社Scientific,1987)中记载的方法,从经过出生后3周以上的嵌合体小鼠得到尾部,利用Puregene DNA分离试剂盒(Qiagen)提取基因组DNA。根据上述G.2记载的引物和PCR条件进行PCR分析,确认携带IGHK-NAC。
进一步,从嵌合体小鼠采血,然后进行细胞固定,制作标本,通过将人cot-1和小鼠小卫星DNA作为探针进行FISH分析,在染色体水平确认携带IGHK-NAC的细胞。
[H.3]携带IGHK-NAC的ES细胞来源的嵌合体小鼠中的人IGM表达评价
HKD31小鼠ES细胞的小鼠Igh、Igk基因被破坏。B淋巴细胞产生所必需的抗体μ链基因敲除小鼠由于缺失负责体液免疫的成熟B淋巴细胞而无法产生抗体。因此,HKD31小鼠ES细胞在嵌合体小鼠中不能成为成熟B细胞。关于用于嵌合体小鼠制作的携带IGHK-NAC的HKD31小鼠细胞,如果由IGHK-NAC表达人IGM,则可以补救该缺失,能够检测到GFP阳性的B细胞。由此,可以间接检验IGHK-NAC上的IGM基因的功能性表达。从嵌合体小鼠采集血液,使用针对小鼠CD45R(B220)的抗体染色,通过流式细胞仪检测小鼠B细胞。分析是否存在CD45R和GFP均为阳性的细胞,由此能确认IGHK-NAC来源的IGM的功能性表达。使用针对人IGM与小鼠CD45R(B220)的抗体,将血液细胞染色,确认人IGM、CD45R、GFP为阳性的细胞。采集外周血,将血液转移到放入有肝素PBS的试管中,翻转混合,进行冰冷。在离心(2000rpm,3分钟,4℃)后,除去上清然后添加各种抗体,在4℃反应30分钟,利用添加有5%胎牛血清的PBS(5%FBS/PBS)进行清洗。在最后的离心后,向沉淀物加入1.2%葡聚糖/生理盐水,轻打后,在室温静置45分钟,使红细胞自然沉降。将上清液转移到新离心管,在4℃、以2000rpm离心3分钟后除去上清,向沉淀物加入室温的溶血剂(0.17M NH4Cl),静置5分钟。在4℃、以2000rpm离心3分钟,用5%FBS/PBS清洗,然后用500μl的5%FBS/PBS悬浮,将所得物作为分析样品,用流式细胞仪进行分析。
[H.4]嵌合体小鼠血清中的人抗体检测
在嵌合体小鼠中,为了确认人抗体基因轻链、重链、各种同种型表达,使用酶联免疫吸附试验(ELISA)测定血清中的人抗体浓度。ELISA按照如下记载的方法进行。富山、安东,单克隆抗体实验手册,讲谈社,1987;安东、千叶,单克隆抗体实验操作入门,讲谈社,1991;石川、超高灵敏度酶免疫测定法,学会出版中心,1993:Ed Harlow and David Lane,Antibodies A Laboratory Manual,Cold Spring Harbor Laboratory,1988;A.Doyle andJ.B.Griffiths,Cell&Tissue Culture:Laboratory Procedures,John Wiley&Sons Ltd.,1996。参考这些文献所述的方法,根据测定体系,将反应时间、温度改良为在4℃过夜进行等。对于特定的抗体检测,使用试剂盒进行。测定人抗体(hγ、hμ、hκ、hγ1、hγ2、hγ3、hγ4、hα、hε、hδ)的表达和血清中的浓度。基本的操作如下所示。
将待测定的针对人免疫球蛋白的抗体稀释,4℃过夜包被ELISA酶标板。血清试样的测定中使用封闭剂、试样和在稀释标记抗体时添加5%胎牛血清的PBS。清洗包被的培养板,然后进行1小时以上的封闭。清洗培养板,然后加入试样,温育30分钟以上,在清洗培养板后,加入稀释的酶标记抗人和小鼠免疫球蛋白抗体,温育1小时以上,然后清洗培养板,加入底物液使其显色。此外,根据测定体系,以基本相同的操作,使用生物素标记的抗体,清洗培养板后,向其加入抗生物素蛋白-酶复合物进行温育,然后清洗加入底物液。用微孔板读板仪测定吸光度。血清中的浓度的测定是将浓度已知的标准品进行系列稀释,与样品同时进行ELISA,绘制标准曲线进行分析,由此可以确定浓度。
[H.5]人抗体的表达分析和序列鉴定
由嵌合体大鼠脾脏来源的RNA合成cDNA,进行人抗体基因可变区的克隆与碱基序列确定。方法可与国际公布WO98/37757号中记载的方法相同地进行,由此进行分析和评价。
[H.6]产生抗原特异性人抗体的应答的评价
对于嵌合体小鼠,评价能否能确认到抗原特异性人抗体滴度的增加。方法与专利(国际公布WO98/37757号)中记载的方法相同的人血清白蛋白进行免疫,分析抗体效价的上升。
[I]从携带IGHK-NAC的嵌合体小鼠的IGHK-NAC的后代传递
[I.1]IGHK-NAC后代传递
使上述[H]中制作的雌嵌合体小鼠(嵌合体率约100%)与ICR雄小鼠交配,对于出生的仔小鼠,观察来源于ES细胞的IGHK-NAC的显性遗传性状即GFP的荧光。如果观察到GFP的荧光,则能确认小鼠个体中IGHK-NAC传递给后代,并稳定地携带。IGHK-NAC传递给后代的小鼠品系称为mTC(IGHK-NAC)。
[I.2]携带IGHK-NAC的小鼠的携带IGHK-NAC的确认
对于mTC(IGHK-NAC),通过进行与(实施例7)[H.2]同样的分析,可以详细地确认IGHK-NAC的后代传递。
[I.3]携带IGHK-NAC的小鼠的产生人抗体能力评价
对于mTC(IGHK-NAC),进行与(实施例7)[H.4][H.5][H.6]相同的评价。
[J]携带IGHK-NAC的大鼠的制作
制作和分析携带IGHK-NAC的大鼠。对于过程中得到的嵌合体也进行分析。
[J.1]嵌合体大鼠的制作
使用在上述实施例7[G]中得到的携带IGHK-NAC的大鼠ES细胞克隆,按照Hirabayashi等人的方法(Mol Reprod Dev.2010Feb;77(2):94.doi:10.1002/mrd.21123.)制作嵌合体大鼠。作为宿主,使用通过Crlj:WI大鼠(白色,从Charles River公司购入)雌雄交配得到的胚囊期胚胎。将注入胚移植到寄养母体,结果所出生的仔大鼠可以通过毛色判定是否为嵌合体。还在刚出生时观察来源于ES细胞的IGHK-NAC的显性遗传性状即GFP的荧光,确认ES细胞的作用。
[J.2]携带IGHK-NAC的ES细胞来源的嵌合体大鼠的携带IGHK-NAC的确认
进行与上述[H.2]同样的分析,更详细地确认携带IGHK-NAC。对于血液细胞,将人cot-1和小鼠cot-1DNA用作探针,进行FISH分析。
[J.3]嵌合体大鼠的产生人抗体能力评价
对于嵌合体大鼠,进行与(实施例7)[A.4][A.5][A.6]相同的评价。
[K]从携带IGHK-NAC的嵌合体大鼠的IGHK-NAC的后代传递
[K.1]从携带IGHK-NAC的嵌合体大鼠的IGHK-NAC的后代传递
使上述[J]中制作的嵌合体大鼠(嵌合体率约100%)与Crlj:WI大鼠交配,对于所出生的仔大鼠,观察来源于ES细胞的IGHK-NAC的显性遗传性状即GFP的荧光。观察到GFP的荧光,则能确认在大鼠个体中IGHK-NAC传递给后代,并稳定地携带。IGHK-NAC传递给后代的大鼠品系称为rTC(IGHK-NAC)。
[K.2]携带IGHK-NAC的大鼠的携带IGHK-NAC的确认
对于rTC(IGHK-NAC),进行与[J.2]同样的分析,由此可以详细地确认IGHK-NAC的后代传递。
[K.3]携带IGHK-NAC的大鼠的产生人抗体能力评价
对于rTC(IGHK-NAC),进行与(实施例7)[H.4][H.5][H.6]相同的评价。
[实施例8]使用新型小鼠人工染色体载体,制作产生人抗体的(IGHL-NAC)小鼠和大鼠
通过向人抗体基因(IGH、IGL)搭载NAC,构建IGHL-NAC,制作携带IGHL-NAC的产生人抗体的小鼠和大鼠(图21)。
[A]修饰的人22号染色体向携带NAC的CHO细胞的转移
通过修饰的人22号染色体携带CHO细胞,将修饰的人22号染色体导入到携带NAC的CHO细胞。
[A.1]利用微核细胞融合法的修饰的人22号染色体向携带NAC的Hprt缺陷CHO细胞的转移
将供体细胞即修饰的人22号染色体CHO细胞(CHO hChr22LF)在细胞培养皿培养,达到汇合时,更换为添加有20%FBS、0.1μg/ml秋水仙酰胺的F12培养基,再培养48小时培养后,培养基更换为添加有20%FBS、0.1μg/ml秋水仙酰胺的F12培养基,再温育过夜以形成微细胞。除去培养液,在离心用烧瓶中装满预先在37℃保温的细胞松弛素B(10μg/ml,SIGMA)溶液,在34℃,以8000rpm离心1小时。将微核(也称为“微细胞”)悬浮于无血清DMEM培养基,用8μm、5μm、3μm过滤器进行纯化。纯化后,将微细胞悬浮于用DMEM制备的0.05mg/ml PHA-P(SIGMA)溶液2mL中,在除去培养液后添加到在6cm细胞培养皿达到汇合时的受体即携带NAC的Hprt缺陷CHO细胞株。温育15分钟,使微核贴附到CHO细胞。然后,用PEG1000(Wako)溶液[将5g的PEG1000完全溶解于6mL的无血清DMEM培养基,添加1ml二甲基亚砜并进行过滤灭菌]以1ml准确融合1分钟。用5mL的无血清DMEM进行4次清洗操作以除去PEG,然后添加CHO培养液。24小时后,将细胞接种到10块10cm细胞培养皿,添加8μg/mL杀稻瘟菌素S,选择培养10天。对于得到的耐药株,进行下面的分析。
[A.2]利用PCR分析的耐药克隆的筛选
提取杀稻瘟菌素S抗性株的基因组,作为模板进行PCR分析,确认修饰的人2号染色体的携带。其引物序列如下所示:
确认修饰的人22号染色体loxP序列的引物:
22CeT La L:5’-CCTGCCTTCTTGTTTCAGCTCTCAACTG-3’(SEQ ID NO:87)
22CeT La R:5’-GACGTGCTACTTCCATTTGTCACGTCCT-3’(SEQ ID NO:88)
22CeT Ra L:5’-ATCCCCATGTGTATCACTGGCAAACTGT-3’(SEQ ID NO:89)
22CeT Ra R:5’-ACACTTTAGTCCCTGTCCCCTCAACGAG-3’(SEQ ID NO:90)
PCR按照推荐的条件进行,作为热循环仪使用Takara社制造的TP600,PCR酶使用KOD FX(TOYOBO),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品。温度、循环条件是在98℃1分钟的热变性后,按98℃15秒、68℃5分钟进行35个循环。
确认修饰的人22号染色体FRT序列的引物:
22TeT La L:5’-TGCAGGTATCTGTTGGTGTCCCTGTTTT-3’(SEQ ID NO:91)
22TeT La R:5’-GACGTGCTACTTCCATTTGTCACGTCCT-3’(SEQ ID NO:92)
22TeT Ra L:5’-AGCAGAGCTCGTTTAGTGAACCGTCAGA-3’(SEQ ID NO:93)
22TeT Ra R:5’-CTGTCCTATCCTTGCAGCTGTCTTCCAG-3’(SEQ ID NO:94)
PCR按照推荐的条件进行,作为热循环仪使用Takara社制造的TP600,PCR酶使用KOD FX(TOYOBO),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品。温度、循环条件是在98℃1分钟的热变性后,按98℃15秒、68℃5分钟进行35个循环来确认重组。
除此之外,确认在人2号染色体上是否携带有使用引物的区域。其引物序列如下所示:
553P-F:5’-AGATCTCTTGAGCCCAGCAGTTTGA-3’(SEQ ID NO:95)
553P-R:5’-TGAAGTTAGCCGGGGATACAGACG-3’(SEQ ID NO:96)
PPM1F L:5’-AACGGCAGCCAAACCAAAGA-3’(SEQ ID NO:97)
PPM1F R:5’-ACCAGGACTGGCTGGGCATA-3’(SEQ ID NO:98)
IGLVI-70L:5’-AGTCTGCGCTGACCCAGGAA-3’(SEQ ID NO:99)
IGLVI-70R:5’-TTGAGCCAGAGAAGCGGTCA-3’(SEQ ID NO:100)
GNAZ L:5’-TCCACTTGGGGGTCTGCATT-3’(SEQ ID NO:101)
GNAZ R:5’-TGGTGCTGAGCAGCTGTGTG-3’(SEQ ID NO:102)
LIF L:5’-TGGGACTTAGGTGGGCCAGA-3’(SEQ ID NO:103)
LIF R:5’-GCCTCCCCAAGAGCCTGAAT-3’(SEQ ID NO:104)
hVpreB1-F:5’-TGTCCTGGGCTCCTGTCCTGCTCAT-3’(SEQ ID NO:105)
hVpreB1-Rm:5’-GGCGGCGACTCCACCCTCTT-3’(SEQ ID NO:106)
hVpreB3-F:5’-CACTGCCTGCCCGCTGCTGGTA-3’(SEQ ID NO:107)
hVpreB3-R:5’-GGGCGGGGAAGTGGGGGAGAG-3’(SEQ ID NO:108)
hL5-F:5’-AGCCCCAAGAACCCAGCCGATGTGA-3’(SEQ ID NO:109)
hL5-R:5’-GGCAGAGGGAGTGTGGGGTGTTGTG-3’(SEQ ID NO:110)
344-F:5’-ATCATCTGCTCGCTCTCTCC-3’(SEQ ID NO:111)
344-R:5’-CACATCTGTAGTGGCTGTGG-3’(SEQ ID NO:112)
350P-F:5’-ACCAGCGCGTCATCATCAAG-3’(SEQ ID NO:113)
350P-R:5’-ATCGCCAGCCTCACCATTTC-3’(SEQ ID NO:114)
IgL-F:5’-GGAGACCACCAAACCCTCCAAA-3’(SEQ ID NO:115)
IgL-Rm:5’-GAGAGTTGGAGAAGGGGTGACT-3’(SEQ ID NO:116)
SERPIND1 L:5’-ACCTAGAGGGTCTCACCTCC-3’(SEQ ID NO:117)
SERPIND1 R:5’-CCCTGGACATCAAGAATGG-3’(SEQ ID NO:118)
使用这些引物的PCR按照推荐的条件进行,Taq聚合酶使用Ampli Taq Gold(Applied Biosystems),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品。温度、循环条件在95℃10分钟的热变性后,按95℃30秒、63、62、60、56、55、50℃中任一温度30秒、72℃1分钟进行35个循环。
对于PCR阳性克隆,进行下面的分析。
[A.3]双色FISH分析
对于PCR分析阳性克隆,将人cot-1DNA和小鼠cot-1DNA作为探针进行FISH分析,筛选独立携带NAC与修饰的人22号染色体的阳性细胞。
[B]利用易位克隆的人22号染色体IGL区域向小鼠人工染色体载体(NAC)的搭载
在携带NAC的CHO细胞中,将人22号染色体上IGK区域易位克隆到NAC。易位克隆中使用Cre/loxP系统,通过使人2号染色体与NAC相互易位,将IGL区域搭载到NAC(图22)。
[B.1]利用Cre表达的HAT抗性染色体重组体的获得
NAC中搭载有loxP位点,在Cre重组酶存在下与修饰的人2号染色体的loxP位点发生重组。此外,发生重组时成为副产物的NAC中未搭载的人2号染色体区域的5’HPRT与成为副产物的NAC末端的3’HPRT相连接,发生HPRT基因的重构,CHO(hprt-/-)获得HAT抗性。
对于携带修饰的人2号染色体和NAC的Hprt缺陷CHO细胞,在10cm细胞培养皿中达到汇合时,使用Lipofectamine2000(Thermo Fisher Scientific),参照制造商的操作步骤加入18μg的Cre表达质粒(载体名称:pBS185)。添加后经过6小时,更换培养液,24小时后,接种到10块10cm细胞培养皿,用1×HAT(SIGMA)、4μg/mL杀稻瘟菌素进行药剂选择。将得到的耐药株用于下面的分析。
[B.2]利用PCR分析的耐药克隆的筛选
为了将提取HAT抗性株的基因组DNA作为模板筛选相互易位克隆,使用以下引物进行PCR,确认在人2号染色体片段和NAC上发生染色体相互易位。其引物序列如下所示:
TRANS L1(同上)
TRANS R1(同上)
KJneo(同上)
PGKr-2(同上)
关于这些引物,使用LA taq(Takara),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品,按照推荐的条件进行。温度、循环条件是在98℃1分钟的热变性后,按94℃10秒、60℃30秒、72℃3分钟进行30个循环。
用于确认是否保持有FRT插入位点的引物如下所示:
22TeT La L(同上)
22TeT La R(同上)
22TeT Ra L(同上)
22TeT Ra R(同上)
PCR按照推荐的条件进行,作为热循环仪使用Takara社制造的TP600,PCR酶使用KOD FX(TOYOBO),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品。温度、循环条件是在98℃1分钟的热变性后,按98℃15秒、68℃5分钟进行35个循环。
另外,对于人22号染色体区域进行PCR分析。序列如下所示。
553P-F(同上)
553P-R(同上)
PPM1F L(同上)
PPM1F R(同上)
IGLVI-70L(同上)
IGLVI-70R(同上)
GNAZ L(同上)
GNAZ R(同上)
LIF L(同上)
LIF R(同上)
hVpreB1-F(同上)
hVpreB1-Rm(同上)
hVpreB3-F(同上)
hVpreB3-R(同上)
hL5-F(同上)
hL5-R(同上)
344-F(同上)
344-R(同上)
350P-F(同上)
350P-R(同上)
IgL-F(同上)
IgL-Rm(同上)
SERPIND1 L(同上)
SERPIND1 R(同上)
使用这些引物的PCR按照推荐的条件进行,Taq聚合酶使用Ampli Taq Gold(Applied Biosystems),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品。温度、循环条件在95℃10分钟的热变性后,按95℃30秒、63、62、60、56、55、50℃中任一温度30秒、72℃1分钟进行35个循环。
[B.3]双色FISH分析
将人cot-1DNA和小鼠cot-1DNA作为探针进行FISH分析,确认NAC与修饰的人22号染色体发生相互易位,且IGL区域搭载在NAC上的IGL-NAC、副产物独立携带。对于筛选的阳性细胞(命名为CHO IGL-NAC),进行下面的实验。
[C]IGL-NAC向修饰的人14号染色体携带CHO(hprt-/-)细胞株的转移
将制作的IGL-NAC装入携带修饰的人14号染色体的CHO(hprt-/-)细胞株,使发生利用FRT/Flp系统的重组,将IGH区域搭载到IGL-NAC,制作IGHL-NAC。
[C.1]微核细胞融合与耐药克隆的分离
将供体细胞即CHO IGL-NAC在细胞培养皿培养,达到汇合时,更换为添加有20%FBS、0.1μg/ml秋水仙酰胺的F12培养基,再培养48小时培养后,培养基更换为添加有20%FBS、0.1μg/ml秋水仙酰胺的F12培养基,再温育过夜以形成微细胞。除去培养液,在离心用烧瓶中装满预先在37℃保温的细胞松弛素B(10μg/ml,SIGMA)溶液,在34℃,以8000rpm离心1小时。将微核(也称为“微细胞”)悬浮于无血清DMEM培养基,用8μm、5μm、3μm过滤器进行纯化。纯化后,将微细胞悬浮于用DMEM制备的0.05mg/ml PHA-P(SIGMA)溶液2mL中,在除去培养液后添加到在6cm细胞培养皿达到汇合时的受体即CHO hprt-/-14FRT(记载于PCT/JP2017/039441)。温育15分钟,使微核贴附到CHO细胞。然后,用PEG1000(Wako)溶液[将5g的PEG1000完全溶解于6mL的无血清DMEM培养基,添加1ml二甲基亚砜并进行过滤灭菌]以1ml准确融合1分钟。用5mL的无血清DMEM进行4次清洗操作以除去PEG,然后添加CHO培养液。24小时后,将细胞接种到10块10cm细胞培养皿,添加600μg/mL G418和6μg/mL杀稻瘟菌素,选择培养10天。对于得到的耐药株,进行下面的分析。
[C.2]利用PCR分析的耐药克隆的筛选
为了确认IGL-NAC是否被转移到携带修饰的人14号染色体的CHO(hprt-/-)株,是否保持有修饰的人14号染色体,进行PCR分析。使用的引物如下所示。
IGL-NAC的确认引物:
KJneo(同上)
PGKr-2(同上)
关于这些引物,使用LA taq(Takara),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品,按照推荐的条件进行。温度、循环条件是在98℃1分钟的热变性后,按94℃10秒、60℃30秒、72℃3分钟进行30个循环。
用于确认是否保持有FRT插入位点的引物如下所示:
22TeT La L(同上)
22TeT La R(同上)
22TeT Ra L(同上)
22TeT Ra R(同上)
PCR按照推荐的条件进行,作为热循环仪使用Takara社制造的TP600,PCR酶使用KOD FX(TOYOBO),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品。温度、循环条件是在98℃1分钟的热变性后,按98℃15秒、68℃5分钟进行35个循环。
另外,对于人22号染色体区域进行PCR分析。序列如下所示。
553P-F(同上)
553P-R(同上)
PPM1F L(同上)
PPM1F R(同上)
IGLVI-70L(同上)
IGLVI-70R(同上)
GNAZ L(同上)
GNAZ R(同上)
LIF L(同上)
LIF R(同上)
hVpreB1-F(同上)
hVpreB1-Rm(同上)
hVpreB3-F(同上)
hVpreB3-R(同上)
hL5-F(同上)
hL5-R(同上)
344-F(同上)
344-R(同上)
350P-F(同上)
350P-R(同上)
IgL-F(同上)
IgL-Rm(同上)
SERPIND1 L(同上)
SERPIND1 R(同上)
使用这些引物的PCR按照推荐的条件进行,Taq聚合酶使用Ampli Taq Gold(Applied Biosystems),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品。温度、循环条件在95℃10分钟的热变性后,按95℃30秒、63、62、60、56、55、50℃中任一温度30秒、72℃1分钟进行35个循环。
人14号染色体区域的确认引物:
MTA1-F3(同上)
MTA1-R3(同上)
ELK2P2-F(同上)
ELK2P2-R(同上)
g1(g2)-F(同上)
g1(g2)-R(同上)
VH3-F(同上)
VH3-R(同上)
CH3F3(同上)
CH4R2(同上)
使用这些引物的PCR按照推荐的条件进行,Taq聚合酶使用Ampli Taq Gold(Applied Biosystems),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品。温度、循环条件在95℃10分钟的热变性后,按95℃30秒、60℃30秒或者56℃30秒、72℃1分钟进行35个循环。
修饰的人14号染色体上FRT插入位点的确认引物:
14TarC_La F(同上)
14TarC_La R(同上)
14TarC_Ra F(同上)
14TarC_Ra R(同上)
关于这些引物,使用KOD FX(TOYOBO),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品,按照推荐的条件进行。温度、循环条件是在98℃1分钟的热变性后,按98℃15秒、68℃6分钟进行35个循环。
根据该结果,筛选克隆,进行下面的实验。
[C.3]双色FISH分析
将人cot-1DNA和小鼠cot-1DNA作为探针进行FISH分析,确认IGL-NAC与修饰的人14号染色体独立地各保持单拷贝的克隆。选择阳性细胞(命名为CHO#14IGL-NAC)进行下面的实验。
[D]使用FRT/Flp重组系统的IGHL-NAC的构建
通过利用FRT/Flp系统将IGL-NAC与修饰的人14号染色体进行相互易位,在IGL-NAC上将人14号染色体来源的IGH区域进行易位克隆,构建IGHL-NAC(图23)。
[D.1]利用FLP表达的HAT抗性染色体重组体的获得
使用IGL-NAC上的FRT位点与修饰的人14号染色体上的FRT位点,在FLPo重组酶存在下使其发生相互易位。此外,发生重组时,在IGHL-NAC上,5’HPRT与3’HPRT相连接,发生HPRT基因的重构,获得HAT抗性。对于CHO#14IGL-NAC,在10cm细胞培养皿中达到汇合时,使用Lipofectamine2000(Thermo Fisher Scientific),参照制造商的操作步骤加入18μg的FLP表达质粒。添加后经过6小时,更换培养液,24小时后,接种到10块10cm细胞培养皿,用1×HAT、8μg/mL杀稻瘟菌素进行药剂选择。
将得到的HAT抗性克隆用于下面的分析。
[D.2]利用PCR分析的耐药克隆的筛选
使用FRT/FLP系统发生期待的相互易位,为了确认是否构建IGHK-NAC,提取耐药克隆的DNA,作为模板进行PCR分析。使用的引物如下所示:
相互易位连接位点的确认引物:
TRANS L1(同上)
TRANS R1(同上)
CMVr-1(同上)
PGKr-2(同上)
KJneo(同上)
PGKr-2(同上)
关于这些引物,使用LA taq(Takara),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品,按照推荐的条件进行。温度、循环条件是在98℃1分钟的热变性后,按94℃10秒、60℃30秒、72℃3分钟进行30个循环。
对于人22号染色体区域进行PCR分析。序列如下所示。
553P-F(同上)
553P-R(同上)
PPM1F L(同上)
PPM1F R(同上)
IGLVI-70L(同上)
IGLVI-70R(同上)
GNAZ L(同上)
GNAZ R(同上)
LIF L(同上)
LIF R(同上)
hVpreB1-F(同上)
hVpreB1-Rm(同上)
hVpreB3-F(同上)
hVpreB3-R(同上)
hL5-F(同上)
hL5-R(同上)
344-F(同上)
344-R(同上)
350P-F(同上)
350P-R(同上)
IgL-F(同上)
IgL-Rm(同上)
使用这些引物的PCR按照推荐的条件进行,Taq聚合酶使用Ampli Taq Gold(Applied Biosystems),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品。温度、循环条件在95℃10分钟的热变性后,按95℃30秒、63、62、60、56、55、50℃中任一温度30秒、72℃1分钟进行35个循环。
人14号染色体区域的确认引物:
MTA1-F3(同上)
MTA1-R3(同上)
ELK2P2-F(同上)
ELK2P2-R(同上)
g1(g2)-F(同上)
g1(g2)-R(同上)
VH3-F(同上)
VH3-R(同上)
CH3F3(同上)
CH4R2(同上)
使用这些引物的PCR按照推荐的条件进行,Taq聚合酶使用Ampli Taq Gold(Applied Biosystems),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品。温度、循环条件在95℃10分钟的热变性后,按95℃30秒、60℃30秒或者56℃30秒、72℃1分钟进行35个循环。
通过PCR结果筛选克隆,用于下面的实验。
[D.3]双色FISH分析
作为探针,使用BAC克隆CH17-424L4(IGL区域)与CH17-262H11(IGH区域)与CH17-95F2(IGL区域)与CH17-212P11(IGH区域)的组合进行双色FISH分析,详细分析是否构建实际IGHL-NAC。将在NAC上分别观察到显示存在IGL区域与IGH区域的信号的作为阳性,确认IGHL-NAC被构建(命名为CHO IGHL-NAC)并筛选克隆,进行下面的实验。
[E]IGHL-NAC向CHO K1细胞株的转移
在IGHK-NAC和用于构建IGHK-MAC的相互易位时形成的副产物这两者中均携带Neo抗性基因,在通过微核细胞融合法转移到目标细胞时,当用G418进行药剂筛选时,会获得转移了单独的IGHK-NAC或副产物或这两者的细胞。由于在NAC上搭载有EGFP,因此能确认IGHK-NAC是否转移到目标细胞,但为了以有效进行染色体导入的供体细胞制作仅携带IGHK-NAC的细胞,将IGHL-NAC转移到CHO K1细胞株。
[E.1]微核细胞融合与耐药克隆的分离:IGHL-NAC向CHO K1株的转移
将供体细胞即CHO IGHL-NAC在细胞培养皿培养,达到汇合时,更换为添加有20%FBS、0.1μg/ml秋水仙酰胺的F12培养基,再培养48小时培养后,培养基更换为添加有20%FBS、0.1μg/ml秋水仙酰胺的F12培养基,再温育过夜以形成微细胞。除去培养液,在离心用烧瓶中装满预先在37℃保温的细胞松弛素B(10μg/ml,SIGMA)溶液,在34℃,以8000rpm进行1小时的离心。将微核(也称为“微细胞”)悬浮于无血清DMEM培养基,用8μm、5μm、3μm过滤器进行纯化。纯化后,将微细胞悬浮于用DMEM制备的0.05mg/ml PHA-P(SIGMA)溶液2mL中,在除去培养液后添加到在6cm细胞培养皿达到汇合时的受体即CHO K1细胞株。温育15分钟,使微核贴附到CHO细胞。然后,用PEG1000(Wako)溶液[将5g的PEG1000完全溶解于6mL的无血清DMEM培养基,添加1ml二甲基亚砜并进行过滤灭菌]以1ml准确融合1分钟。用5mL的无血清DMEM进行4次清洗操作以除去PEG,然后添加CHO培养液。24小时后,将细胞接种到10块10cm细胞培养皿,添加800μg/mL G418,选择培养10天。对于得到的耐药株,进行下面的分析。
[E.2]利用PCR分析的耐药克隆的筛选
为了确认IGHL-NAC转移到CHO K1细胞株,提取耐药克隆的DNA,并将其作为模板进行PCR分析。使用的引物如下所示:
相互易位连接位点确认引物:
TRANS L1(同上)
TRANS R1(同上)
CMVr-1(同上)
PGKr-2(同上)
KJneo(同上)
PGKr-2(同上)
关于这些引物,使用LA taq(Takara),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品,按照推荐的条件进行。温度、循环条件是在98℃1分钟的热变性后,按94℃10秒、60℃30秒、72℃3分钟进行30个循环。
对于人22号染色体区域进行PCR分析。序列如下所示。
553P-F(同上)
553P-R(同上)
PPM1F L(同上)
PPM1F R(同上)
IGLVI-70L(同上)
IGLVI-70R(同上)
hVpreB1-F(同上)
hVpreB1-Rm(同上)
hVpreB3-F(同上)
hVpreB3-R(同上)
hL5-F(同上)
hL5-R(同上)
344-F(同上)
344-R(同上)
350P-F(同上)
350P-R(同上)
IgL-F(同上)
IgL-Rm(同上)
使用这些引物的PCR按照推荐的条件进行,Taq聚合酶使用Ampli Taq Gold(Applied Biosystems),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品。温度、循环条件在95℃10分钟的热变性后,按95℃30秒、63、62、60、56、55、50℃中任一温度30秒、72℃1分钟进行35个循环。
人14号染色体区域的确认引物:
MTA1-F3(同上)
MTA1-R3(同上)
ELK2P2-F(同上)
ELK2P2-R(同上)
g1(g2)-F(同上)
g1(g2)-R(同上)
VH3-F(同上)
VH3-R(同上)
CH3F3(同上)
CH4R2(同上)
使用这些引物的PCR按照推荐的条件进行,Taq聚合酶使用Ampli Taq Gold(Applied Biosystems),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品。温度、循环条件在95℃10分钟的热变性后,按95℃30秒、60℃30秒或者56℃30秒、72℃1分钟进行35个循环。对于PCR分析阳性细胞株进行下面的分析。
[E.3]双色FISH分析
将人cot-1DNA和小鼠cot-1DNA作为探针进行FISH分析,确认以单拷贝独立携带IGHL-NAC。
进一步,作为探针,使用BAC克隆CH17-424L4(IGL区域)与CH17-262H11(IGH区域)和CH17-95F2(IGL区域)与CH17-212P11(IGH区域)的组合进行双色FISH分析,详细分析IGHL-NAC的结构。将在NAC上分别观察到显示存在IGL区域与IGH区域的信号的作为阳性(命名为CHO K1 IGHL-NAC),用于下面的实验。
[F]IGHL-NAC向小鼠ES细胞的转移
为了制作产生人抗体的小鼠,需要将IGHL-NAC转移到小鼠ES细胞,在受精卵8细胞期进行注射,制作嵌合体小鼠,使IGHL-NAC传递给后代。
[F.1]微核细胞融合与耐药克隆的分离
供体细胞使用CHO K1 IGHL-NAC。使用与实施例7[F.1]相同的方法进行微核细胞融合,获得EGFP阳性且耐药株,进行下面的分析。
[F.2]利用PCR分析的耐药克隆的筛选
为了确认IGHL-NAC转移到小鼠ES细胞株,提取耐药克隆的DNA,并将其作为模板进行PCR分析。使用的引物如下所示:
相互易位连接位点确认引物:
TRANS L1(同上)
TRANS R1(同上)
KJneo(同上)
PGKr-2(同上)
关于这些引物,使用LA taq(Takara),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品,按照推荐的条件进行。温度、循环条件是在98℃1分钟的热变性后,按94℃10秒、60℃30秒、72℃3分钟进行30个循环。
对于人22号染色体区域进行PCR分析。序列如下所示。
553P-F(同上)
553P-R(同上)
PPM1F L(同上)
PPM1F R(同上)
IGLVI-70L(同上)
IGLVI-70R(同上)
hVpreB1-F(同上)
hVpreB1-Rm(同上)
hVpreB3-F(同上)
hVpreB3-R(同上)
hL5-F(同上)
hL5-R(同上)
344-F(同上)
344-R(同上)
350P-F(同上)
350P-R(同上)
IgL-F(同上)
IgL-Rm(同上)
使用这些引物的PCR按照推荐的条件进行,Taq聚合酶使用Ampli Taq Gold(Applied Biosystems),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品。温度、循环条件在95℃10分钟的热变性后,按95℃30秒、63、62、60、56、55、50℃中任一温度30秒、72℃1分钟进行35个循环。
人14号染色体区域的确认引物:
MTA1-F3(同上)
MTA1-R3(同上)
ELK2P2-F(同上)
ELK2P2-R(同上)
g1(g2)-F(同上)
g1(g2)-R(同上)
VH3-F(同上)
VH3-R(同上)
CH3F3(同上)
CH4R2(同上)
使用这些引物的PCR按照推荐的条件进行,Taq聚合酶使用Ampli Taq Gold(Applied Biosystems),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品。温度、循环条件在95℃10分钟的热变性后,按95℃30秒、60℃30秒或者56℃30秒、72℃1分钟进行35个循环。对于PCR分析阳性细胞株进行下面的分析。
[F.3]双色FISH分析
将人cot-1DNA和小鼠cot-1DNA作为探针进行FISH分析,确认只携带IGHL-NAC,保持小鼠ES的正常核型。
作为探针,使用BAC克隆CH17-95F2(IGL区域)与CH17-262H11(IGH区域)和CH17-424L4(IGL区域)与CH17-212P11(IGH区域)的组合进行双色FISH分析,详细分析是否构建实际IGHL-NAC。将在NAC上分别显示存在IGL区域与IGH区域的信号所期待的位置观察到的作为阳性细胞株(HKD31 IGHL-NAC),用于注射。
[G]向大鼠ES细胞的IGHL-NAC的转移
为了制作产生人抗体的大鼠,需要将IGHL-NAC转移到大鼠ES细胞,在受精卵8细胞期进行注射,制作嵌合体小鼠,使IGHL-NAC传递给后代。
[G.1]微核细胞融合与耐药克隆的分离
使用与实施例7[F.1]所述的向小鼠ES细胞的微核细胞融合法相同的方法,进行向大鼠ES细胞的IGHL-NAC的导入。供体细胞使用CHO K1IGHL-NAC。融合后,温育过夜,加入G418使达到150μg/mL,选择培养3~4周时间。将结果为GFP阳性且耐药的克隆用于下面的分析。
[G.2]利用PCR分析的耐药克隆的筛选
为了确认IGHL-NAC转移到大鼠ES细胞株,提取耐药克隆的DNA,并将其作为模板进行PCR分析。使用的引物如下所示:
相互易位连接位点确认引物:
TRANS L1(同上)
TRANS R1(同上)
KJneo(同上)
PGKr-2(同上)
关于这些引物,使用LA taq(Takara),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品,按照推荐的条件进行。温度、循环条件是在98℃1分钟的热变性后,按94℃10秒、60℃30秒、72℃3分钟进行30个循环。
对于人22号染色体区域进行PCR分析。序列如下所示。
553P-F(同上)
553P-R(同上)
PPM1F L(同上)
PPM1F R(同上)
IGLVI-70L(同上)
IGLVI-70R(同上)
hVpreB1-F(同上)
hVpreB1-Rm(同上)
hVpreB3-F(同上)
hVpreB3-R(同上)
hL5-F(同上)
hL5-R(同上)
344-F(同上)
344-R(同上)
350P-F(同上)
350P-R(同上)
IgL-F(同上)
IgL-Rm(同上)
使用这些引物的PCR按照推荐的条件进行,Taq聚合酶使用Ampli Taq Gold(Applied Biosystems),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品。温度、循环条件在95℃10分钟的热变性后,按95℃30秒、63、62、60、56、55、50℃中任一温度30秒、72℃1分钟进行35个循环。
人14号染色体区域确认引物:
MTA1-F3(同上)
MTA1-R3(同上)
ELK2P2-F(同上)
ELK2P2-R(同上)
g1(g2)-F(同上)
g1(g2)-R(同上)
VH3-F(同上)
VH3-R(同上)
CH3F3(同上)
CH4R2(同上)
使用这些引物的PCR按照推荐的条件进行,Taq聚合酶使用Ampli Taq Gold(Applied Biosystems),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品。温度、循环条件在95℃10分钟的热变性后,按95℃30秒、60℃30秒或者56℃30秒、72℃1分钟进行35个循环。对于PCR分析阳性细胞株进行下面的分析。
[G.3]双色FISH分析
将人cot-1DNA和小鼠cot-1DNA作为探针进行FISH分析,确认以单拷贝独立携带IGHL-NAC,保持大鼠ES的正常核型(42条)。作为探针,使用BAC克隆CH17-95F2(IGL区域)与CH17-262H11(IGH区域)与CH17-424L4(IGL区域)与CH17-212P11(IGH区域)的组合进行双色FISH分析,进一步详细分析IGHL-NAC的结构。将在NAC上分别显示存在IGL区域与IGH区域的信号所期待的位置观察到的作为阳性细胞株(命名为rESIGHL-NAC),用于注射。
[H]携带IGHL-NAC的小鼠和大鼠的制作与后代传递个体的制作
使用携带IGHL-NAC的小鼠和大鼠ES细胞,通过进行与(实施例7)[H][I][J][K]相同的操作,可以制作携带IGHL-NAC的后代传递小鼠和大鼠。对于后代传递小鼠、大鼠和在过程中得到的嵌合体小鼠,也进行与(实施例7)[H.4][H.5][H.6]同样的分析,确认携带IGHL-NAC和抗体表达(也含hλ)。制作的携带IGHL-NAC的小鼠和大鼠品系分别称为mTC(IGHL-NAC)、rTC(IGHL-NAC)。
[实施例9]产生完全人抗体的小鼠的制作
使携带IGHK-NAC和IGHL-NAC的小鼠与小鼠Igh和Igk基因被破坏且具有Igl突变(具有Igl表达下降的突变)的小鼠交配,制作产生人抗体的小鼠。
[A]Igh和Igk基因缺失、Igl低表达小鼠的制作
为了制作产生人抗体的小鼠,制作使小鼠抗体基因缺失或低表达的小鼠。
[A.1]Igh和Igk基因缺失、Igl低表达小鼠的制作
使通过HKD31(小鼠Igh、Igk的基因破坏被破坏)小鼠ES得到的小鼠品系与具有小鼠Igl低表达的突变的CD-1(ICR,由Charles River公司购入)交配,制作Igh和Igk基因缺失、Igl低表达小鼠。
来源于CD-1的小鼠Iglc突变通过PCR-RFLP分析进行确认。
使用以下的引物进行PCR。
mIglc1VnC L:5’-CCTCAGGTTGGGCAGGAAGA-3’(SEQ ID NO:119)
J3C1:5’-GACCTAGGAACAGTCAGCACGGG-3’(SEQ ID NO:120)
Taq聚合酶使用Ampli Taq Gold(Applied Biosystems),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品,按照推荐的条件进行。温度、循环条件在95℃10分钟的热变性后,按95℃30秒、60℃30秒、72℃1分钟进行35个循环。
用KpnI-HF(NEB)处理PCR产物,电泳后,将未确认到PCR产物裂解的所得物判定为携带突变等位基因。利用交配得到在两个等位基因中确认到Igλ突变的小鼠(称为“LD品系”)。
[A.2]小鼠抗体基因的表达评价
通过流式细胞仪(FCM)分析和ELISA,来评价小鼠抗体表达消失和基本未表达。
如实施例7[H.3]中所述的,如果Igh基因被破坏,Igμ的表达消失,则不能得到B细胞,可以通过判定B细胞的有无来评价Igh基因缺失。FCM分析与以前的报告(Proc NatlAcad Sci U S A.2000Jan 18;97(2):722-7.)同样地进行,对于确认到B细胞缺失的个体,判定为小鼠Igh缺失。使认为小鼠Igh、Igκ被破坏的小鼠(称为HKD品系)与Igλ突变小鼠进行交配,得到Igh、Igκ被破坏且在两个等位基因具有Igλ突变的小鼠(称为HKLD品系)。
另外,对于得到的小鼠,除了小鼠Igh之外,Igk、Igl的表达也与以前的报告(ProcNatl Acad Sci U S A.2000Jan 18;97(2):722-7)同样地进行ELISA,确认表达消失和低表达。
[A.3]产生人抗体的小鼠的制作
使携带IGHK-NAC的小鼠或者携带IGHL-NAC的小鼠与小鼠IghKO、IgkKO、Igl突变小鼠交配,制作完全产生人抗体的小鼠。
[B]产生完全人抗体的小鼠的评价
[B.1]FACS分析
为了确认携带IGHK-NAC或IGHL-NAC的B细胞的存在,进行流式细胞仪分析。使用针对人IGM与小鼠CD45R(B220)的抗体,将血液细胞染色,确认人IGM、CD45R、GFP为阳性的细胞。使用肝素化毛细管,由眼窝采血,将血液转移到放入有肝素PBS的试管中,翻转混合,进行冰冷。在4℃,以2000rpm离心3分钟,然后除去上清然后添加各种抗体,在4℃反应30分钟,利用添加有5%胎牛血清的PBS(5%FBS/PBS)进行清洗。在最后的离心后,向沉淀物加入1.2%葡聚糖/生理盐水,轻打后,在室温静置45分钟,使红细胞自然沉降。将上清液转移到新离心管,在4℃、以2000rpm离心3分钟后除去上清,向沉淀物加入室温的溶血剂(0.17MNH4Cl),静置5分钟。在4℃、以2000rpm离心3分钟,用5%FBS/PBS清洗,然后用500μl的5%FBS/PBS悬浮,将所得物作为分析样品,用流式细胞仪进行分析。
[B.2]人抗体的表达分析
为了确认人抗体基因轻链、重链、各种同种型表达,利用ELISA进行测定。与(实施例7)[H.4]中记载的方法相同地也包括确认小鼠的抗体表达的有无,测定小鼠抗体(mγ、mμ、mκ、mλ)、人抗体(hγ、hμ、hκ、hλ、hγ1、hγ2、hγ3、hγ4、hα、hε、hδ)的表达和血清中的浓度。
[B.3]人抗体的表达分析和序列鉴定
由产生人抗体的小鼠脾脏来源的RNA合成cDNA,进行人抗体基因可变区的克隆与碱基序列确定。可以通过与(实施例7)[H.5]中记载的方法相同地进行,由此进行分析和评价。
[B.4]产生抗原特异性人抗体的应答的评价
对于产生人抗体的小鼠,评价是否能确认到产生抗原特异性人抗体的应答。以与(实施例7)[H.6]中记载的方法相同的人血清白蛋白进行免疫,分析抗体效价的上升。
[B.5]从产生人抗体的小鼠获得产生人抗体的杂交瘤
可以与专利(国际公布WO98/37757号)中记载方法相同的获得产生人抗体的杂交瘤。
[实施例10]产生完全人抗体的大鼠的制作
使携带IGHK-NAC和IGHL-NAC的大鼠与大鼠Igh、Igk、Igl被破坏的KO大鼠交配,制作产生人抗体的大鼠。
[A]产生人抗体的大鼠的制作和评价
[A.1]产生人抗体的大鼠的制作
通过使携带IGHK-NAC或IGHL-NAC的大鼠品系与大鼠Igh、Igκ、Igλ基因被破坏的大鼠品系交配,可以制作产生人抗体的大鼠。
[A.2]FACS分析
进行携带IGHK-NAC或IGHL-NAC的B细胞的确认。用与(实施例14)[B.1]相同的方法实施,抗体使用抗大鼠CD45R(B220)抗体,溶血剂使用0.15M NH
[A.3]人Ig的表达分析
作为为了利用ELISA的人抗体基因轻链、重链、各种同种型表达的确认,可以通过进行与(实施例7)[H.4]]同样的分析来评价产生人抗体。使用抗大鼠免疫球蛋白抗体也可以对大鼠抗体(rγ、rμ、rκ、rλ)的表达进行评价。
[A.4]人抗体的表达分析和基因序列鉴定
可以使用与上述的(实施例7)[H.5]相同的方法,进行抗体基因序列确定、分析、评价。
[A.5]产生抗原特异性人抗体的应答的评价
与(实施例7)[H.6]的记载相同地实施,能够进行评价。
[A.6]从产生人抗体的大鼠获得产生人抗体的杂交瘤
可以与(实施例9)[B.5]的记载相同的方法实施,获得产生人抗体的杂交瘤。
[实施例11]小鼠人工染色体载体10MAC2、10MAC3的构建
通过在小鼠人工染色体10MAC插入作为DNA插入序列的PGKneo-5’HPRT-loxP和GFP-PGKneo-5’HPRT-loxP型的loxP序列来各自构建小鼠人工染色体载体10MAC2、10MAC3,导入到为了经由环状DNA进行基因搭载的hprt缺陷CHO细胞株,并进行验证。
[A]利用PGKneo-5’HPRT-loxP和GFP-PGKneo-5’HPRT-loxP型的loxP序列向小鼠人工染色体10MAC插入的小鼠人工染色体载体10MAC2、10MAC3的构建
构建在小鼠人工染色体10MAC仅搭载基因搭载位点loxP后的10MAC和搭载基因搭载位点loxP与能够监视其存在的GFP表达单元后的10MAC。
[A.1]PGKneo-5’HPRT-loxP和GFP-PGKneo-5’HPRT-loxP型的loxP打靶载体的制作
用于在DT40(10MAC)插入loxP序列的基本质粒中使用V907(Lexicon genetics)。loxP插入位点即小鼠10号染色体的DNA序列从GenBank数据库得到(NC_000076.6)。从耐药克隆提取基因组DNA作为模板,用于同源重组的二个靶序列扩增的引物的序列如下所示:
NotI_m10 LA F:
5’-TCGAGCGGCCGCTCTAAGTCAGGGAAAGATCCCCTTCTTG-3’(SEQ ID NO:121)
SalI_m10 LA R:
5’-TCGAGTCGACGACCATGAAGATGGTCCAACTAAAGCAA-3’(SEQ ID NO:122)
ClaI_m10 RA F:
5’-TCGAATCGATCACTGCTCTTTCTTTAGTTACATGCAGCCC-3’(SEQ ID NO:123)
ClaI_m10 RA R:
5’-TCGAATCGATATTCTTGCCAAGCTACTCTTCCGAGCTA-3’(SEQ ID NO:124)
PCR按照推荐的条件进行,作为热循环仪使用Perkin-Elmer公司制造的GeneAmp9600,Taq聚合酶使用KOD FX(TOYOBO),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品。温度、循环条件是在98℃1分钟的热变性后,按98℃15秒、68℃3分钟和5分钟进行35个循环。
PGKneo-5’HPRT-loxP的打靶载体的制作。
将PGKneo克隆到V907的EcoRI位点。在AscI位点与ClaI位点插入5’HPRT-loxP。用ClaI(NEB)消化ClaI m10 RA F与ClaI m10 RA R的PCR产物,并利用琼脂糖凝胶进行分离纯化,然后克隆到V907的ClaI位点(载体名称:V907-PGKneo-5’HPRT-loxP-m10RA)。用NotI(NEB)与SalI(NEB)消化NotI_m10 LA F与SalI_m10 LA R的PCR产物,并利用琼脂糖凝胶进行分离纯化,然后克隆到V907-PGKneo-5’HPRT-loxP-m10RA的NotI/SalI位点(载体名称:p10MAC2)。
GFP-PGKneo-5’HPRT-loxP的打靶载体的制作。将CAG-EGFP克隆到V907-PGKneo-5’HPRT-loxP―m10RA的NotI/SalI位点。用NotI(NEB)与PspOMI(NEB)消化NotI_m10 LA F与SalI_m10 LA R的PCR产物,并利用琼脂糖凝胶进行分离纯化,然后克隆到V907-EGFP-PGKneo-5’HPRT-loxP―m10RA的NotI位点(载体名称:p10MAC3)。
打靶载体、靶序列和通过同源重组产生的染色体等位基因如图24、25所示。
[A.2]转染和G418抗性克隆的分离
鸡DT40细胞的培养在添加有10%胎牛血清(Gibco,以下记为“FBS”)、1%鸡血清(Gibco)、10
[A.3]同源重组体的筛选
[A.3.1]PCR分析
为了提取G418抗性株的基因组DNA作为模板来筛选重组体,使用以下引物进行PCR,确认在小鼠10号染色体上是否发生位点特异性地重组。其引物序列如下所示:
DT40(10MAC2)
m10 F1(同上)
NAC R1:5’-CTCTTCAGCAATATCACGGGTAGCCAAC-3’(SEQ ID NO:125)
NAC F1:5’-TGCTTGCATTGTATGTCTGGCTATTCTG-3’(SEQ ID NO:126)
m10 R2(同上)
m10 F6(同上)
Puro I(同上)
DT40(10MAC3)
m10 F1(同上)
EGFP-R:5’-TGCTCAGGTAGTGGTTGTCG-3’(SEQ ID NO:127)
NAC F1(同上)
m10 R2(同上)
m10 F6(同上)
Puro I(同上)
PCR按照推荐的条件进行,作为热循环仪使用Perkin-Elmer公司制造的GeneAmp9600,Taq聚合酶使用KOD FX(TOYOBO),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品。温度、循环条件是在98℃1分钟的热变性后,按98℃15秒、68℃5分或者6分钟进行35个循环。
其结果提示对于DT40(10MAC2)和DT40(10MAC3),对于各14个、11个克隆进行了目的重组。
[A.3.2]双色FISH分析
在由上述得到的DT40(10MAC2)和DT40(10MAC3)中,按照松原等人(FISH实验方案,秀润社,1994)进行双色FISH分析。将小鼠cot-1DNA和5’-HPRT-loxP(X6.1)盒作为探针进行FISH分析,结果在打靶有loxP序列的小鼠10号染色体片段的着丝粒附近检测到探针来源的FITC信号,因此可视觉确认发生位点特异性地重组(图26、27)。由这些结果可得到如下结论:得到携带小鼠人工染色体载体10MAC2和10MAC3的DT40细胞克隆。在下面的步骤中,采用DT40(10MAC2)#8、DT40(10MAC3)#12的各1个克隆。
[B]从含有小鼠人工染色体载体10MAC2和10MAC3的DT40细胞向CHO细胞导入10MAC2和10MAC3
为了在CHO细胞内经由小鼠人工染色体载体10MAC2或者10MAC3的DNA序列插入位点即loxP序列插入携带目的基因(组)的环状DNA,或者为了经由CHO细胞将搭载有目的基因的小鼠人工染色体载体10MAC2或者10MAC3导入到小鼠ES细胞等,将10MAC2或者10MAC3导入到CHO细胞。
[B.1]微核细胞融合与耐药克隆的分离
使用供体细胞即DT40(10MAC2)和DT40(10MAC3),与上述同样地对CHO hprt缺陷细胞(由日本健康科学研究资源库(HSRRB)获得,登录号JCRB0218)即CHO(HPRT
[B.2]耐药克隆的筛选
[B.2.1]PCR分析
为了提取G418抗性株的基因组DNA作为模板来筛选重组体,使用以下引物进行PCR,确认小鼠人工号染色体载体10MAC2和10MAC3是否能导入到CHO细胞。其引物序列如下所示:
CHO(HPRT
m10 F1(同上)
NAC R1(同上)
NAC F1(同上)
m10 R2(同上)
m10 F6(同上)
Puro I(同上)
CHO(HPRT
m10 F1(同上)
EGFP-R(同上)
NAC F1(同上)
m10 R2(同上)
m10 F6(同上)
Puro I(同上)
PCR按照推荐的条件进行,作为热循环仪使用Perkin-Elmer公司制造的GeneAmp9600,Taq聚合酶使用KOD FX(TOYOBO),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品。温度、循环条件是在98℃1分钟的热变性后,按98℃15秒、68℃5分钟或者6分钟进行35个循环。为了得到PCR阳性的克隆各12个、10个克隆,进行下面的分析。
[B.2.2]单色FISH分析
关于在上述得到的CHO(HPRT
[C]确认环状DNA向MAC2和MAC3的插入
检验制作的环状DNA向MAC2和MAC3的重组插入是否确实。
[C.1]利用Cre/loxP系统的环状DNA向MAC2和MAC3的插入
将CHO(HPRT
[C.2]耐药克隆的分析
发生期待的位点特异性重组,如果插入携带LoxP-3’HPRT的环状DNA,则在10MAC2和10MAC3发生HPRT基因的重构,形成HAT抗性。从耐药克隆提取DNA,进行检测该重组接头的PCR。使用的引物如下所示:
TRANS L1(同上)
TRANS R1(同上)
关于这些引物,使用LA taq(Takara),缓冲液和dNTPs(dATP、dCTP、dGTP、dTTP)使用附带品,按照推荐的条件进行。温度、循环条件是在98℃1分钟的热变性后,按94℃10秒、60℃30秒、72℃3分钟进行30个循环。其结果中,对利用位点特异性重组的环状DNA的插入效率进行评价。
工业上的可利用性
本发明的小鼠人工染色体载体能够用于例如表达外源基因的细胞和有用的非人动物的制作、人抗体等蛋白的制造等各种用途或目的。
保藏编号
本说明书中所述的下述(1)和(2)的细胞株分别是通过电穿孔将来源于小鼠10号染色体的和来源于小鼠16号染色体的小鼠人工染色体导入到鸡B前体细胞株DT40的动物细胞株,这2个动物细胞株均基于布达佩斯条约的规定,在2018年2月28日国际保藏于国际保藏机构的日本独立行政法人产品评价技术基础机构的专利微生物保藏中心(NPMD)(邮政编码292-0818日本千叶县木更津市上总镰足2-5-8,122号室)。
(1)DT40(10MAC)T5-26的保藏编号:NITE BP-02656
(2)DT40(16MAC)T1-14的保藏编号:NITE BP-02657
序列表自由文本
SEQ ID NO:1~127:引物
本说明书中所引用的所有出版物、专利和专利申请均通过直接引用并入本说明书中。
序列表
<110> 国立大学法人鸟取大学
染色体移入股份公司
<120> 小鼠人工染色体载体及其用途
<130> PH-7860-PCT
<150> JP 2018-050178
<151> 2018-03-16
<160> 127
<170> PatentIn version 3.5
<210> 1
<211> 14
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 1
aattcggcgc gccg 14
<210> 2
<211> 40
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 2
tcgaggcgcg ccagccttct agggaacagg agatgttcaa 40
<210> 3
<211> 38
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 3
tcgaggatcc gccttgagtg gggttctagt catctttc 38
<210> 4
<211> 28
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 4
aactacccag ttctgcattt ggtgtgag 28
<210> 5
<211> 28
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 5
atcagtcatc agtaccccca acctctct 28
<210> 6
<211> 24
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 6
gagctgcaag aactcttcct cacg 24
<210> 7
<211> 21
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 7
tggatgggtt tcaatgccac t 21
<210> 8
<211> 20
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 8
ggcattctcc cctgttgtgg 20
<210> 9
<211> 20
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 9
acccctcgaa cccctattgc 20
<210> 10
<211> 20
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 10
cacgccatcg gtgatggata 20
<210> 11
<211> 21
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 11
tgggatgacc cccacttctt t 21
<210> 12
<211> 20
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 12
ttttggcctc ttgccccata 20
<210> 13
<211> 38
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 13
tcgaggtacc tctaagtcag ggaaagatcc ccttcttg 38
<210> 14
<211> 38
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 14
tcgactcgag gaccatgaag atggtccaac taaagcaa 38
<210> 15
<211> 40
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 15
tcgagtcgac cactgctctt tctttagtta catgcagccc 40
<210> 16
<211> 40
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 16
tcgagcggcc gcattcttgc caagctactc ttccgagcta 40
<210> 17
<211> 28
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 17
tgagaaatac cgaatggcag agaaacac 28
<210> 18
<211> 20
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 18
cctgaagttc atctgcacca 20
<210> 19
<211> 26
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 19
catcgccttc tatcgccttc ttgacg 26
<210> 20
<211> 28
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 20
gagaggaggg aagcttgatg agaaaatg 28
<210> 21
<211> 22
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 21
tggaggccat aaacaagaag ac 22
<210> 22
<211> 21
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 22
ccccttgacc cagaaattcc a 21
<210> 23
<211> 38
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 23
tcgaggatcc gggagtaatt ttcaatcctt gaggcaga 38
<210> 24
<211> 38
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 24
tcgaagatct catcagtgta caccacaatc ccatctgt 38
<210> 25
<211> 28
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 25
cctcttcttg acggttacca cattttgc 28
<210> 26
<211> 21
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 26
tgaattggtc cctcctgctc a 21
<210> 27
<211> 20
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 27
caggccatga gacccagaca 20
<210> 28
<211> 20
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 28
tcctcggaga atggctcctg 20
<210> 29
<211> 20
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 29
ggcaggttga tgggaactgg 20
<210> 30
<211> 20
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 30
aaccagggtc cccatcctgt 20
<210> 31
<211> 20
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 31
tgggctggtt tcaatgctga 20
<210> 32
<211> 20
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 32
gtgtggccat ggctggagta 20
<210> 33
<211> 20
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 33
tgttcctctg ctgccactcg 20
<210> 34
<211> 20
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 34
acccagccac tcccaccata 20
<210> 35
<211> 20
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 35
aagggcatgg ctatcccaca 20
<210> 36
<211> 38
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 36
tcgaggtacc gggagtaatt ttcaatcctt gaggcaga 38
<210> 37
<211> 38
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 37
tcgactcgag tggcactgac cccttaatta cgtacaga 38
<210> 38
<211> 37
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 38
tcgagtcgac aaagatttgc atccttggcc atgactc 37
<210> 39
<211> 40
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 39
tcgagcggcc gccatcagtg tacaccacaa tcccatctgt 40
<210> 40
<211> 38
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 40
tcgaggtacc aagaacaagc ttcagaacac agccagac 38
<210> 41
<211> 39
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 41
tcgactcgag aacttgtcac acagatccta ctggaggtg 39
<210> 42
<211> 39
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 42
tcgagtcgac ccacagactg aagcaattga cctcaaaag 39
<210> 43
<211> 40
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 43
tcgagcggcc gcaaagcagt tatccgctat ttgggacctt 40
<210> 44
<211> 28
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 44
catgcacatt tgcttacaca cagaggtt 28
<210> 45
<211> 28
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 45
atctgggcac tggggtacaa ctgttaat 28
<210> 46
<211> 29
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 46
ctgagaagag tcattgttta tggtagact 29
<210> 47
<211> 28
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 47
atccccatgt gtatcactgg caaactgt 28
<210> 48
<211> 28
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 48
ggggaataaa caccctttcc aaatcctc 28
<210> 49
<211> 28
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 49
accaagtaac cgatcaaacc aacccttg 28
<210> 50
<211> 28
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 50
tgagaacaca ggggtctcca ttctgact 28
<210> 51
<211> 28
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 51
acaatcaaca gcatccccat ctctgaag 28
<210> 52
<211> 28
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 52
gacgtgctac ttccatttgt cacgtcct 28
<210> 53
<211> 28
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 53
tggtcactga agctttccat ctgctctt 28
<210> 54
<211> 20
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 54
agctcagaga cacctctcca 20
<210> 55
<211> 25
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 55
ctgtattagg atacttggct attga 25
<210> 56
<211> 25
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 56
tatcaagggg gtgtcggaaa tcgtg 25
<210> 57
<211> 25
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 57
actgggcctg ggagaacctg agact 25
<210> 58
<211> 22
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 58
aggtgctgct gggtggtcaa gt 22
<210> 59
<211> 23
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 59
gctcctgcaa atgtctcctg tca 23
<210> 60
<211> 24
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 60
cttacccagg ctccaggctc tatt 24
<210> 61
<211> 25
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 61
ctctacctcc ctaccccatc atcac 25
<210> 62
<211> 20
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 62
tggaaggtgg ataacgccct 20
<210> 63
<211> 22
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 63
tcattctcct ccaacattag ca 22
<210> 64
<211> 20
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 64
agtcagggca ttagcagtgc 20
<210> 65
<211> 20
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 65
gctgctgatg gtgagagtga 20
<210> 66
<211> 20
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 66
ctctcctgca gggccagtca 20
<210> 67
<211> 22
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 67
tgctgatggt gagagtgaac tc 22
<210> 68
<211> 25
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 68
ctctaactga atcaagggaa tgaac 25
<210> 69
<211> 24
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 69
agcagtttga gtttaggatg aagg 24
<210> 70
<211> 26
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 70
catcgccttc tatcgccttc ttgacg 26
<210> 71
<211> 28
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 71
atctgcacga gactagtgag acgtgcta 28
<210> 72
<211> 28
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 72
agcaattagg gcctgtgcat ctcacttt 28
<210> 73
<211> 28
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 73
ccagctcatt cctcccactc atgatcta 28
<210> 74
<211> 28
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 74
catctggagt cctattgaca tcgccagt 28
<210> 75
<211> 28
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 75
cttattcctc cttctgccca cccttcat 28
<210> 76
<211> 24
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 76
agcactttac gcatcccagc atgt 24
<210> 77
<211> 24
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 77
ccaagagagt agtcgtgccc ctca 24
<210> 78
<211> 20
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 78
cccactttac cgtgctcatt 20
<210> 79
<211> 20
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 79
atgaaggtcc gtgactttgg 20
<210> 80
<211> 25
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 80
accccaaagg ccaaactctc cactc 25
<210> 81
<211> 24
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 81
cacttgtact ccttgccatt cagc 24
<210> 82
<211> 20
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 82
agtgagataa gcagtggatg 20
<210> 83
<211> 20
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 83
cttgtgctac tcccatcact 20
<210> 84
<211> 20
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 84
aggccagcat ctgcgaggat 20
<210> 85
<211> 20
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 85
gtggcagcaa gtagacatcg 20
<210> 86
<211> 28
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 86
cctattggcg ttactatggg aacatacg 28
<210> 87
<211> 28
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 87
cctgccttct tgtttcagct ctcaactg 28
<210> 88
<211> 28
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 88
gacgtgctac ttccatttgt cacgtcct 28
<210> 89
<211> 28
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 89
atccccatgt gtatcactgg caaactgt 28
<210> 90
<211> 28
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 90
acactttagt ccctgtcccc tcaacgag 28
<210> 91
<211> 28
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 91
tgcaggtatc tgttggtgtc cctgtttt 28
<210> 92
<211> 28
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 92
gacgtgctac ttccatttgt cacgtcct 28
<210> 93
<211> 28
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 93
agcagagctc gtttagtgaa ccgtcaga 28
<210> 94
<211> 28
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 94
ctgtcctatc cttgcagctg tcttccag 28
<210> 95
<211> 25
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 95
agatctcttg agcccagcag tttga 25
<210> 96
<211> 24
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 96
tgaagttagc cggggataca gacg 24
<210> 97
<211> 20
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 97
aacggcagcc aaaccaaaga 20
<210> 98
<211> 20
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 98
accaggactg gctgggcata 20
<210> 99
<211> 20
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 99
agtctgcgct gacccaggaa 20
<210> 100
<211> 20
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 100
ttgagccaga gaagcggtca 20
<210> 101
<211> 20
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 101
tccacttggg ggtctgcatt 20
<210> 102
<211> 20
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 102
tggtgctgag cagctgtgtg 20
<210> 103
<211> 20
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 103
tgggacttag gtgggccaga 20
<210> 104
<211> 20
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 104
gcctccccaa gagcctgaat 20
<210> 105
<211> 25
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 105
tgtcctgggc tcctgtcctg ctcat 25
<210> 106
<211> 20
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 106
ggcggcgact ccaccctctt 20
<210> 107
<211> 22
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 107
cactgcctgc ccgctgctgg ta 22
<210> 108
<211> 21
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 108
gggcggggaa gtgggggaga g 21
<210> 109
<211> 25
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 109
agccccaaga acccagccga tgtga 25
<210> 110
<211> 25
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 110
ggcagaggga gtgtggggtg ttgtg 25
<210> 111
<211> 20
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 111
atcatctgct cgctctctcc 20
<210> 112
<211> 20
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 112
cacatctgta gtggctgtgg 20
<210> 113
<211> 20
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 113
accagcgcgt catcatcaag 20
<210> 114
<211> 20
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 114
atcgccagcc tcaccatttc 20
<210> 115
<211> 22
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 115
ggagaccacc aaaccctcca aa 22
<210> 116
<211> 22
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 116
gagagttgga gaaggggtga ct 22
<210> 117
<211> 20
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 117
acctagaggg tctcacctcc 20
<210> 118
<211> 19
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 118
ccctggacat caagaatgg 19
<210> 119
<211> 20
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 119
cctcaggttg ggcaggaaga 20
<210> 120
<211> 23
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 120
gacctaggaa cagtcagcac ggg 23
<210> 121
<211> 40
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 121
tcgagcggcc gctctaagtc agggaaagat ccccttcttg 40
<210> 122
<211> 38
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 122
tcgagtcgac gaccatgaag atggtccaac taaagcaa 38
<210> 123
<211> 40
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 123
tcgaatcgat cactgctctt tctttagtta catgcagccc 40
<210> 124
<211> 38
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 124
tcgaatcgat attcttgcca agctactctt ccgagcta 38
<210> 125
<211> 28
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 125
ctcttcagca atatcacggg tagccaac 28
<210> 126
<211> 28
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 126
tgcttgcatt gtatgtctgg ctattctg 28
<210> 127
<211> 20
<212> DNA
<213> 人工的(Artificial)
<220>
<223> 引物
<400> 127
tgctcaggta gtggttgtcg 20
PCT/RO/134表
- 小鼠人工染色体载体及其用途
- 小鼠人工染色体载体