宽分子量硫酸软骨素制备工艺
文献发布时间:2023-06-19 09:30:39
技术领域
本发明涉及一种硫酸软骨素的制备方法,尤其涉及一种宽分子量硫酸软骨素制备工艺。
背景技术
硫酸软骨素是从动物的软骨组织中提取的一种糖胶聚糖,化学结构复杂, 糖链中编码着大量的生物信息。硫酸软骨素通过与许多功能蛋白质相互作用, 参与细胞分裂、分化、组织器官形成和中枢神经系统发育等许多细胞事件和生理过程,在医药领域中具有广阔应用前景。
分子量是高分子化合物的基本参数之一,硫酸软骨素的分子量是区分不同物种硫酸软骨素的重要参考标准,这是在原料药检查中区分物种来源的方法之一。现行硫酸软骨素中国药品标准中,测量分子量的SEC-HPLC法采用的对照品(右旋糖酣) 不合理致使其测定结果与SEC-MALLS法的结果相差较大。为能够采用SEC-HPLC法准确测定硫酸软骨素的分子量及分子量分布,需要筛选分子量对照品。由于SEC-HPLC法不是个绝对分子量的测定方法,对给定的色谱柱需要用分子量标准样品来标定其淋出体积和分子尺寸大小间的关系。
通过市售品硫酸软骨素制备宽分子量硫酸软骨素有较大的难度,因为硫酸软骨素在生物体内合成后,并没有像肝素一样经过酶降解,所以硫酸软骨素的分子量分布要明显窄于肝素, 无法使用简单的纯化或分离的方法获得宽分子量样品。
发明内容
发明目的:为了克服现有技术中存在的不足,本发明提供一种宽分子量硫酸软骨素制备工艺。
技术方案:一种宽分子量硫酸软骨素制备工艺,包括以下步骤:
(S1)收集动物软骨,将收集的动物软骨进行酶解,酶解后进行纯化,得到高分子量硫酸软骨素;
(S2)将步骤(S1)中得到的高分子量硫酸软骨素,加入缓冲溶液,制成10%(W/V)溶液;
(S3)将步骤(S2)中得到的溶液利用硫酸软骨素裂解酶降解;
(S4)向步骤(S3)中得到的溶液中依次加入1.0%(W/V)的氯化钠、1.5倍(V/V)95%乙醇,随后进行沉淀、干燥,得到宽分子量硫酸软骨素中间体;
(S5)将步骤(S4)最后得到的宽分子量硫酸软骨素中间体溶解成2%(W/V)溶液,向溶液中加入1.0%(W/V)的氯化钠后,加入1.5倍(V/V)95%乙醇沉淀,干燥后再次溶解成2%(W/V)水溶液,加入2.0%(W/V)的氯化钠后,加入1.5倍(V/V)95%乙醇沉淀,干燥得到宽分子量硫酸软骨素。
进一步地,步骤(S1)中使用的酶为碱性蛋白酶。
进一步地,步骤(S1)中酶解后,用盐酸溶液调节pH6.0,离心后取上清液;向上清液中加入3.0%(W/V)的氯化纳后,加入2.0倍(V/V)95%乙醇,静置30min得到的沉淀弃去上层乙醇;重新溶解成原体积的1/3,加入0.5%(W/V)氯化纳溶解后,过滤澄清,经过沉淀、脱水、烘干后得高分子量硫酸软骨素。
进一步地,步骤(S2)中的缓冲溶液含有三羟甲基氨基甲烷、醋酸钠、水和稀盐酸,且缓冲溶液的PH值为8.0。
进一步地,步骤(S3)使用的裂解酶为硫酸软骨素AC酶,所述AC酶的用量为每克高分子量硫酸软骨素0.1IU,反应时温度为37℃,反应时间为4小时。
有益效果:将利用本发明所述方案制备得到的宽分子量硫酸软骨素与市场上常见的硫酸软骨素进行对比具有如下优点:(1)分布系数宽,能够满足涵盖所测样品的分子量范围;(2)峰位保留时间与样品的保留时间相差不大;(3)峰形饱满;基于此,由本发明制得的宽分子量硫酸软骨素更符合分子量标准品的要求。
附图说明
图1为本发明所述的高分子量硫酸软骨素、市售品硫酸软骨素、肝素标准品的分子量峰形分布对比,其中曲线A表示的是高分子量硫酸软骨素的分子量,曲线B表示的是市售品硫酸软骨素的分子量,曲线C表示的是肝素标准品的分子量;
图2为本发明所述的宽分子量硫酸软骨素中间体的分子量分布峰型对比;
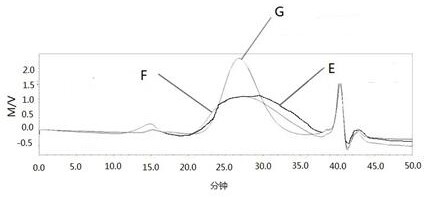
图3为本发明所述的宽分子量硫酸软骨素经过乙醇沉淀前后以及市售品硫酸软骨素的分子量峰形分布对比,曲线E表示的是乙醇沉淀前的宽分子量硫酸软骨素的分子量,曲线F表示的是经过乙醇沉淀后的宽分子量硫酸软骨素的分子量,曲线G表示的是市售品的硫酸软骨素的分子量。
具体实施方式
以下结合附图1对本发明的原理和特征进行描述,所举实例只用于解释本发明,并非用于限定本发明的范围。
(S1)高分子量硫酸软骨素制备;将500g猪鼻干软骨加入10倍体积(W/V)饮用水,升温至90℃,溶胀3h,切碎,控制温度55-58℃,用氢氧化纳溶液调节料液的PH值8.0-8.5;加入猪鼻干软骨重量0.4%的碱性蛋白酶(诺维信生产的3.0T型号),酶解4小时,且酶解过程中保证PH值8.0-8.5,控制温度为55-58℃;
随后,用盐酸溶液调节pH6.0,离心后得上清液;上清液中加入3.0%(W/V)的氯化纳后,加入2.0倍(V/V)95%乙醇,静置30min得到的沉淀弃去上层乙醇;重新溶解成原体积的1/3,加入0.5%(W/V)氯化纳溶解后,过滤澄清,加入3.0倍(V/V) 95%乙醇沉淀;用95%乙醇脱水2h,60℃烘干4h得高分子量硫酸软骨素,其绝对分子量为Mw46000(利用SEC-MALLS方法测得),约为硫酸软骨素原料药市售品(Mw22000-24000,利用SEC-MALLS方法测得)的两倍,是由核心蛋白连接两个硫酸软骨素分子形成的双聚体。
以肝素药典分子量标准品(Mw16000)作为对照例1,与得到的高分子量硫酸软骨素的分子量进行对比,检测方法为SEC-MALLS,如附图1所示,其中曲线A表示的是高分子量硫酸软骨素,曲线B表示的是市售品硫酸软骨素,曲线C表示的是肝素标准品。
(S2)高分子量硫酸软骨素被裂解酶降解;将步骤(S1)中得到的高分子量硫酸软骨素40g,加入400ml缓冲溶液溶解。所述缓冲溶液的制备:称取三控甲基氨基甲院15.15g与醋酸纳16.34g,加水2250ml使之溶解,用稀盐酸调节pH值至8.0,并用水稀释至2500mL)。
(S3)加入4IU硫酸软骨素AC酶于37℃降解8h。
(S4)每小时取样,向取样中加入1.0%(W/V)的氯化纳后,再加入1.5 倍(V/V) 95%乙醇沉淀、离心、脱水、烘干,得到宽分子量硫酸软骨素中间体。
采用SEC-MALLS检测方法,所得宽分子量硫酸软骨素中间体的分子量分布峰型对比见附图2。从峰型可以判断出,裂解酶降解1-2h时,组分中大分子偏多;裂解酶降解3-4h时,峰型比较对称;降解5h及以上时,小分子偏多。根据后续的硫酸软骨素峰型调整步骤发现,裂解酶降解4h时,尽管小分子部分稍微偏多,但是经过乙醇分级沉淀,最终可以调整出对称度很好的峰型。
(S5)宽分子量硫酸软骨素峰型调整;取裂解酶降解4h的宽分子量硫酸软骨素中间体9g,加入450ml水溶解;向料液中加入1.0%(W/V)的氯化钠后,加入1.5倍(V/V)95%乙醇沉淀;干燥后再次溶解成2%(W/V)水溶液,加入2.0% (W/V)的氯化纳后,加入1.5倍(V/V)95%乙醇沉淀、干燥,得到宽分子量硫酸软骨素。
将本发明所述的宽分子量硫酸软骨素在经过步骤(S5)乙醇沉淀前后的产品以及硫酸软骨素市售品分别进行分子量检测,检测方法为SEC-MALLS,检测结果如附图3所示,其中曲线E表示的是乙醇沉淀前的宽分子量硫酸软骨素,曲线F表示的是经过乙醇沉淀后的宽分子量硫酸软骨素,曲线G表示的是市售品的硫酸软骨素。具体地:
供试品溶液:样品加超纯水溶解,制成浓度约为10mg/mL的溶液;
流动相:称取无水硫酸纳113.6g,加3900mL超纯水溶解,用稀硫酸调节pH至5.0, 定容至4L。
色谱柱:TSKgel G4000SWXL 7.8*300 mm
流动相:0.2mol/L 硫酸纳溶液(pH=5.0)
流速:0.5mL/min
检测器:示差折光检测器
柱温:35℃
进样体积:25µL。
由图3可知,经过乙醇分级沉淀后,宽分子量硫酸软骨素的峰型更加对称饱满,并且高分子量组分和低分子量组分均比市售品更宽。
以上所述仅为本发明的较佳实施例,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。