一种mRNA 3`末端测序文库快速构建方法及应用
文献发布时间:2023-06-19 19:28:50
技术领域
本发明涉及高通量测序文库及文库构建领域,更具体地,涉及一种mRNA′3′末端测序文库快速构建方法及应用。
背景技术
分子生物学的中心法则解释了遗传信息从DNA到功能性蛋白的流动,大多数情况下,mRNA在丰度、中间体和功能产物之间存在线性关系。对于mRNA来说,信使RNA前体(Pre-mRNAs)剪接加工为成熟mRNA是转录过程中的重要步骤,对mRNA功能及稳定性有关键作用。
大多数人类基因的mRNA加工过程是以某种条件或某种细胞类型的相关方式表达不同的转录异构体。替代启动子和转录本终止位点分别决定了5′和3′非翻译区(UTR)的边界,UTR包含大量影响mRNA稳定性、局部化和翻译率的序列和结构原件,即使是有相同编码区但不同UTR异构体可能产生不同的功能结果,也可能会将该蛋白质定位到不同的亚细胞区室,这些都是造成亚型及功能区别的主要原因。
目前对人类多聚腺苷酸化(APA)组织图谱分析表明,一般基因都有多个Poly A位点,在Pre–mRNA加工过程中,交替切割和多聚腺苷酸化会产生编码序列或不同的3′UTR异构体,APA可保护Pre-mRNA核糖核酸免受酶降解,并促进核输出和翻译。因此3′末端加工的缺陷会影响对Poly-A位点选择,对3′UTR更深入的理解对治疗发展具有重要意义。
最近的几项研究报告了APA调节广泛存在于免疫系统、神经发育和肿瘤发生以及诱导多能干细胞的产生。有趣的是,细胞的整体APA谱与其增殖和分化状态也密切相关。例如,近端poly(A)位点倾向于用于增殖或未分化细胞,导致产生具有较短3′UTR的mRNA,而远端poly(A)位点倾向于用于分化细胞,导致mRNA具有较长的3′UTR。越来越多的实验说明APA比以前预期要普遍得多,并且APA调节在各种生理和病理过程中起着重要作用。
APA产生不同3′UTR的mRNA,可通过多种机制影响基因表达。如3′UTR富含AU、GU的元件和microRNA靶位点以及RNA结合蛋白(RBP)位点。大多数癌症表达的转录本比相应的正常组织的3′UTR短,针对miRNA的抑制作用,癌基因的短3′UTR同种型比长3′UTR更稳定,事实表明通过APA缩短3′UTR可导致癌细胞中致癌基因的激活,因此miRNA在3′UTR的靶向位点会改变疾病基因的转化,有利于发现治疗癌症、心血管病等疾病治疗靶标。除此,RBP可以识别特定的基元、简并位点或二级结构,3′UTR的APA会引起RBP结合基序的数量改变,可以介导mRNA的稳定性、定位和效率,从而对基因转录有增强或衰减的调控作用。
另一方面,3′UTR影响基因表达的相关性与Poly(A)位点的选择有关,有许多例子说明mRNA 3′末端的加工在人类血液学、免疫和神经疾病以及癌症中经常发生失调。如单个poly(A)位点的损失、获得或突变已在血液病中观察到,如多内分泌病肠病X-连锁综合征(IPEX)主要是由于维持T细胞调节的转录调节因子FOXP3的突变,其PAS(Poly A site,AATAAA到AATGAA)中的A-G突变使FOXP3转录本的非特异性降解,导致FOXP3表达减少,从而不能正常维持T细胞调节。还有一些转录本3′末端因单核苷酸多态性(SNP)导致受损,增加癌症的易感性。由此,了解APA如何在时间和空间上受到调节至关重要。
现有许多3′UTR-seq测序手段,对3′末端的的序列进行一系列基因表达分析,以便更好研究APA研究中miRNA的调控,mRNA的稳定、定位及翻译。
尽管目前的技术取得了许多进展,但综合来看,绝大多数利用oligo dT引物两步反应生成双链cDNA,需单独反应加接头操作,这在一定程度上降低了效率;在去剔除rRNA这方面,有些利用探针结合磁珠剔除,有些利用RNaseH降解DNA探针-rRNA杂交链,但仍会有残留rRNA影响测序深度及探针影响;针对模板片段化,大多技术使用超声打断、二价金属离子试剂盒等,成本高、操作复杂、副产物多;且大多对片段化及建库样品使用醇沉回收,耗时且损失大,增加操作复杂性,不适合批量样品建库操作,不利于整个方法流程自动化。
特别是,一般建库中常使用的末端修复加接头操作,这存在着不同程度的问题。比如接头效率非常有限,会导致部分信息丢失;平末端的双链效率中等,但又易形成接头自连;Y型结构双链接头效率偏低,制作工艺需要额外添加退火步骤,而且退火前常因为单链比例失衡而不稳定;U型结构单链接头由于单链结构,天然形成部分双链茎环结构,效率虽高,但是需要在文库PCR前使用酶将U型结构打开,增加实验步骤和试剂成本。现阶段,接头设计序列仍然受到测序上游公司制约,接头效率参差,影响最终文库得率。
有鉴于此,针对mRNA 3′末端测序,有必要提供一种快速构建高效建库的方法,有必要构建一种用于mRNA 3′末端测序文库构建的高效稳定试剂盒。
发明内容
鉴于上述内容,本发明目的在于提供一种mRNA 3′末端测序文库快速构建方法及其应用。
为实现上述目的,本发明采用如下技术方案:
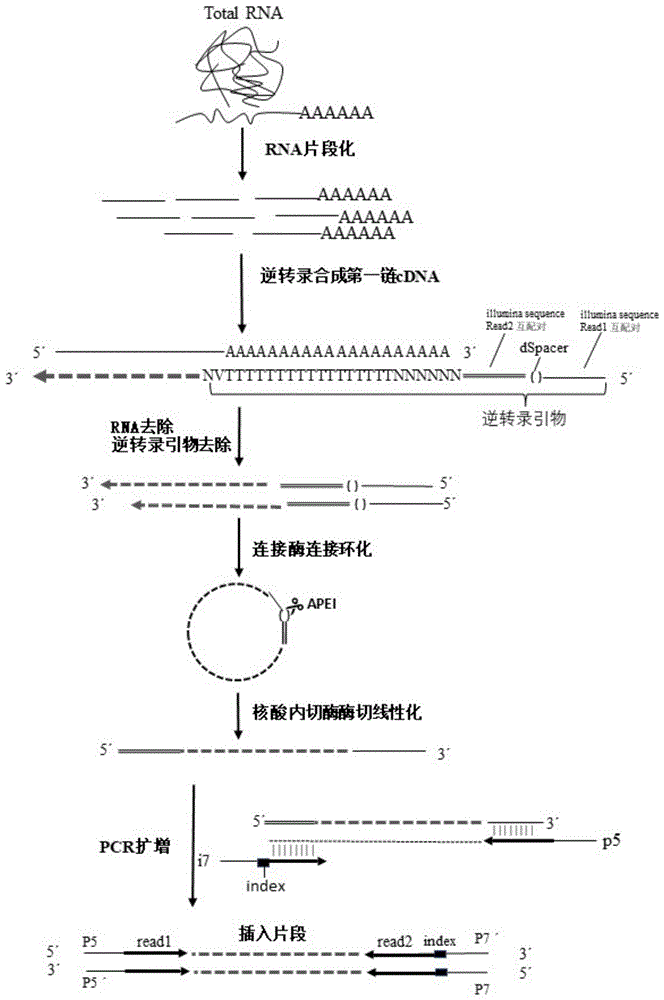
一种mRNA 3′末端测序文库的构建方法,包括如下步骤:采用逆转录引物对片段化的RNA进行逆转录,获得cDNA第一链产物;cDNA第一链使用连接酶连接环化,环化产物再用核酸内切酶酶切进行线性化,线性化产物进行PCR扩增,得到mRNA 3′末端测序文库。
所述的逆转录引物从5′端至3′端依次包括:与测序引物配对的序列、一个脱碱基位点dSpacer、与测序引物配对的序列、UMI、与mRNA poly A尾配对的oligo dT、两个脱氧核苷酸VN,5′末端带有PO
所述的连接酶优选为ssDNA/RNA环化连接酶。
所述的核酸内切酶优选为APE1酶。
进一步的,所述的mRNA 3′末端测序文库的构建方法,包括如下步骤:
1)提取样品总RNA;
2)将1)所得RNA高温变性后,用NaOH强碱,在一定条件下水解,获得片段化产物,长度100-500nt,去除基因组DNA,磁珠纯化RNA;
3)将2)所得产物用逆转录引物进行逆转录,反应生成cDNA第一链产物;
4)高效去除逆转录引物及RNA,采用磁珠分选纯化100-500nt范围的第一链cDNA片段;
5)cDNA第一链使用连接酶连接环化,环化产物再用核酸内切酶酶切进行线性化,将本在同侧的接头分于cDNA两侧,获得建库模板;
6)以线性化产物为模板,使用带有index引物PCR扩增,磁珠纯化250-500bp PCR产物,即获得mRNA 3′末端测序文库。
作为一种优选,进一步的技术方案是:
本发明优选地采用Trizol法进行样品的总RNA提取,该方法特别适合组织和细胞样品;
优选地,在所述步骤2)中,NaOH水解反应体系中NaOH终浓度范围0.02M-0.03M,更优选地使用0.03M NaOH终浓度;体系反应时间优选地20min-35min,更优选时间为30min;体系反应温度优选27℃-37℃,更优选地使用27℃。水解反应后,及时使用HCl中和NaOH。步骤2)的优选反应体系如下:
优选地;在所述步骤3)中,RNA逆转录生成第一链cDNA是使用逆转录引物为特殊设计序列oligo dT(含UMI),序列如下:逆转录引物5′端至3′端依次包括5′-AGATCGGAAG
优选地,所述步骤4)中,高效去除逆转录引物使用的是3μL Exonuclease,优选反应条件37℃,1h;高效去除rRNA、mRNA及其他RNA,采用的是终浓度0.04M NaOH(新鲜配制),反应条件98℃,20min。
优选地,所述步骤4)中,纯化第一链cDNA是用诺唯赞RNA clean beads纯化,体积调整RNA clean beads比例是产物体积的0.9~1.8倍,更优化使用1.2倍,使纯化cDNA片段大小在250-500nt。
优选地,所述步骤5)中,单链cDNA在热稳定连接酶的作用下,将第一链cDNA的5′-磷酸基团和3′-OH基团通过磷酸二酯键连接起来,使得逆转录引物设计的5′端接头能与cDNA链3′端相连,再将环化产物在指定位置切开线性化。
优选地,所述步骤5)中,将环化产物进行线性化使用APE1酶,使得本在同侧的两个接头分别位于cDNA的5′和3′两端。线性化缓冲液的成分包括240mM C4H6MgO4和25mM DTT。反应先使用1μL APE 1,37℃孵育1h,再补加0.5μL APE1,37℃孵育30min,65℃条件孵育20min失活。
优选地,所述步骤6)中,使用带有index的引物将片段PCR扩增,放大。优选操作步骤先选择1μL作为模板,预PCR 18X循环,分析预PCR产物在聚丙烯酰胺凝胶电泳条带亮度确定,以便选择更合适的建库PCR循环数;
优选地,所述步骤6)中,磁珠纯化PCR产物是用诺唯赞DNAclean beads,需调整磁珠比例0.70X~1.5X,最优选择0.75X(珠子体积=0.75×PCR产物体积)珠子纯化,操作按参考说明书进行,最后用水洗脱,上机测序。
本发明所提供的这种高效稳定构建mRNA 3′末端测序文库的方法,可应用在mRNA3′末端研究;可开发成一种快速简便的试剂盒,操作简单,条件优化,高效快速,是一种研究mRNA 3′末端的有效工具。
本发明方法专门针对100-500中长度mRNA的3′末端测序文库的高效构建方法,能高效去除逆转录引物、rRNA,不影响后期对数据的深度测序;不需要合成双链cDNA及接头反应,仅通过合成第一链cDNA,逆转录引物特殊设计带有接头,cDNA环化在指定位置线性化切开,两端接头分于两侧。本发明效率更高,操作简单,减少对样品损耗,能以低浓度样品得高质量的clean reads,有效实现样品测序文库在基础研究、临床诊断、药物研发等领域有广泛应用,具有良好的开发和应用前景。
与现有的各种技术相比,本发明具有以下优点和效果:
(1)本发明在RNA片段化过程区别于现有的技术,现有许多使用超声打断、二价金属离子试剂盒等,成本高、操作极其复杂、副产物多和易造成RNA的降解,且大多技术需使用醇沉回收纯化样品才能进行逆转录,仅该过程就需要2天,减慢实验速度,增加操作复杂性,不适合批量样品建库操作,不利于整个方法流程自动化;而本发明使用NaOH在合适的反应条件下,仅需30分钟,对mRNA片段化,操作简单,大小均匀,适合批量建库。
(2)本发明利用特殊设计逆转录引物,一步反应生成单链cDNA,不需要两步反应合成双链cDNA,在一个反应管中完成,简便快捷。
(3)现有技术建库中常使用的加接头或末端修复在进行扩增,都存在着不同程度的问题,如接头效率非常有限或不稳定等。本发明通过特殊设计的逆转录引物,序列设计包含接头,环化使得5′端接头能与cDNA链5′端相连,线性化两个接头分别位于cDNA的5′和3′两端,该过程仅需加入一种酶,不用添加其他化学试剂及接头片段等,效率高,副产物少。
(4)由于本发明测序集中于转录本的3′末端,因此需要的读长的reads少,从而降低了成本,每次测序样本数量可以更多。取消接头连接步骤,可以在低测序深度上实现较好的灵敏水平,因而可以实现多个文库同步测序。
(5)现有的大多技术是利用生物素化探针结合磁珠剔除rRNA或利用RNaseH降解DNA探针-rRNA杂交链,但仍会有残留rRNA及探针影响测序深度,且两种方法都会残留许多的副产物,操作麻烦,试剂成本高。本发明采用强碱高温水解rRNA及逆转录反应过后的的mRNA模板,试剂便宜,去除彻底且无副产物。
(6)相比于现有的胶回收DNA片段方法,在PCR建库环节,使用聚丙烯酰胺凝胶电泳胶回收纯化目的片段,操作繁琐、耗时且损失大。本发明采用磁珠分选250-500bp DNA片段,纯化时间仅需15min,操作简单,大大缩短时长,且产物损失较少。
附图说明
图1为本发明对mRNA 3′末端测序文库快速构建的构建示意图。
图2为本发明使用的逆转录引物的设计示意图。引物5′末端带有PO
图3为实施例1、2的1μg-2μg总RNA片段化及去基因组的RNA片段在聚丙烯酰胺尿素变性胶的检测图。泳道1-3分别是实施例1的RNA片段化产物、RNA去基因组产物、RNA cleanbeads洗脱产物;泳道4:50bp marker;泳道5-7分别是实施例2的RNA片段化产物、RNA去基因组产物、RNA clean beads洗脱产物。
图4为实施例1的RNA逆转录合成cDNA各过程中的产物在聚丙烯酰胺尿素变性胶的检测图。泳道1:50bp marker;泳道2:RNA片段化产物;泳道3:RNA clean beads洗脱产物;泳道4:RNA逆转录合成第一链cDNA产物;泳道5:去除逆转录引物后的产物;泳道6:去除RNA后的产物。
图5为实施例1、2线性化后的DNA预PCR18个循环产物的聚丙烯酰胺胶检测图。泳道1:50bp marker;泳道2-3:实施例1的两个重复组;泳道4-5:实施例2的两个重复组。
图6为实施案例1建库PCR产物使用磁珠分选的聚丙烯酰胺胶检测图。泳道1:50bpmarker;泳道2:1μL模板19个PCR循环产物;泳道3:1μL模板19个PCR循环后0.75倍DNA cleanbeads纯化产物。
图7为本发明对实施例1与2混合文库的Agilent 2100Bioanalyzer核酸检测峰图。
图8为实施例1映射到注释基因的远端polyA位点的整体分析。A:文库测序后的reads在全基因视图的分布,横坐标0表示基因的转录延伸终止点(TES),纵坐标表示归一化后reads高峰值;B:测序reads的基序分析,横坐标0表示reads末端的位置,纵坐标为基序频率。
图9为实施例1检测独特和替代的多聚腺苷酸化位点映射到人类基因组的两个基因示例。
具体实施方式
下面结合具体实施例,进一步阐述本发明,应当理解,这些实施例仅用于说明本发明而不用于限制本发明要求保护的范围,下列实施例中未注明的具体实验条件和方法,通常按照常规条件所建议的方法。
除非特别说明,本发明采用的试剂、方法和设备为本技术领域常规试剂、方法和设备。除非特别说明,以下实施例所用试剂和材料均为市购。未注明具体条件的实验方法,通常按照常规条件,或制造厂商所建议条件实施。
下面结合实施例对本发明做详细的说明。
实施例1:
1、细胞RNA提取
利用Trizol法从人源HepG2细胞抽提总RNA,用聚丙烯酰胺尿素变性胶或者安捷伦2100Bioanalyzer检测RNA质量。电泳结果能清晰看出28S rRNA和18S rRNA条带,RNA没有降解,质量完好。
2、RNA片段化
使用总RNA 1μg在65℃条件下变性5min,迅速放置冰上,变性后将RNA用新鲜配制的NaOH水解片段化,反应体系中NaOH终浓度范围0.02M-0.03M,本实施例采用0.03M NaOH终浓度;体系反应时间30min,体系反应温度为27℃。片段化反应后,立即使用等量HCl中和NaOH(图3为实施例1、2的1μg及2μg总RNA片段化及去基因组的RNA片段在聚丙烯酰胺尿素变性胶的检测图),反应体系如下:
3、去除基因组DNA
采用RQ1 DNase(Promega,M6101)去除样品中的基因组DNA,在37℃条件,反应15min,体系如下:
去除基因组后的产物加水稀释至40-50μL,使用
4、逆转录生成第一链cDNA
RNA逆转录生成第一链cDNA使用的逆转录引物为特殊设计引物(含UMI),序列如下:5′-
RNA逆转录生成第一链cDNA是使用
/>
高效去除逆转录引物使用的是3μL Exonuclease(NEB,M0293S),缓冲液2.5μL,优选反应条件37℃,1h;高效去除rRNA、mRNA及其他RNA,采用的是终浓度0.04M NaOH(新鲜配制),反应条件98℃、20min,碱水解反应去除RNA,减少副产物生成,反应后加入等量HCl中和碱。
5、纯化第一链cDNA
纯化第一链cDNA使用
6、单链cDNA环化
单链cDNA模板环化使用ssDNA/RNA环化连接酶(ssDNA/RNA CircLigase)(Haigene,D2602)。单链cDNA在ssDNA/RNA CircLigase热稳定连接酶的作用下,将单链cDNA的5′-磷酸基团和3′-OH基团通过磷酸二酯键连接起来,在60℃条件下反应1h,80℃条件灭活10min,具体反应体系如下:
将环化产物进行线性化使用APE1酶,0.5μL的线性化缓冲液的成分包括240mMC4H6MgO4和25mM DTT。反应先使用1μL APE1,37℃孵育1h,再加0.5μL APE1,再37℃孵育30min,65℃条件孵育20min失活。
7、PCR扩增,建库
选择1μL作为模板,使用带有index的引物扩增,预PCR 18个循环,分析预结果。预PCR体系如下:
/>
反应条件如下:
建库PCR循环最终通过预PCR产物在聚丙烯酰胺凝胶电泳条带亮度(图5)确定建库PCR循环数为15个,反应体系如下:
反应条件如下:
磁珠纯化PCR产物是使用VAHTS DNA Clean Beads(Vazyme,N411),调整磁珠比例,选择0.75X(珠子体积=0.75×PCR产物体积)珠子纯化,操作按参考说明书进行,最后用水洗脱,上机测序(图6为实施例1建库PCR产物使用磁珠分选的聚丙烯酰胺胶检测图)。
图7为本发明对实施例1与2混合文库的的Agilent 2100Bioanalyzer核酸检测峰图。由图所示,混合文库的峰集中在346nt左右,质检结果符合要求,可进行测序。
为了系统统计mRNA 3′末端相对于转录延伸终止位点TES的位置,对实施例1所有reads进行分析,发现在TES上游200nt内出现最大峰值(图8A),与已知一致;通常mRNA 3′末端含有共识序列AWTAAA,为了验证本文库测序reads特征,对实施例1测序reads基序分析,结果显示位于read的0-30nt范围内,其包含的序列大部分含有这AWTAAA共识基序,与mRNA3′末端包含的基序特征有相似性(图8B)。
经过基因组比对track(图9)分析,可以看出测出的本实施例测出的mRNA末端3′UTR建库测序的poly(A)位点与现有的数据库对poly A的注释大多匹配,除此以外还有出现一些在3′UTR未注释的poly A位点。
实施例2
实施例2除使用总RNA量与实施例1不同,为2μg,其余操作条件与实施例1完全相同。
- 一种针对FFPE样本的mRNA二代测序文库构建的试剂盒及其应用
- 一种构建杂交捕获测序文库的方法及应用
- 一种测序文库构建的方法、建库试剂及其应用
- 一种基于动物粪便样品构建测序文库的方法及其应用
- 一种快速多时间生物素修饰核酸转录连缀测序文库的构建方法及其应用
- 一种转录组测序文库快速构建方法及其应用