蒽光二聚体衍生物、其制备、手性胺基蒽光二聚体衍生物、其制备及应用
文献发布时间:2024-04-18 19:48:15
技术领域
本发明涉及手性有机化合物技术领域。更具体地,涉及一种蒽光二聚体衍生物、其制备、手性胺基蒽光二聚体衍生物、其制备及应用。
背景技术
发展新型骨架结构的手性配体或催化剂对过渡金属催化的有机合成方法学的应用具有重要研究意义。因此使得设计与合成新骨架的配体的研究,成为近几十年来合成化学领域的前沿热点,并不断在催化领域的重要创新研究取得新进展。
蒽光二聚体独特的几何结构和X型骨架常常的被应用于主客体识别、分子开关和能量存储材料等领域,并且以蒽光二聚体为核心骨架的手性单膦配体在钯催化的不对称反应中也取得系列重要进展。但其在有机合成中的应用目前仍十分有限,其主要的研究困难在于蒽光二聚体的立体异构体的拆分和分离提纯,以及较差的溶解度等问题。目前获得其骨架的手性配体均为制备色谱拆分,使其合成成本过高,在一定程度上限制了应用。
发明内容
基于以上事实,本发明的目的在于提供一种蒽光二聚体衍生物、其制备、手性胺基蒽光二聚体衍生物、其制备及应用。该蒽光二聚体衍生物具有高效、快速且容易拆分制备得到手性胺基蒽光二聚体衍生物等特点,同时,该拆分的成本低,方法简单。
一方面,本发明提供一种蒽光二聚体衍生物,其具有如下互为非对映异构体的式I-1、或I-2所示的结构:
其中,
所述R
M选自
R
R
n选自1或2。
进一步地,上述技术方案中,包含式I-1、或I-2所示的结构的对映异构体、消旋体或非对映异构体。
又一方面,本发明提供一种蒽光二聚体衍生物的制备方法,该方法包括如下步骤:
将式II所示化合物
得到式IV-1所示化合物
将式IV-1所示化合物或式IV-2所示化合物与式V所示化合物与蒽发生[4+4]光反应,得到具有互为非对映异构体的式I-1、或I-2所示的结构的所述蒽光二聚体衍生物;
其中,所述R
上述制备方法中,制备得到的产物为互为非对映异构体的式I-1、或I-2所示的结构的所述蒽光二聚体衍生物。也即,产物中同时含有互为非对应异构体的两种产物,式I-1和式I-2所示结构的物质的混合。
上述制备方法中,通过对式III-1所示化合物
如需将两种物质进一步分离,可通过柱层析或重结晶的方式进行分离,在此不赘述。
又一方面,本发明提供一种手性胺基蒽光二聚体衍生物,其由如上所述的蒽光二聚体衍生物水解得到,该手性胺基蒽光体二聚衍生物具有如下式VI所示的结构:
其中,
Q
本发明中提供的手性胺基蒽光二聚体衍生物具有X型立体结构。
又一方面,本发明提供如上所述的手性胺基蒽光二聚体衍生物的制备方法,该方法包括如下步骤:
将所述蒽光二聚体衍生物进行水解,得到所述手性胺基蒽光二聚体衍生物。
进一步地,该方法具体包括如下步骤:
在强碱条件下,于溶剂中回流反应,得到所述手性胺基蒽光二聚体衍生物。
进一步地,所述强碱选自叔丁醇钠;所述溶剂选自正丁醇和二甲基亚砜。
又一方面,本发明提供如上所述的手性胺基蒽光二聚体衍生物在制备手性试剂或手性药物中间体中的应用。
进一步地,将所述手性胺基蒽光二聚体衍生物用于制备其它蒽光二聚衍生的平面手性配体或催化剂。
本发明中,蒽光二聚体衍生物的制备方法中,方法采用化学拆分法生成非对映异构体的方式,即蒽光二聚体衍生物与手性试剂作用,化学转变为一对非对映异构体,即制备得到蒽光二聚体衍生物;再利用其极性不同或溶解度差异将其分离,最后通过去保护的手段(水解)以获取对映体纯的手性胺基蒽光二聚体衍生物。
除非另有说明,本申请中基团的上标为基团标记,下标一般指该基团的个数。
“任选……取代”为被任意取代基取代或未取代。
本发明单独使用或用作后缀或前缀的“C
本发明使用的术语“C
本发明使用的术语“3-20元杂环基”指包含3至20个原子的饱和、不饱和或部分饱和的单环、二环或三环(除非另有说明),其中1、2、3、4或5个环原子选自氮、硫或氧,除非另有说明,其可通过碳或氮来连接,其中-CH
术语“C
术语“5-20元杂芳基”应理解为包括这样的一价单环、双环或三环芳族环系:其具有5~20个环原子且包含1-5个独立选自N、O和S的杂原子,例如“5-14元杂芳基”。术语“5-14元杂芳基”应理解为包括这样的一价单环、双环或三环芳族环系:其具有5、6、7、8、9、10、11、12、13或14个环原子,特别是5或6或9或10个碳原子,且其包含1-5个,优选1-3个独立选自N、O和S的杂原子并且,另外在每一种情况下可为苯并稠合的。特别地,杂芳基选自噻吩基、呋喃基、吡咯基、噁唑基、噻唑基、咪唑基、吡唑基、异噁唑基、异噻唑基、噁二唑基、三唑基、噻二唑基、噻-4H-吡唑基等以及它们的苯并衍生物,例如苯并呋喃基、苯并噻吩基、苯并噁唑基、苯并异噁唑基、苯并咪唑基、苯并三唑基、吲唑基、吲哚基、异吲哚基等;或吡啶基、哒嗪基、嘧啶基、吡嗪基、三嗪基等,以及它们的苯并衍生物,例如喹啉基、喹唑啉基、异喹啉基等;或吖辛因基、吲嗪基、嘌呤基等以及它们的苯并衍生物;或噌啉基、酞嗪基、喹唑啉基、喹喔啉基、萘啶基、蝶啶基、咔唑基、吖啶基、吩嗪基、吩噻嗪基、吩噁嗪基等。
术语“C1-20酰基”应理解为具有酰基结构的另一端连接有C1-19的烷基、环烷基、芳基、杂芳基的单元。优选地,比如苯二甲酰基、乙酰基等。
术语“C1-20磺酰基”应理解为具有磺酰基结构的另一端连接有C1-20的烷基、环烷基、芳基、杂芳基的单元。优选地,比如对甲苯磺酰基等。
术语“C1-C20苄基”应理解为具有苄基各种取代芳基结构的苄基。优选地,比如苄基、对甲氧基苄基等。在本发明中,芳基包含取代和未取代的芳基,其中取代是指基团上的一个或多个氢原子被选自下组的取代基取代:C1-C4烷基、C3-C10环烷基、卤素、羟基、羧基、醛基、酰基、胺基。代表性的芳基包括带有给电子和/或吸电子取代基芳基,如对甲苯基、对甲氧基苯基、五氟苯基等。
术语“C1-20烷氧基羰基”应理解为具有烷氧基羰基结构的另一端连接有C1-19的烷基、环烷基、芳基、杂芳基的单元。优选地,比如叔丁氧羰基、苄氧羰基、笏甲氧羰基等。
除非另有说明,杂环基、杂芳基或亚杂芳基包括其所有可能的异构形式,例如其位置异构体。因此,对于一些说明性的非限制性实例,吡啶基或亚吡啶基包括吡啶-2-基、亚吡啶-2-基、吡啶-3-基、亚吡啶-3-基、吡啶-4-基和亚吡啶-4-基;噻吩基或亚噻吩基包括噻吩-2-基、亚噻吩-2-基、噻吩-3-基和亚噻吩-3-基。
本发明中,当所述结构具有手性时,除非明确说明是光学纯外,所涉及的化合物以及所画结构均指外消旋物,或者手性异构体的混合物。
本发明的有益效果如下:
本发明提供的蒽光二聚体衍生物因其独特的结构使得其具有更容易拆分成手性胺基蒽光二聚体衍生物(现有技术中获得手性胺基蒽光二聚体的方法需要使用制备型手性液相色谱进行分离,成本高)的特点。本发明的制备方法中,以廉价易得商业可用的起始原料和手性拆分试剂,快速地开发了一条制备蒽光二聚体衍生物以及采用该手性蒽光二聚体衍生物制备得到手性胺基蒽光二聚体衍生物的新路径。利用此平面手性的胺基蒽光二聚体衍生物,并结合其独特的立体骨架,可设计与合成出新型平面手性配体或催化剂。这将有助于开发更加友好、更加原子经济性的反应新体系,对社会发展具有重大的研究意义。
附图说明
下面结合附图对本发明的具体实施方式作进一步详细的说明。
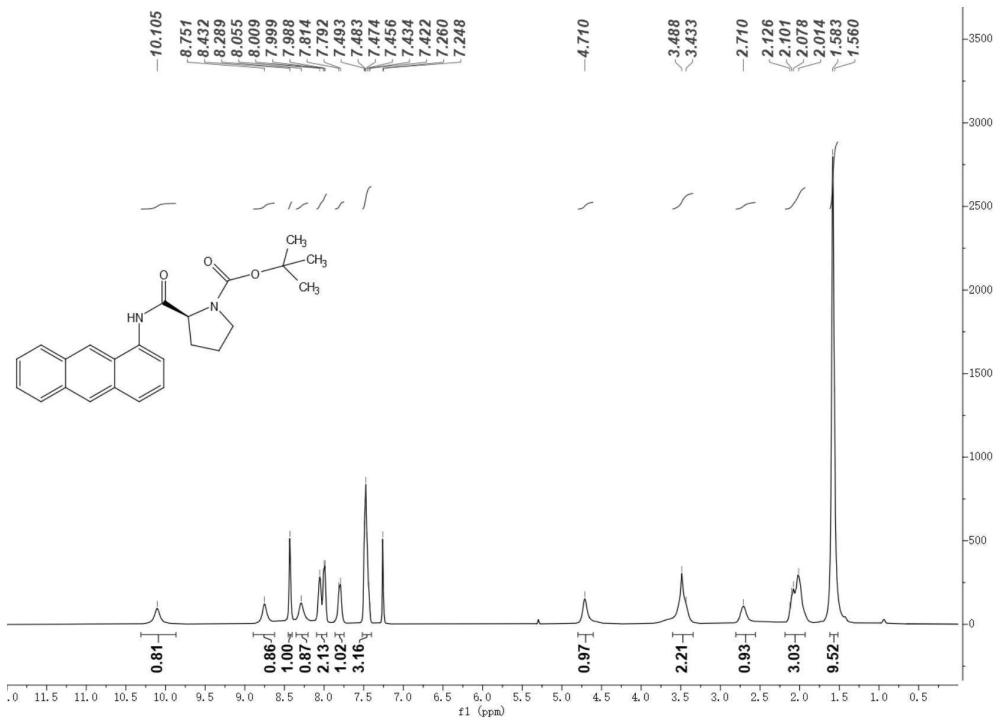
图1示出实施例1制备得到的黄色固体粉末3的核磁谱图。
图2示出实施例1制备得到的化合物4a的核磁谱图。
图3示出实施例1制备得到的化合物4b的核磁谱图。
图4示出实施例2中制备得到的化合物(S
图5示出实施例2制备得到的化合物(S
图6示出实施例2中制备得到的化合物(R
图7示出实施例3制备得到的白色固体7的核磁谱图。
图8示出实施例3制备得到的化合物8a和8b混合物的核磁谱图。
图9示出实施例4制备得到的化合物5的核磁谱图。
图10示出实施例5制备得到的化合物9的核磁谱图。
图11示出实施例6制备得到的化合物10的核磁谱图。
具体实施方式
为了更清楚地说明本发明,下面结合优选实施例和附图对本发明做进一步的说明。附图中相似的部件以相同的附图标记进行表示。本领域技术人员应当理解,下面所具体描述的内容是说明性的而非限制性的,不应以此限制本发明的保护范围。
以下,对各实施例中需要用到的原料进行如下说明:
实施例1:
茄形瓶中称取Boc-L-脯氨酸2(6.70g,31.0mmol,1.5equiv),加入二氯甲烷(62mL)进行溶解。在0℃下依次加入氯甲酸叔丁酯(4.20g,31.0mmol,1.5equiv)和三乙胺(3.10g,31.0mmol,1.5equiv),在0℃下搅拌20分钟。在0℃下加入1-氨基蒽1(4.00g,20.7mmol,1.0equiv)到该反应混合物中,搅拌1-2分钟后将反应恢复室温,搅拌10小时。将反应液转移到分液漏斗,依次加入饱和碳酸氢钠溶液、饱和氯化铵溶液洗涤,水相用二氯甲烷萃取3次。合并有机相,用无水硫酸钠干燥,减压除去溶剂,得到粗产物。用石油醚对该粗产物进行重结晶,得到黄白色固体粉末3(7.82g,97%产率)。
在充满氮气的手套箱中,将蒽(2.30g,12.8mmol,10.0equiv),化合物3(500mg,1.28mmol,1.0equiv)和脱气的甲苯(102mL)加入到厚壁耐压瓶中。将瓶盖拧紧,移出手套箱,在氙灯光源照射下室温搅拌24小时,混合物用硅藻土过滤,随后通过柱层析和重结晶分离得白色固体化合物4a(309mg,42%收率,98%纯度)和白色固体化合物4b(202mg,28%收率,96%纯度)。固体化合物4a和固体化合物4b容易进一步分离。
化合物4a:
化合物4b:
采用类似如上所述的方法用一些环状手性氨基酸生成制备如下表1所示的一些蒽光二聚体衍生物非对映异构体,其质量比也如下表1所示。
表1
实施例2
茄形瓶中加入4a(250mg,0.44mmol,1.0equiv),叔丁醇钠(861.6mg,8.79mmol,20.0equiv),正丁醇(40mL)和二甲基亚砜(2.35mL),在回流的条件下剧烈搅拌反应24h。在减压移除溶剂后,用少量二氯甲烷溶解瓶中残留固体将其倒入干净的分液漏斗,随后加入2M盐酸萃取,水相用二氯甲烷萃取三次,合并有机相,无水硫酸钠干燥,浓缩。残余固体在石油醚的条件下进行重结晶得黄色粉末(146mg,89%产率,99%纯度)。
使用Daicel AS柱的HPLC分析确定对映体(分析条件:正己烷/i-PrOH=95:5)。结果如图4所示。
根据上述制备(S
使用Daicel AS柱的HPLC分析确定对映体(分析条件:正己烷/i-PrOH=95:5),结果如图6所示。
实施例3
茄形瓶中加入6(636mg,2.4mmol,1.2equiv),1-氨基蒽1(386mg,2.0mmol,1.0equiv),1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(460mg,2.2mmol,1.1equiv),1-羟基苯并三唑(460mg,2.2mmol,1.1equiv),在室温下搅拌24小时。将反应液转移到分液漏斗,加入少量水萃取,水相用二氯甲烷萃取3次。合并有机相,用无水硫酸钠干燥,减压除去溶剂,得到粗产物。粗产物用柱层析方法进一步纯化得到白色固体7(312mg,35%产率)。
在充满氮气的手套箱中,将蒽(1.78g,10.0mmol,10.0equiv),化合物7(440mg,1.0mmol,1.0equiv)和脱气的甲苯(68mL)加入到厚壁耐压瓶中。将瓶盖拧紧,移出手套箱,在氙灯光源照射下室温搅拌24小时,混合物用硅藻土过滤,随后通过柱层析分离得白色固体8a和8b的混合物(327mg,53%总产率)。8a和8b较难继续分离。
采用类似如上所述的方法用一些开链手性氨基酸生成制备如下表2所示的一些蒽光二聚体衍生物非对映异构体,其质量比也如下表2所示。
表2
实施例4
茄形瓶中加入8(61.8mg,0.10mmol,1.0equiv),浓盐酸(1mL)和乙醇(2mL),在80℃加热的条件下剧烈搅拌反应12h。待反应结束,NaOH中和该反应液,水相用二氯甲烷萃取三次,合并有机相,在减压移除溶剂后,通过柱层析分离得目标产物(27mg,36%产率)。
一些应用实施例:
实施例5
反应瓶中加入化合物5(100mg,0.27mmol,1.0equiv),亚硝酸钠(93mg,1.35mmol,5.0equiv),一溴三氯甲烷(107mg,0.54mmol,2.0equiv),二氯甲烷(5.0mL)和水(5.0ml),在室温下搅拌5分钟。随后加入乙酸(323mg,5.38mmol,20.0equiv),将该反应在室温下搅拌1小时。将反应液倒入分液漏斗,水相用二氯甲烷萃取3次,合并有机相,用饱和食盐水洗,无水硫酸钠干燥,减压下移除溶剂。粗产物用柱层析方法进一步纯化得到白色固体9(77.4mg,66%产率)。
从依据本方法拆分后的化合物5出发,可以得到光学纯的化合物9,化合物9的核磁谱图如图10所示。进而可根据参考文献(Org.Lett.2019,21,8158)制备光学纯的单膦配体diAnthPhos。
实施例6
在空气中,化合物5(104mg,0.28mmol),浓硫酸(2.0mL)和醋酸(4.0mL)分别加入到40mL反应瓶中,于0℃搅拌5分钟。随后在0℃下10分钟内加入亚硝酸钠的水溶液(38mg,0.58mmol,2.0equiv,2.0mL H
从依据本方法拆分后的化合物5出发,可以得到光学纯的化合物10,化合物10的核磁谱图如图11所示。进而可根据参考文献(Org.Lett.2021,23,5485)制备光学纯单膦配体DapPhos。
显然,本发明的上述实施例仅仅是为清楚地说明本发明所作的举例,而并非是对本发明的实施方式的限定,对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动,这里无法对所有的实施方式予以穷举,凡是属于本发明的技术方案所引伸出的显而易见的变化或变动仍处于本发明的保护范围之列。
- 共轭炔基蒽类衍生物、制备方法及其应用
- 蒽光二聚体衍生物、其制备、手性胺基蒽光二聚体衍生物、其制备及应用
- 一类9,10-二苯基蒽-蒽二聚体及其制备方法