一种苯并五元氮环类化合物、其制备方法及医药用途
文献发布时间:2024-04-18 19:58:26
本申请是以CN申请号为202211040448.7,申请日为2022年8月29日的申请为基础,并主张其优先权,该CN申请的公开内容在此作为整体引入本申请中。
技术领域
本申请涉及药物化学领域,具体涉及一种苯并五元氮环类化合物、其制备方法及医药用途。
背景技术
肾脏是人体的重要器官,除了具有泌尿系统的基本功能外,还能够分泌促红细胞生成素(erythropoietin,EPO)促进红细胞生成,是EPO生成的主要场所。慢性肾病(Chronickidney disease,CKD)在全球范围内的发病率超过10%,已成为全球性公共健康问题。肾性贫血是CKD的主要并发症之一,主要由于肾损伤后EPO产出不足所致。目前,肾性贫血的主要治疗方式为注射重组人EPO(recombinant human EPO,rhEPO)及其相关产品,即促红细胞生成药物(erythropoiesis-stimulating agents,ESAs)。ESAs能够增加肾性贫血病人的血红蛋白水平,降低病人输血需求,极大的改善病人的生活质量。然而,临床研究表明对CKD病人注射rhEPO能够导致较高的心血管疾病风险以及死亡率,且rhEPO价格较高,其应用具有局限性。因此,寻找口服易吸收、价格低廉且副作用低的ESAs具有重要意义。
治疗肾性贫血的另一种策略是通过脯氨酸羟化酶(prolyl hydroxylase domainproteins,PHD)的小分子抑制剂抑制缺氧诱导因子(hypoxia-inducible factor,HIF)2α的泛素化途径从而促进EPO的表达。研究发现,CKD病人中HIF的活性可能受到氧化应激及尿毒症等因素的抑制,从而导致肾性贫血。临床数据显示,Roxadustat、Vadadustat及Daprodustat等PHD抑制剂均能够有效增加病人血液中血红蛋白的水平。目前,Roxadustat已作为I类新药获批上市。由于PHD抑制剂大多为α-酮戊二酸(2-oxoglutarate,2-OG)的结构类似物,很可能影响体内其它依赖于2-OG的酶的活性,因此这类化合物其临床药理机制还需要进一步研究。
已报道的HIF-2α激动剂(Wu DL et al.Nat.Chem.Biol.2019,15,367),可直接作用于HIF-2α靶点,激活HIF-2通路,正向调节下游基因EPO的蛋白表达水平。但已知激动剂仅在较高的化合物浓度下才能激活HIF-2通路。因此,开发具有更好活性、更理想的HIF-2α激动剂具有重要临床价值和意义。
发明内容
本申请提供式I所示苯并五元氮环类化合物、或其药学上可接受的盐、异构体、同位素标记的化合物、前药、多晶型物或溶剂化物及其制备方法。所述化合物能够激活HIF-2α信号通路,对HIF-2α下游基因(例如,EPO、VGEF、Glut1、NDRG1)的转录或表达具有显著调控作用。
具体地,在第一个方面,本申请提供一种如式I所示的苯并五元氮环类化合物、或其药学上可接受的盐、异构体、同位素标记的化合物、前药、多晶型物或溶剂化物:
其中,
X、Y和Z各自独立地选自C、N、O和S(O)
各R
R和R’各自独立地选自氢、卤素、C
R
R
n=1、2、3、4或5。
在一些实施方案中,式I中X为N。
在一些实施方案中,式I中X为N;R
在一些实施方案中,式I中-X(R
在一些实施方案中,各R
在一些实施方案中,各R
在一些实施方案中,各R
在一些实施方案中,各R
在一些实施方案中,各R
在一些实施方案中,R
在一些实施方案中,R
在一些实施方案中,R
在一些实施方案中,R
在一些实施方案中,R
在一些实施方案中,R
在一些实施方案中,R
在一些实施方案中,所述化合物具有式II所示结构:
其中,各基团如前文任一项中所定义。
在一些实施方案中,X选自N、S和O;
各R
R
R
n=1、2、3、4或5。
在一些实施方案中,式II化合物中X为N。
在一些实施方案中,式II化合物中各R
在一些实施方案中,式II化合物中各R
在一些实施方案中,式II化合物中各R
在一些实施方案中,式II化合物中各R
在一些实施方案中,式II化合物中R
在一些实施方案中,式II化合物中R
在一些实施方案中,式II化合物中R
在一些实施方案中,式II化合物中R
在一些实施方案中,式II化合物中R
在一些实施方案中,式II化合物中R
在一些实施方案中,式II化合物中R
在一些实施方案中,式II化合物中R
在一些实施方案中,所述化合物具有式III所示结构:
各基团如前文任一项中所定义。
在一些实施方案中,R
在一些实施方案中,各R
R和R’各自独立地选自氢、卤素、C
n=1、2或3。
在一些实施方案中,各R
在一些实施方案中,各R
在一些实施方案中,各R
在一些实施方案中,式III中X为N;R
在一些实施方案中,式III中-X(R
在一些实施方案中,X选自N、S和O;
各R
R
R
n=1、2、3、4或5。
在一些实施方案中,所述化合物选自:
/>
/>
在第二个方面,本申请提供制备前文任一项所述苯并氮杂环类化合物、或其药学上可接受的盐、异构体、同位素标记的化合物、前药、多晶型物或溶剂化物的方法。
当Y=O、X=N、Z=C时,所述方法包括以下步骤:
步骤1:将化合物1a与盐酸羟胺反应得到化合物1b;
步骤2:将化合物1b卤化得到化合物1c;
步骤3:将化合物1c与化合物B反应得到化合物1d;
步骤4:将化合物1d环化得到所述式II’化合物;
其中,R
在一些实施方案中,本申请提供制备式I’所示的苯并异噁唑类化合物(即X-R
步骤1:将化合物1a与盐酸羟胺反应得到化合物1b;
步骤2:将化合物1b卤化得到化合物1c;
步骤3:将化合物1c与化合物B反应得到化合物1d;
步骤4:将化合物1d环化得到所述式I’化合物;
其中,R
在一些具体的实施方案中,步骤1包括以下操作:
将化合物1a(5mmol)溶于17mL的乙醇溶液中,随后向反应体系中加入盐酸羟胺的水溶液(5mL),反应液在室温的条件下反应过夜,反应完毕后减压浓缩部分溶剂,加入50mL的水稀释反应液并用乙酸乙酯萃取,有机层依次用清水洗涤,随后用饱和NaCl溶液洗涤、无水硫酸钠干燥、在真空下减压浓缩,得到无色油状液体。
在一些具体的实施方案中,步骤2包括以下操作:
将化合物1b(5mmol)溶于无水DMF(12mL)中,随后向反应体系中加入NCS(10mmol)(分两次加入,间隔10min),在室温下反应,约0.5h反应完毕,反应结束后向反应液中加入80mL水稀释反应液,并用乙酸乙酯萃取,有机层用大量清水洗涤、饱和氯化钠溶液洗涤,并用无水硫酸钠干燥,在真空下减压浓缩得白色固体。
在一些具体的实施方案中,步骤3包括以下操作:
第三步:将化合物1c(4.6mmol)溶于无水THF(14mL)中,随后加入取代苯胺(6.9mmol),使反应液回流并搅拌,约48h后TLC监测无原料剩余,反应结束后,向反应液中加入60mL水稀释反应液,并用乙酸乙酯萃取,有机层用饱和氯化钠溶液洗涤,无水硫酸钠干燥,减压浓缩后采用硅胶柱层析分离,得固体化合物1d。
在一些具体的实施方案中,步骤4包括以下操作:
第四步:将化合物1d(0.71mmol)溶于NMP(7mL)中,加入叔丁醇钾,在100℃下搅拌,约2.5h反应完毕,反应结束后向反应液中加入40mL水稀释反应液,并用乙酸乙酯萃取,有机层用饱和氯化钠溶液洗涤,无水硫酸钠干燥,真空下减压浓缩后,采用硅胶柱层析分离,得固体化合物,即苯并异噁唑类化合物。
当R
步骤1:将化合物2a与硝酸钠、氯化铜、二氧化硫反应得到化合物2b;
步骤2:将化合物2b氨化得到化合物2c;
步骤3:将化合物2c氯化得到化合物2d;
步骤4:将化合物2d与化合物B’反应所述式III'化合物;
其中,R
在一些具体的实施方案中,步骤1包括以下操作:
向2-氨基-6-氟苯甲酸酯的溶液(1.8克,10.6毫摩尔)中加入醋酸(6毫升)和盐酸(20毫升),在0℃下加入硝酸钠(1.46克,21.2毫摩尔)的水溶液(5.0毫升),混合物在0℃下搅拌1小时后,在0℃加入二氯化铜(1.86克,13.7毫摩尔),再加入二氧化硫的醋酸溶液中搅拌(31.8毫升,63.6毫摩尔,2.0摩尔/升),将混合物逐渐温升至25℃,并在此温度下搅拌12小时。将混合物倒入冰水中(500毫升),用乙醚萃取两次,每次250毫升,有机层被浓缩后,残留物通过硅胶色谱法进行纯化,用乙醚/石油醚洗脱液(从0到10%的乙醚在20分钟内),得到黄色固体2-(氯磺酰)-6-氟苯甲酸盐(1.5克,52%的产率)。
在一些具体的实施方案中,步骤2包括以下操作:
将2-(氯磺酰)-6-氟苯甲酸酯(1.5克,5.9毫摩尔),氨水(2.07克,59毫摩尔)和四氢呋喃(40.0毫升)的混合物加热到40℃,并在该温度下搅拌12小时,混合物被浓缩并用1N盐酸(20毫升)洗涤之后进行过滤,得到白色固体4-氟苯并[d]异噻唑-3(2H)-酮1,1-二氧化物(980毫克,83%产率)。
在一些具体的实施方案中,步骤3包括以下操作:
向4-氟苯并[d]异噻唑-3(2H)-酮1,1-二氧化物的溶液(980毫克,4.87毫摩尔)和N,N-二甲基甲酰胺(73.0毫克,1.0毫摩尔)中加入二氧六环(20.0毫升),然后在25℃下滴加氯化亚砜(869毫克,7.3毫摩尔),将混合物加热至100℃,并在此温度下搅拌12小时,反应混合物在减压下浓缩,残渣通过重新结晶法进行纯化,使用石油醚/二氯甲烷(PE/DCM=5∶1,40毫升),得到黄色固体3-氯-4-氟苯并[d]异噻唑1,1-二氧化物(720毫克,64%收率)。
在一些具体的实施方案中,步骤4包括以下操作:
先将3-氯-4-氟苯并[d]异噻唑1,1-二氧化物(1,5克,6,83毫摩尔)溶解在20mL的二氯甲烷中并且将溶液降温到0摄氏度,然后将2-(吡咯烷-1-基)苯胺(1.11克,6.83毫摩尔)和乙基二异丙胺(1.32克,10.25毫摩尔)分别缓慢加入到上述溶液中并升温到室温,反应在室温下搅拌2小时,液质监控原料完全转化,将反应液中加入10mL蒸馏水,分液后再用蒸馏水洗涤有机相(5mL*2),收集有机相并用无水硫酸钠干燥,减压蒸除溶剂得到粗产物,粗产物经过硅胶柱色谱分离提纯得到4-氟-3-((2-(吡咯烷-1-基)苯基)氨基)苯并[d]异噻唑1,1-二氧化物(1,32g,53%产率)。
在第三个方面,本申请提供一种药物组合物,其含有第一方面任一项所述的苯并五元氮环类化合物或其药学上可接受的盐、异构体、同位素标记的化合物、前药、多晶型物或溶剂化物,以及任选的载体和/或赋型剂。
所述药物组合物中,所述化合物可以是单一有效成分,也可以与其他活性组分进行组合,构成联合制剂。所述其他活性组分可以是其他各种可以用于慢性肾病、肾贫血、血脂异常和高胆固醇症状的药物。组合物中活性组分的含量通常为安全有效量,所述安全有效量对于本领域技术人员来说应该是可以调整的。在一些实施方案中,所述药物组合物中进一步含有脯氨酰羟化酶抑制剂。在一些实施方案中,所述脯氨酰羟化酶抑制剂选自罗沙司他、伐度司他、达度司他、恩那司他和莫立司他。
在第四个方面,本申请提供一种疫苗佐剂,其含有第一方面任一项所述的苯并五元氮环类化合物或其药学上可接受的盐、异构体、同位素标记的化合物、前药、多晶型物或溶剂化物、或第三方面任一项所述的药物组合物。
在第五个方面,本申请提供一种免疫原性或免疫刺激组合物,其含有第四方面任一项所述的疫苗佐剂。
本申请的化合物或其药学上可接受的盐,及其药物组合物对缺氧诱导因子HIF-2下游的EPO、VGEF、Glut1、NDRG1等mRNA和蛋白的表达具有显著的增强活性。因此,本申请进一步提供所述化合物在医药中的应用。
在另一个方面,本申请提供第一方面任一项所述的苯并五元氮环类化合物或其药学上可接受的盐、异构体、同位素标记的化合物、前药、多晶型物或溶剂化物、或第三方面任一项所述的药物组合物在制备药物中的用途,所述药物用作缺氧诱导因子HIF-2激动剂、免疫调节剂、或用于治疗和/或预防下述疾病和/或症状:
(1)HIF-2信号通路异常相关疾病;优选的,所述HIF-2信号通路异常相关疾病选自慢性肾病、血脂异常、高胆固醇、或与EPO或EPO受体活性低下相关、或以EPO缺乏或红细胞缺少或缺陷为特征的疾病和/或病症;优选的,所述与EPO或EPO受体活性低下相关、或以EPO缺乏或红细胞缺少或缺陷为特征的疾病和/或病症为缺血性疾病,例如,肾性贫血、慢性肾贫血、红细胞生成减少性贫血、缺血引起的脑卒中或心肌缺血;
(2)肾衰竭、高血压、冠心病、或衰老。
在另一个方面,本申请提供一种激活受试者细胞HIF-2信号通路的方法,包括将所述细胞与第一方面任一项所述的苯并五元氮环类化合物或其药学上可接受的盐、异构体、同位素标记的化合物、前药、多晶型物或溶剂化物、或第三方面任一项所述的药物组合物接触的步骤。
在另一个方面,本申请提供一种免疫调节方法,其包括向有此需要的受试者施用有效量的第一方面任一项所述的苯并五元氮环类化合物或其药学上可接受的盐、异构体、同位素标记的化合物、前药、多晶型物或溶剂化物、或第三方面任一项所述的药物组合物。
在另一个方面,本申请提供治疗和/或预防疾病和/或症状的方法,其包括向有此需要的受试者施用有效量的第一方面任一项所述的苯并五元氮环类化合物或其药学上可接受的盐、异构体、同位素标记的化合物、前药、多晶型物或溶剂化物、或第三方面任一项所述的药物组合物,所述疾病和/或症状如前文所述。
在第另一个方面,本申请提供第一方面任一项所述的苯并五元氮环类化合物或其药学上可接受的盐、异构体、同位素标记的化合物、前药、多晶型物或溶剂化物、或第三方面任一项所述的药物组合物作为疫苗佐剂或者在制备疫苗佐剂中的用途。
在一些实施方案中,所述化合物或其药学上可接受的盐、异构体、同位素标记的化合物、前药、多晶型物或溶剂化物刺激(例如,引发或增强)受试者的免疫应答。
在一些实施方案中,所述免疫应答为非特异性免疫应答。
在一些实施方案中,所述免疫应答是抗原特异性免疫应答。
在一些实施方案中,所述免疫应答包括B细胞的活化、T细胞的活化、抗体的产生和/或细胞因子的释放。
在另一个方面,本申请提供第五方面任一项所述的免疫原性或免疫刺激组合物用作疫苗或在制备疫苗中的用途。
术语定义
在本发明中,除非另有说明,否则本文中使用的科学和技术名词具有本领域技术人员所通常理解的含义。并且,本文中所用的细胞培养、生物化学、核酸化学、免疫学等实验操作除非特殊说明为相应领域内常规使用的操作。同时,为了更好地理解本发明,下面提供相关术语的定义和解释。
如本文中所使用的,术语“盐”应当被理解为由本发明使用的任何形式的活性化合物,其中所述化合物可以为离子形式或带电荷或被偶联到反离子(阳离子或阴离子)或在溶液中。术语“药学上可接受的盐”通常指当以适当的方式用于治疗时(特别是在人类和/或哺乳动物中应用或使用时)在生理学上可耐受的任何盐。本发明的化合物的药学上可接受的盐包括其酸加成盐及碱加成盐。适合的酸加成盐由形成药学可接受盐的酸来形成。适合的碱加成盐由形成药学可接受盐的碱来形成。实例包括盐酸盐、三氟醋酸盐及其它类似的盐。适合的盐的综述参见Stahl及Wermuth的“Handbook of Pharmaceutical Salts:Properties,Selection,and Use”(Wiley-VCH,2002)。用于制备本发明的化合物的药学上可接受的盐的方法为本领域技术人员已知的。
如本文中所使用的,术语“异构体”是指分子式相同、结构不同的化合物,包括构造异构体、立体异构体和电子互变异构体。应当理解,本发明的范围涵盖任意比例(例如60%、65%、70%、75%、80%、85%、90%、95%、96%、97%、98%、99%)的所述异构体或其混合物。
本发明还包括所述化合物药学上可接受的同位素标记的化合物,其与本发明的化合物相同,除了一个或多个原子被具有相同原子序数但原子质量或质量数不同于在自然界中占优势的原子质量或质量数的原子替代。适合包含入本发明的化合物中的同位素的实例包括(但不限于)氢的同位素(例如氘(
本发明在其范围内进一步包括所述化合物的前药,其为自身可具有较小药理学活性或无药理学活性的本发明所述化合物的某些衍生物当被给药至身体中或其上时可通过例如水解裂解转化成具有期望活性的本发明的化合物。术语“前药”以其最广泛的意义使用,并且包括在体内可以转化为本发明的化合物的那些衍生物。通常这样的前药会是所述化合物的官能团衍生物,其易于在体内转化成期望的治疗活性化合物。关于前药的使用的其他信息可参见“Pro-drugs as Novel Delivery Systems”,第14卷,ACS SymposiumSeries(T.Higuchi及V.Stella)。本发明的前药可例如通过用本领域技术人员已知作为“前-部分(pro-moiety)(例如“Design of Prodrugs”,H.Bundgaard(Elsevier,1985)中所述)”的某些部分替代本发明的化合物中存在的适当官能团来制备。
本发明涵盖所述化合物的所有可能的结晶形式或多晶型物,其可为单一多晶型物或多于一种多晶型物的任意比例的混合物。
如本文中所使用的,术语“溶剂化物”通常指任何形式的根据本发明的活性化合物通过非共价键与另一分子(通常为极性溶剂)相结合,所获得的物质,具体可以是包括但不限于水化物和醇化物,例如甲醇化物。
如本文中所使用的,术语“烷基”通常指饱和脂肪族烃基,它们可以是直链或支链。例如,C
如本文中所使用的,术语“卤代烷基”通常指前文所述的烷基被一个或多个(例如,1个、2个、3个、4个、5个或6个等)卤素(例如,氟、氯、溴或碘)取代后得到的基团。例如,卤代C
如本文中所使用的,术语“烷氧基”是指具有烷基-O-结构的基团,其中烷基如前文定义。例如,C
如本文中所使用的,术语“卤代烷氧基”通常指前文所述的烷氧基被一个或多个(例如,1个、2个、3个、4个、5个或6个等)卤素(例如,氟、氯、溴或碘)取代后得到的基团。例如,卤代C
如本文中所使用,术语“环烷基”指饱和的单环或多环(诸如双环)烃基。例如,“C
如本文中所使用,术语“杂环基”是指饱和或不饱和的环状结构,其环原子由碳原子以及至少一个(例如1、2或3个)选自氮、氧和硫的杂原子构成。术语“3-8元杂环基”是指具有3至8个环原子的杂环基,包括3元杂环基、4元杂环基、5元杂环基、6元杂环基、7元杂环基或8元杂环基,包括氮杂环基、氧杂环基等。具体实例包括但不限于吡咯烷基、哌啶基、吗啉基、丁内酰胺基、丁二酰亚胺基、4H-[1,2,4]三氮唑基或1,3-氧氮杂己环-2-酮等。
如本文中所使用,术语“芳基”指单环或稠合多环芳烃失去一个氢原子形成的基团。例如,“6-10元芳基”。具体实例包括但不限于苯基或萘基。
如本文中所使用,术语“取代”指所指定的原子上的一个或多个(例如一个、两个、三个或四个)氢被从所指出的基团的选择代替,条件是未超过所指定的原子在当前情况下的正常原子价并且所述取代形成稳定的化合物。取代基和/或变量的组合仅仅当这种组合形成稳定的化合物时才是允许的。如果取代基被描述为“任选地被……取代”,则取代基可(1)未被取代或(2)被取代。如果取代基的碳被描述为任选地被取代基列表中的一个或多个取代,则碳上的一个或多个氢(至存在的任何氢的程度)可单独和/或一起被独立地选择的任选的取代基替代。如果取代基的氮被描述为任选地被取代基列表中的一个或多个取代,则氮上的一个或多个氢(至存在的任何氢的程度)可各自被独立地选择的任选的取代基替代。如果取代基被描述为“独立地选自”一组基团,则各取代基独立于另一者被选择。因此,各取代基可与另一(其他)取代基相同或不同。
如本文中所使用,术语“一个或多个”意指在合理条件下的1个或超过1个,例如2个、3个、4个、5个或10个。
如本文中所使用的,术语“药学上可接受的载体或赋形剂”是指,在药理学和/或生理学上与受试者和活性成分相容的载体和/或赋形剂,其是本领域公知的(参见例如Remington′s Pharmaceutical Sciences.Edited by Gennaro AR,19thed.Pennsylvania:Mack Publishing Company,1995),并且包括但不限于:pH调节剂,表面活性剂,离子强度增强剂,维持渗透压的试剂,延迟吸收的试剂,稀释剂,佐剂,防腐剂等。例如,pH调节剂包括但不限于磷酸盐缓冲液。表面活性剂包括但不限于阳离子,阴离子或者非离子型表面活性剂,例如Tween-80。离子强度增强剂包括但不限于氯化钠。维持渗透压的试剂包括但不限于糖、NaCI及其类似物。延迟吸收的试剂包括但不限于单硬脂酸盐和明胶。稀释剂包括但不限于水,水性缓冲液(如缓冲盐水),醇和多元醇(如甘油)等。佐剂包括但不限于铝佐剂(例如氢氧化铝),弗氏佐剂(例如完全弗氏佐剂)等。防腐剂包括但不限于各种抗细菌试剂和抗真菌试剂,例如硫柳汞,2-苯氧乙醇,对羟苯甲酸酯,三氯叔丁醇,苯酚,山梨酸等。在某些实施方案中,所述药学上可接受的载体或赋形剂是无菌等渗水性或非水性溶液(例如,平衡盐溶液或生理盐水)、分散液、悬浮液或乳液。
如本文中所使用的,术语“疫苗”是被施用以产生或人工增加对特定抗原的免疫的组合物。如本文中所使用的,术语“免疫原性组合物”、“免疫刺激组合物”是当施用于个体时能够在体内产生免疫应答的组合物。因此,应理解术语“免疫原性组合物”、“免疫刺激组合物”和“疫苗”是同义术语。在一些实施方案中,所述个体优选为哺乳动物,更优选为人,当然也可以是其它哺乳动物,例如所述组合物可在牛(cattle)(包括乳牛(cow))、绵羊、山羊或马中诱导免疫,或者宠物,如狗或猫。
如本文中所使用的,术语“疫苗佐剂”、“佐剂”是指能够修饰或增强对抗原的免疫应答的物质。换言之,针对抗原的免疫应答在佐剂存在时可以高于或不同于当佐剂不存在时(包括当应答被修饰时,例如,佐剂存在时活化的T细胞子集与不存在佐剂时活化的子集不同)。
如本文中所使用的,术语“预防”包括抑制和延迟疾病的发作,并且不仅包括在发展疾病之前的预防,还包括在治疗后预防疾病的复发。
如本文中所使用的,术语“治疗”意指逆转、减轻或清除这样的术语所应用的病症或病况或者这样的病症或病况的一或多种症状的进展。
如本文中所使用的,术语“有效量”是指足以实现所需预防或治疗效果的量,例如,实现减轻与待治疗疾病相关的一或多种症状的量。
可调整给药方案以提供最佳所需响应。例如,可给药单次推注,可随时间给药数个分剂量,或可如治疗情况的急需所表明而按比例减少或增加剂量。要注意,剂量值可随要减轻的病况的类型及严重性而变化,且可包括单次或多次剂量。要进一步理解,对于任何特定受试者,具体的给药方案应根据受试者需要及给药组合物或监督组合物的给药的人员的专业判断来随时间调整。
给药量取决于所治疗的受试者、病症或病况的严重性、给药的速率、化合物的处置及处方医师的判断。一般而言,有效剂量在每日每kg体重约0.0001至约50mg,例如约0.01至约10mg/kg/日(单次或分次给药)。对70kg的人而言,这会合计为约0.007mg/日至约3500mg/日,例如约0.7mg/日至约700mg/日。在一些情况下,不高于前述范围的下限的剂量水平可以是足够的,而在其它情况下,仍可在不引起任何有害副作用的情况下采用较大剂量,条件是首先将所述较大剂量分成数个较小剂量以在一整天中给药。
如本文中所使用的,术语“受试者”包括人或非人动物。示例性人受试者包括患有疾病(例如本文所述的疾病)的人受试者(称为患者)或正常受试者。本发明中“非人动物”包括所有脊椎动物,例如非哺乳动物(例如鸟类、两栖动物、爬行动物)和哺乳动物,例如非人灵长类、家畜和/或驯化动物(例如绵羊、犬、猫、奶牛、猪等)。
本发明所提供的化合物可以适应于任何形式的给药方式,可以是口服或胃肠外给药,例如,可以是经肺、经鼻、经直肠和/或静脉注射,更具体可以是真皮内、皮下、肌内、关节内、腹膜内、肺部、口腔、舌下含服、经鼻、经皮、阴道、口服或胃肠外给药。
本发明的有益效果
所述苯并五元氮环类化合物相较化合物对照化合物,具有更好的HIF-2激动活性,对HIF-2下游的EPO、VGEF、Glut1、NDRG1等mRNA和蛋白的表达具有显著的增强活性,具有良好的产业化前景。
附图说明
此处的附图用来提供对本发明的进一步理解,构成本发明的一部分,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。在附图中:
图1显示本发明化合物对786-O细胞系HIF-2α下游基因NDRG1转录的影响。
图2显示本发明化合物对786-O细胞系HIF-2α下游基因VEGFA转录的影响。
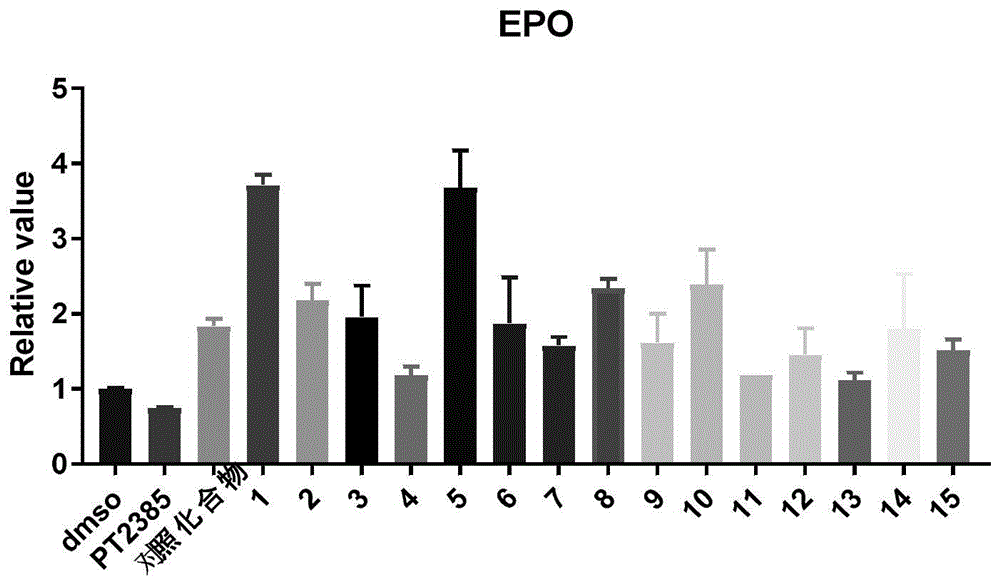
图3显示本发明化合物对HIF-2α下游基因EPO表达的影响。
具体实施方式
下面将结合实施例对本发明的实施方案进行详细描述,但是本领域技术人员将会理解,下列实施例仅用于说明本发明,而不应视为限定本发明的范围。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规产品。
实施例1 N-(3,5-双(三氟甲基)苯基)苯并[d]异噁唑-3-胺(1)的制备
化合物1b的合成:将化合物1a(621mg,5mmol)溶于17mL的乙醇溶液中,随后向反应体系中加入盐酸羟胺的水溶液(3.48g,5mL)。反应液在室温的条件下反应过夜,反应完毕后减压浓缩部分溶剂。加入50mL的水稀释反应液并用乙酸乙酯萃取。有机层依次用清水洗涤,随后用饱和NaCl溶液洗涤、无水硫酸钠干燥、在真空下减压浓缩,得到无色油状液体(700mg,产率100%)。
化合物1c的合成:将化合物1b(700mg,5mmol)溶于无水DMF(12mL)中,随后向反应体系中加入NCS(1.34g,10mmol)(分两次加入,间隔10min),在室温下反应,约0.5h反应完毕。反应结束后向反应液中加入80mL水稀释反应液,并用乙酸乙酯萃取。有机层用大量清水洗涤、饱和氯化钠溶液洗涤,并用无水硫酸钠干燥,在真空下减压浓缩得白色固体(800mg,产率92%)。
化合物1d的合成:将化合物1c(800mg,4.6mmol)溶于无水THF(14mL)中,随后加入3,5-双三氟甲基苯胺(1.58g,6.9mmol),使反应液回流并搅拌,约48h后TLC监测无原料剩余。反应结束后,向反应液中加入60mL水稀释反应液,并用乙酸乙酯萃取,有机层用饱和氯化钠溶液洗涤,无水硫酸钠干燥,减压浓缩后采用硅胶柱层析分离(PE∶EA=50∶1-20∶1),得固体化合物1d,产率16%。
化合物1的合成:将化合物1d(260mg,0.71mmol)溶于NMP(7mL)中,加入叔丁醇钾,在100℃下搅拌,约2.5h反应完毕。反应结束后向反应液中加入40mL水稀释反应液,并用乙酸乙酯萃取,有机层用饱和氯化钠溶液洗涤,无水硫酸钠干燥,真空下减压浓缩后,采用硅胶柱层析分离(PE∶EA=50∶1),得白色固体化合物(272mg,产率57%)。
实施例2 N-(3,5-二甲氧基苯基)苯并[d]异噁唑-3-胺(2)的制备
化合物2d的合成:将化合物1c(865mg,5mmol)溶于无水THF(14mL)中,随后加入3,5-二甲氧基苯胺(1.1g,7.5mmol),使反应液回流并搅拌,约48h后TLC监测无原料剩余。反应结束后,向反应液中加入60mL水稀释反应液,并用乙酸乙酯萃取,有机层用饱和氯化钠溶液洗涤,无水硫酸钠干燥,减压浓缩后采用硅胶柱层析分离,得固体化合物2d。
化合物2的合成:将化合物2d(290mg,1mmol)溶于NMP(7mL)中,加入叔丁醇钾,在100℃下搅拌,约2.5h反应完毕。反应结束后向反应液中加入40mL水稀释反应液,并用乙酸乙酯萃取,有机层用饱和氯化钠溶液洗涤,无水硫酸钠干燥,真空下减压浓缩后,采用硅胶柱层析分离(PE∶EA=50∶1),得白色固体化合物(162mg,产率60%)。
实施例3 N-(3,5-二氟苯基)苯并[d]异噁唑-3-胺(3)的制备
化合物3d的合成:将化合物1c(865mg,5mmol)溶于无水THF(14mL)中,随后加入3,5-二氟苯胺(0.97g,7.5mmol),使反应液回流并搅拌,约48h后TLC监测无原料剩余。反应结束后,向反应液中加入60mL水稀释反应液,并用乙酸乙酯萃取,有机层用饱和氯化钠溶液洗涤,无水硫酸钠干燥,减压浓缩后采用硅胶柱层析分离,得固体化合物3d,产率24%。
化合物3的合成:将化合物3d(532mg,2mmol)溶于NMP(15mL)中,加入叔丁醇钾,在100℃下搅拌,约2.5h反应完毕。反应结束后向反应液中加入40mL水稀释反应液,并用乙酸乙酯萃取,有机层用饱和氯化钠溶液洗涤,无水硫酸钠干燥,真空下减压浓缩后,采用硅胶柱层析分离,得固体化合物(290mg,产率60%)。
实施例4 N-(3-三氟甲基苯基)苯并[d]异噁唑-3-胺(4)的制备
化合物4d的合成:将化合物1c(800mg,4.6mmol)溶于无水THF(14mL)中,随后加入3-三氟甲基苯胺(1.1g,6.9mmol),使反应液回流并搅拌,约48h后TLC监测无原料剩余。反应结束后,向反应液中加入60mL水稀释反应液,并用乙酸乙酯萃取,有机层用饱和氯化钠溶液洗涤,无水硫酸钠干燥,减压浓缩后采用硅胶柱层析分离,得固体化合物4d,产率23%。
化合物4的合成:将化合物4d(298mg,1mmol)溶于NMP(10mL)中,加入叔丁醇钾,在100℃下搅拌,约2.5h反应完毕。反应结束后向反应液中加入40mL水稀释反应液,并用乙酸乙酯萃取,有机层用饱和氯化钠溶液洗涤,无水硫酸钠干燥,真空下减压浓缩后,采用硅胶柱层析分离,得白色固体化合物(164mg,产率59%)。
实施例5 N-(3-氰基苯基)苯并[d]异噁唑-3-胺(5)的制备
化合物5d的合成:将化合物1c(0.5g,3mmol)溶于无水THF(20mL)中,随后加入3-氰基苯胺(0.53g,4.5mmol),使反应液回流并搅拌,约48h后TLC监测无原料剩余。反应结束后,向反应液中加入60mL水稀释反应液,并用乙酸乙酯萃取,有机层用饱和氯化钠溶液洗涤,无水硫酸钠干燥,减压浓缩后采用硅胶柱层析分离,得固体化合物5d(153mg),产率20%。
化合物5的合成:将化合物5d(153mg,0.6mmol)溶于NMP(5mL)中,加入叔丁醇钾,在100℃下搅拌,约2.5h反应完毕。反应结束后向反应液中加入40mL水稀释反应液,并用乙酸乙酯萃取,有机层用饱和氯化钠溶液洗涤,无水硫酸钠干燥,真空下减压浓缩后,采用硅胶柱层析分离,得白色固体化合物(80mg,产率57%)。
实施例6 3-(苯并[d]异噁唑-3-氨基)-5-(三氟甲基)苯腈(6)的制备
化合物6d的合成:将化合物1c(865mg,5mmol)溶于无水THF(20mL)中,随后加入3-氰基-5-三氟甲基苯胺(1.4g,7.5mmol),使反应液回流并搅拌,约48h后TLC监测无原料剩余。反应结束后,向反应液中加入水稀释反应液,并用乙酸乙酯萃取,有机层用饱和氯化钠溶液洗涤,无水硫酸钠干燥,减压浓缩后采用硅胶柱层析分离,得固体化合物6d(306mg),产率19%。
化合物6的合成:将化合物6d(306mg,0.95mmol)溶于NMP(10mL)中,加入叔丁醇钾,在100℃下搅拌,约2.5h反应完毕。反应结束后向反应液中加入水稀释反应液,并用乙酸乙酯萃取,有机层用饱和氯化钠溶液洗涤,无水硫酸钠干燥,真空下减压浓缩后,采用硅胶柱层析分离,得白色固体化合物(138mg,产率48%)。
实施例7 N-(3,5-双(三氟甲基)苯基)-N-甲基苯并[d]异噁唑-3-胺(7)的制备
化合物7a的合成:3,5-二三氟甲基苯胺(0.65g,4mmol)溶于DMF(20mL)中,加入碳酸钾(1.38g,10mmol)和碘甲烷(0.25mL,4mmol),在室温搅拌2h后,70℃搅拌24h。薄层层析(TLC)(PE∶EtOAc=15∶1)确定反应完毕后,冷却到室温,倒入80mL水中,用150mL DCM萃取三次,有机相使用饱和食盐水洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩后柱层析(PE∶EtOAc=300∶1)得到目标中间体7a(0.29g),黄色油状物,产率30%。
化合物7b的合成:将化合物1c(138mg,0.8mmol)溶于无水THF(5mL)中,随后加入化合物7a(0.29g,1.2mmol),使反应液回流并搅拌,约48h后TLC监测无原料剩余。反应结束后,向反应液中加入60mL水稀释反应液,并用乙酸乙酯萃取,有机层用饱和氯化钠溶液洗涤,无水硫酸钠干燥,减压浓缩后采用硅胶柱层析分离,得固体化合物7b,产率30%。
化合物7的合成:将化合物7b(91mg,0.24mmol)溶于NMP(3mL)中,加入叔丁醇钾,在100℃下搅拌,约2.5h反应完毕。反应结束后向反应液中加入10mL水稀释反应液,并用乙酸乙酯萃取,有机层用饱和氯化钠溶液洗涤,无水硫酸钠干燥,真空下减压浓缩后,采用硅胶柱层析分离,得白色固体化合物(53mg,产率62%)。
实施例8 N-(3,5-双(三氟甲基)苯基)-N-环丙基苯并[d]异噁唑-3-胺(8)的制备
化合物8b的合成:化合物8a(200mg,1.2mmol)和3,5-二三氟甲基苯胺(229mg,1mmol)溶于甲醇(4mL)后加入醋酸(5mL),于80℃搅拌2.5h,室温加入氰基硼氢化钠(126mg,2mmol),于80℃搅拌3h;反应完毕后,冷却到室温,使用饱和碳酸氢钠溶液淬灭,然后用乙酸乙酯萃取,有机相用饱和食盐水洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩后柱层析(PE∶EtOAc=120∶1)得到黄色油状物,产率为55%。
化合物8c的合成:将化合物1c(76mg,0.44mmol)溶于无水THF(3mL)中,随后加入化合物8b(177mg,0.66mmol),使反应液回流并搅拌,约48h后TLC监测无原料剩余。反应结束后,向反应液中加入水稀释反应液,并用乙酸乙酯萃取,有机层用饱和氯化钠溶液洗涤,无水硫酸钠干燥,减压浓缩后采用硅胶柱层析分离(PE∶EA=50∶1-20∶1),得固体化合物8c,产率25%。
化合物8的合成:将化合物8c(44mg,0.08mmol)溶于NMP(1mL)中,加入叔丁醇钾,在100℃下搅拌,约2.5h反应完毕。反应结束后向反应液中加入水稀释反应液,并用乙酸乙酯萃取,有机层用饱和氯化钠溶液洗涤,无水硫酸钠干燥,真空下减压浓缩后,采用硅胶柱层析分离(PE∶EA=40∶1),得白色固体化合物(22mg,产率67%)。
实施例9 N-(3,5-双(三氟甲基)苯基)-N-苯基苯并[d]异噁唑-3-胺(9)的制备
化合物9a的合成:醋酸钯(11mg,0.05mmol)和DPPE溶于甲苯(39mg,0.1mmol)中,于110℃搅拌十分钟,将碘苯(204mg,1mmol)和3,5-二三氟甲基苯胺(229mg,1mmol)溶于甲苯(5mL)的混合溶液加入,最后加入甲醇钠(59.4mg,1.1mmol),反应24h;反应完毕后,冷却到室温,减压浓缩,柱层析(PE∶EtOAc=120∶1)得到油状物,产率70%。
化合物9b的合成:将化合物1c(81mg,0.47mmol)溶于无水THF(2mL)中,随后加入化合物9a(0.21g,0.7mmol),使反应液回流并搅拌,约48h后TLC监测无原料剩余。反应结束后,向反应液中加入水稀释反应液,并用乙酸乙酯萃取,有机层用饱和氯化钠溶液洗涤,无水硫酸钠干燥,减压浓缩后采用硅胶柱层析分离,得固体化合物9b,产率21%。
化合物9的合成:将化合物9b(44mg,0.1mmol)溶于NMP(1mL)中,加入叔丁醇钾,在100℃下搅拌,约2.5h反应完毕。反应结束后向反应液中加入水稀释反应液,并用乙酸乙酯萃取,有机层用饱和氯化钠溶液洗涤,无水硫酸钠干燥,真空下减压浓缩后,采用硅胶柱层析分离,得白色固体化合物(27mg,产率65%)。
实施例10 N-(3,5-双(三氟甲基)苯基)-5-溴苯并[d]异噁唑-3-胺(10)的制备
化合物10b的合成:将化合物10a(1g,5mmol)溶于20mL的乙醇溶液中,随后向反应体系中加入盐酸羟胺的水溶液(3.48g,5mL)。反应液在室温的条件下反应过夜,反应完毕后减压浓缩部分溶剂。加入50mL的水稀释反应液并用乙酸乙酯萃取。有机层依次用清水洗涤,随后用饱和NaCl溶液洗涤、无水硫酸钠干燥、在真空下减压浓缩,得到无色油状液体(1.1g,产率100%)。
化合物10c的合成:将化合物10b(1.1g,5mmol)溶于无水DMF(12mL)中,随后向反应体系中加入NCS(1.34g,10mmol)(分两次加入,间隔10min),在室温下反应,约0.5h反应完毕。反应结束后向反应液中加入80mL水稀释反应液,并用乙酸乙酯萃取。有机层用大量清水洗涤、饱和氯化钠溶液洗涤,并用无水硫酸钠干燥,在真空下减压浓缩得白色固体(1.1g,产率90%)。
化合物10d的合成:将化合物10c(1.1g,4.5mmol)溶于无水THF(15mL)中,随后加入3,5-双三氟甲基苯胺(1.56g,6.8mmol),使反应液回流并搅拌,约48h后TLC监测无原料剩余。反应结束后,向反应液中加入60mL水稀释反应液,并用乙酸乙酯萃取,有机层用饱和氯化钠溶液洗涤,无水硫酸钠干燥,减压浓缩后采用硅胶柱层析分离,得固体化合物10d,产率19%。
化合物10的合成:将化合物10d(379mg,0.85mmol)溶于NMP(10mL)中,加入叔丁醇钾,在100℃下搅拌,约2.5h反应完毕。反应结束后向反应液中加入40mL水稀释反应液,并用乙酸乙酯萃取,有机层用饱和氯化钠溶液洗涤,无水硫酸钠干燥,真空下减压浓缩后,采用硅胶柱层析分离,得白色固体化合物(183mg,产率54%)。
实施例11 N-(3,5-双(三氟甲基)苯基)-5-氟苯并[d]异噁唑-3-胺(11)的制备
化合物11b的合成:将化合物11a(710mg,5mmol)溶于20mL的乙醇溶液中,随后向反应体系中加入盐酸羟胺的水溶液(3.48g,5mL)。反应液在室温的条件下反应过夜,反应完毕后减压浓缩部分溶剂。加入50mL的水稀释反应液并用乙酸乙酯萃取。有机层依次用清水洗涤,随后用饱和NaCl溶液洗涤、无水硫酸钠干燥、在真空下减压浓缩,得到无色油状液体(780mg,产率100%)。
化合物11c的合成:将化合物11b(780mg,5mmol)溶于无水DMF(12mL)中,随后向反应体系中加入NCS(1.34g,10mmol)(分两次加入,间隔10min),在室温下反应,约0.5h反应完毕。反应结束后向反应液中加入80mL水稀释反应液,并用乙酸乙酯萃取。有机层用大量清水洗涤、饱和氯化钠溶液洗涤,并用无水硫酸钠干燥,在真空下减压浓缩得白色固体(850mg,产率89%)。
化合物11d的合成:将化合物11c(850mg,4.4mmol)溶于无水THF(15mL)中,随后加入3,5-双三氟甲基苯胺(1.5g,6.6mmol),使反应液回流并搅拌,约48h后TLC监测无原料剩余。反应结束后,向反应液中加入60mL水稀释反应液,并用乙酸乙酯萃取,有机层用饱和氯化钠溶液洗涤,无水硫酸钠干燥,减压浓缩后采用硅胶柱层析分离(PE∶EA=50∶1-20∶1),得固体化合物11d,产率25%。
化合物11的合成:将化合物11d(422mg,1.1mmol)溶于NMP(15mL)中,加入叔丁醇钾,在100℃下搅拌,约2.5h反应完毕。反应结束后向反应液中加入50mL水稀释反应液,并用乙酸乙酯萃取,有机层用饱和氯化钠溶液洗涤,无水硫酸钠干燥,真空下减压浓缩后,采用硅胶柱层析分离,得白色固体化合物(216mg,产率54%)。
实施例12 N-(3,5-双(三氟甲基)苯基)-5-甲氧基苯并[d]异噁唑-3-胺(12)的制备
化合物12b的合成:将化合物12a(770mg,5mmol)溶于20mL的乙醇溶液中,随后向反应体系中加入盐酸羟胺的水溶液(3.48g,5mL)。反应液在室温的条件下反应过夜,反应完毕后减压浓缩部分溶剂。加入50mL的水稀释反应液并用乙酸乙酯萃取。有机层依次用清水洗涤,随后用饱和NaCl溶液洗涤、无水硫酸钠干燥、在真空下减压浓缩,得到无色油状液体(845mg,产率100%)。
化合物12c的合成:将化合物12b(845mg,5mmol)溶于无水DMF(15mL)中,随后向反应体系中加入NCS(1.34g,10mmol)(分两次加入,间隔10min),在室温下反应,约0.5h反应完毕。反应结束后向反应液中加入80mL水稀释反应液,并用乙酸乙酯萃取。有机层用大量清水洗涤、饱和氯化钠溶液洗涤,并用无水硫酸钠干燥,在真空下减压浓缩得白色固体(1g,产率98%)。
化合物12d的合成:将化合物12c(1g,4.9mmol)溶于无水THF(15mL)中,随后加入3,5-双三氟甲基苯胺(1.68g,7.35mmol),使反应液回流并搅拌,约48h后TLC监测无原料剩余。反应结束后,向反应液中加入60mL水稀释反应液,并用乙酸乙酯萃取,有机层用饱和氯化钠溶液洗涤,无水硫酸钠干燥,减压浓缩后采用硅胶柱层析分离,得固体化合物12d,产率22%。
化合物12的合成:将化合物12d(396mg,1.0mmol)溶于NMP(10mL)中,加入叔丁醇钾,在100℃下搅拌,约2.5h反应完毕。反应结束后向反应液中加入40mL水稀释反应液,并用乙酸乙酯萃取,有机层用饱和氯化钠溶液洗涤,无水硫酸钠干燥,真空下减压浓缩后,采用硅胶柱层析分离(PE∶EA=50∶1),得白色固体化合物(169mg,产率45%)。
实施例13 N-(3,5-双(三氟甲基)苯基)-6-溴苯并[d]异噁唑-3-胺(13)的制备
化合物13b的合成:将化合物13a(1g,5mmol)溶于20mL的乙醇溶液中,随后向反应体系中加入盐酸羟胺的水溶液(3.48g,5mL)。反应液在室温的条件下反应过夜,反应完毕后减压浓缩部分溶剂。加入50mL的水稀释反应液并用乙酸乙酯萃取。有机层依次用清水洗涤,随后用饱和NaCl溶液洗涤、无水硫酸钠干燥、在真空下减压浓缩,得到无色油状液体(1.08g,产率100%)。
化合物13c的合成:将化合物13b(1.08g,5mmol)溶于无水DMF(15mL)中,随后向反应体系中加入NCS(1.34g,10mmol)(分两次加入,间隔10min),在室温下反应,约0.5h反应完毕。反应结束后向反应液中加入80mL水稀释反应液,并用乙酸乙酯萃取。有机层用大量清水洗涤、饱和氯化钠溶液洗涤,并用无水硫酸钠干燥,在真空下减压浓缩得白色固体(1.15g,产率92%)。
化合物13d的合成:将化合物13c(1.15g,4.6mmol)溶于无水THF(15mL)中,随后加入3,5-双三氟甲基苯胺(1.58g,6.9mmol),使反应液回流并搅拌,约48h后TLC监测无原料剩余。反应结束后,向反应液中加入60mL水稀释反应液,并用乙酸乙酯萃取,有机层用饱和氯化钠溶液洗涤,无水硫酸钠干燥,减压浓缩后采用硅胶柱层析分离(PE∶EA=50∶1-20∶1),得固体化合物13d,产率20%。
化合物13的合成:将化合物13d(408mg,0.92mmol)溶于NMP(7mL)中,加入叔丁醇钾,在100℃下搅拌,约2.5h反应完毕。反应结束后向反应液中加入40mL水稀释反应液,并用乙酸乙酯萃取,有机层用饱和氯化钠溶液洗涤,无水硫酸钠干燥,真空下减压浓缩后,采用硅胶柱层析分离(PE∶EA=40∶1),得白色固体化合物(222mg,产率57%)。
实施例14 N-(3,5-双(三氟甲基)苯基)-6-氟苯并[d]异噁唑-3-胺(14)的制备
化合物14b的合成:将化合物14a(710mg,5mmol)溶于17mL的乙醇溶液中,随后向反应体系中加入盐酸羟胺的水溶液(3.48g,5mL)。反应液在室温的条件下反应过夜,反应完毕后减压浓缩部分溶剂。加入50mL的水稀释反应液并用乙酸乙酯萃取。有机层依次用清水洗涤,随后用饱和NaCl溶液洗涤、无水硫酸钠干燥、在真空下减压浓缩,得到无色油状液体(780mg,产率100%)。
化合物14c的合成:将化合物14b(780mg,5mmol)溶于无水DMF(15mL)中,随后向反应体系中加入NCS(1.34g,10mmol)(分两次加入,间隔10min),在室温下反应,约0.5h反应完毕。反应结束后向反应液中加入80mL水稀释反应液,并用乙酸乙酯萃取。有机层用大量清水洗涤、饱和氯化钠溶液洗涤,并用无水硫酸钠干燥,在真空下减压浓缩得白色固体(850mg,产率89%)。
化合物14d的合成:将化合物14c(850mg,4.4mmol)溶于无水THF(14mL)中,随后加入3,5-双三氟甲基苯胺(1.5g,6.6mmol),使反应液回流并搅拌,约48h后TLC监测无原料剩余。反应结束后,向反应液中加入60mL水稀释反应液,并用乙酸乙酯萃取,有机层用饱和氯化钠溶液洗涤,无水硫酸钠干燥,减压浓缩后采用硅胶柱层析分离(PE∶EA=50∶1-20∶1),得固体化合物14d,产率26%。
化合物14的合成:将化合物14d(439mg,1.14mmol)溶于NMP(10mL)中,加入叔丁醇钾,在100℃下搅拌,约2.5h反应完毕。反应结束后向反应液中加入40mL水稀释反应液,并用乙酸乙酯萃取,有机层用饱和氯化钠溶液洗涤,无水硫酸钠干燥,真空下减压浓缩后,采用硅胶柱层析分离(PE∶EA=50∶1),得白色固体化合物(220mg,产率53%)。
实施例15 N-(3,5-双(三氟甲基)苯基)-6-甲氧基苯并[d]异噁唑-3-胺(15)的制备
化合物15b的合成:将化合物15a(770mg,5mmol)溶于17mL的乙醇溶液中,随后向反应体系中加入盐酸羟胺的水溶液(3.48g,5mL)。反应液在室温的条件下反应过夜,反应完毕后减压浓缩部分溶剂。加入50mL的水稀释反应液并用乙酸乙酯萃取。有机层依次用清水洗涤,随后用饱和NaCl溶液洗涤、无水硫酸钠干燥、在真空下减压浓缩,得到无色油状液体(845mg,产率100%)。
化合物15c的合成:将化合物15b(845mg,5mmol)溶于无水DMF(15mL)中,随后向反应体系中加入NCS(1.34g,10mmol)(分两次加入,间隔10min),在室温下反应,约0.5h反应完毕。反应结束后向反应液中加入80mL水稀释反应液,并用乙酸乙酯萃取。有机层用大量清水洗涤、饱和氯化钠溶液洗涤,并用无水硫酸钠干燥,在真空下减压浓缩得白色固体(964mg,产率95%)。
化合物15d的合成:将化合物15c(964mg,4.6mmol)溶于无水THF(15mL)中,随后加入3,5-双三氟甲基苯胺(1.58g,6.9mmol),使反应液回流并搅拌,约48h后TLC监测无原料剩余。反应结束后,向反应液中加入60mL水稀释反应液,并用乙酸乙酯萃取,有机层用饱和氯化钠溶液洗涤,无水硫酸钠干燥,减压浓缩后采用硅胶柱层析分离(PE∶EA=50∶1-20∶1),得固体化合物15d,产率20%。
化合物15的合成:将化合物15d(364mg,0.92mmol)溶于NMP(10mL)中,加入叔丁醇钾,在100℃下搅拌,约2.5h反应完毕。反应结束后向反应液中加入40mL水稀释反应液,并用乙酸乙酯萃取,有机层用饱和氯化钠溶液洗涤,无水硫酸钠干燥,真空下减压浓缩后,采用硅胶柱层析分离(PE∶EA=60∶1),得白色固体化合物(183mg,产率53%)。
实施例16-实施例46的合成方法同式III'通用合成方法。
实施例16 4-氟-3-((2-(吡咯烷-1-基)苯基)氨基)苯并[d]异噻唑-1,1-二氧化物
先将3-氯-4-氟苯并[d]异噻唑-1,1-二氧化物(1.5克,6.83毫摩尔,参照前文化合物2d制备方法合成得到)溶解在20mL的二氯甲烷中并且将溶液降温到0摄氏度,然后将2-(吡咯烷-1-基)苯胺(1.11克,6.83毫摩尔)和乙基二异丙胺(1.32克,10.25毫摩尔)分别缓慢加入到上述溶液中并升温到室温,反应在室温下搅拌2小时,液质监控原料完全转化。将反应液中加入10mL蒸馏水,分液后再用蒸馏水洗涤有机相(5mL*2)。收集有机相并用无水硫酸钠干燥,减压蒸除溶剂得到粗产物。粗产物经过硅胶柱色谱分离提纯得到4-氟-3-((2-(吡咯烷-1-基)苯基)氨基)苯并[d]异噻唑-1,1-二氧化物(1.32g,53%产率)。
1
实施例17 N-(3,5-双(三氟甲基)苯基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺的合成
使用3-氯-4-氟苯并[d]异噻唑-1,1-二氧化物和3,5-二(三氟甲基)苯胺作为原料,通过步骤4的合成方法,得到N-(3,5-二(三氟甲基)苯基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺。
1H NMR(400MHz,DMSO-d6)δ10.32(s,1H),8.55(d,J=1.6Hz,2H),8.07-7.95(m,3H),7.87-7.76(m,1H).
实施例18 N-(3,5-双(甲氧基)苯基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺的合成
使用3-氯-4-氟苯并[d]异噻唑-1,1-二氧化物和3,5-二甲氧基苯胺作为原料,通过步骤4的合成方法,得到N-(3,5-双(甲氧基)苯基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺。
1H NMR(400MHz,DMSO-d6)δ9.71(s,1H),7.96(q,J=3.1,2.3Hz,2H),7.78(dp,J=13.4,4.5,3.9Hz,1H),7.04(d,J=2.2Hz,2H),6.49-6.43(m,1H),3.78(s,6H).
实施例19 N-(3-三氟甲基-5-甲氧基苯基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺的合成
使用3-氯-4-氟苯并[d]异噻唑-1,1-二氧化物和3-三氟甲基-5-甲氧基苯胺作为原料,通过步骤4的合成方法,得到(3-三氟甲基-5-甲氧基苯基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺。
1H NMR(400MHz,DMSO-d6)δ10.03(s,1H),7.98(q,J=2.9,2.3Hz,2H),7.82(s,1H),7.80(dq,J=9.4,4.8Hz,1H),7.73(s,1H),7.19(s,1H),3.88(s,3H).
实施例20 N-(2-(吡咯烷-1-基)-3-甲基苯基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺的合成
使用3-氯-4-氟苯并[d]异噻唑-1,1-二氧化物和2-(吡咯烷-1-基)-3-甲基苯胺作为原料,通过步骤4的合成方法,得到N-(2-(吡咯烷-1-基)-3-甲基苯基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺。
1H NMR(400MHz,DMSO-d6)δ10.78(d,J=8.7Hz,1H),8.28(d,J=8.1Hz,1H),8.01-7.91(m,2H),7.85-7.76(m,1H),7.29(t,J=7.9Hz,1H),7.10(d,J=7.7Hz,1H),3.33(s,3H),3.20(d,J=12.4Hz,1H),2.34(s,3H),2.06(d,J=12.0Hz,1H),2.06(s,3H).
实施例21 N-(2-(吡咯烷-2-酮)-6-氟苯基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺的合成
使用3-氯-4-氟苯并[d]异噻唑-1,1-二氧化物和2-(吡咯烷-2-酮)-6-氟苯胺作为原料,通过步骤4的合成方法,得到N-(2-(吡咯烷-2-酮)-6-氟苯基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺。
1H NMR(400MHz,DMSO-d6)δ9.66(d,J=5.1Hz,1H),8.03-7.91(m,2H),7.82-7.73(m,1H),7.54(td,J=8.2,5.9Hz,1H),7.39(d,J=8.4Hz,2H),3.93(t,J=6.9Hz,2H),2.46(d,J=7.9Hz,2H),2.10(p,J=7.4Hz,2H).
实施例22 N-(2-(吡咯烷-1-基)-5-三氟甲基苯基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺的合成
使用3-氯-4-氟苯并[d]异噻唑-1,1-二氧化物和2-(吡咯烷-1-基)-5-三氟甲基苯胺作为原料,通过步骤4的合成方法,得到N-(2-(吡咯烷-1-基)-5-三氟甲基苯基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺。
1H NMR(400MHz,DMSO-d6)δ10.15(s,1H),7.96(td,J=7.7,4.1Hz,1H),7.92(dd,J=7.5,1.1Hz,1H),7.76(ddd,J=10.1,7.9,1.2Hz,1H),7.61(d,J=2.4Hz,1H),7.51(dd,J=8.8,2.4Hz,1H),6.95(d,J=8.8Hz,1H),3.38-3.30(m,4H),1.88-1.80(m,4H).
实施例23 N-(2-(吡咯烷-1-基)-3,5-二甲基苯基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺的合成
使用3-氯-4-氟苯并[d]异噻唑-1,1-二氧化物和2-(吡咯烷-1-基)-3,5-二甲基苯胺作为原料,通过步骤4的合成方法,得到N-(2-(吡咯烷-1-基)-3,5-二甲基苯基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺。
1H NMR(400MHz,DMSO-d6)δ10.77(s,1H),8.11(s,1H),7.95(s,2H),7.78(d,J=9.9Hz,1H),6.90(s,1H),3.16(q,J=5.9,4.9Hz,4H),2.31(d,J=15.8Hz,6H),2.03(s,4H).
实施例24 N-(2-(吡咯烷-1-基)-5-甲基苯基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺的合成
使用3-氯-4-氟苯并[d]异噻唑-1,1-二氧化物和2-(吡咯烷-1-基)-5-甲基苯胺作为原料,通过步骤4的合成方法,得到N-(2-(吡咯烷-1-基)-5-甲基苯基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺。
1H NMR(400MHz,DMSO-d6)δ10.00(d,J=7.7Hz,1H),7.92(td,J=3.5,2.1Hz,2H),7.80(d,J=2.1Hz,1H),7.79-7.69(m,1H),7.13(d,J=8.2Hz,1H),7.02(ddd,J=8.1,2.0,0.8Hz,1H),3.08-3.01(m,4H),2.28(s,3H),1.92-1.81(m,4H).
实施例25 N-(2-(甲基异丁基氨基)苯基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺的合成
使用3-氯-4-氟苯并[d]异噻唑-1,1-二氧化物和N-异丁基-N-甲基-1,2-苯二胺作为原料,通过步骤4的合成方法,得到N-(2-(甲基异丁基氨基)苯基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺。
1H NMR(400MHz,DMSO-d6)δ10.10(d,J=7.4Hz,1H),8.40(d,J=7.7Hz,1H),8.03-7.92(m,2H),7.87-7.78(m,1H),7.46(dd,J=7.8,1.6Hz,1H),7.29(pd,J=7.5,1.8Hz,2H),2.81(d,J=7.1Hz,2H),2.62(s,3H),1.65(hept,J=6.8Hz,1H),0.88(d,J=6.6Hz,6H).
实施例26(S)-N-(3-(2-甲基吡咯烷-1-基)吡啶-4-基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺的合成
使用3-氯-4-氟苯并[d]异噻唑-1,1-二氧化物和3-(2-甲基吡咯烷)-4-吡啶作为原料,通过步骤4的合成方法,得到(S)-N-(3-(2-甲基吡咯烷-1-基)吡啶-4-基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺。
1H NMR(400MHz,DMSO-d6)δ8.19(d,J=6.3Hz,2H),7.93(d,J=5.7Hz,1H),7.90-7.78(m,2H),7.63(t,J=7.7Hz,1H),4.04(s,1H),3.47(ddd,J=9.5,7.5,5.8Hz,1H),2.99(q,J=7.9Hz,1H),2.11(dtd,J=12.2,7.6,5.3Hz,1H),1.96-1.74(m,2H),1.50(ddt,J=12.1,8.6,7.0Hz,1H),0.93(d,J=6.0Hz,3H).
实施例27 N-(3,5-二异丙基苯基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺的合成
使用3-氯-4-氟苯并[d]异噻唑-1,1-二氧化物和3,5-二异丙基苯胺作为原料,通过步骤4的合成方法,得到N-(3,5-二异丙基苯基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺。
1H NMR(400MHz,DMSO-d6)δ9.75(d,J=5.0Hz,1H),7.97-7.87(m,2H),7.80-7.68(m,1H),7.41(d,J=1.6Hz,2H),7.03(t,J=1.6Hz,1H),2.88(hept,J=6.9Hz,2H),1.19(d,J=6.9Hz,12H).
实施例28 N-(2,6-二异丙基吡啶-4-基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺的合成
使用3-氯-4-氟苯并[d]异噻唑-1,1-二氧化物和2,6二异丙基-4-氨基吡啶作为原料,通过步骤4的合成方法,得到N-(2,6-二异丙基吡啶-4-基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺。
1H NMR(400MHz,DMSO-d6)δ13.24(s,1H),9.84(s,1H),7.98-7.95(d,1H),7.79(s,1H),7.56(s,1H),3.03(s,2H),1.27(d,J=6.9Hz,12H)
实施例29 N-(3-(吡咯烷-1-基)-4-吡啶-4-基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺的合成
使用3-氯-4-氟苯并[d]异噻唑-1,1-二氧化物和3-吡咯烷-1-基-4-氨基吡啶作为原料,通过步骤4的合成方法,得到N-(3-(吡咯烷-1-基)吡啶-4-基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺。
1H NMR(400MHz,DMSO-d6)δ8.01(s,1H),7.73(d,J=5.9Hz,4H),7.56(s,1H),3.42(s,4H),1.90-1.82(m,4H).
实施例30 N-(3-(吡咯烷-1-基)吡啶-2-基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺的合成
使用3-氯-4-氟苯并[d]异噻唑-1,1-二氧化物和3-(吡咯烷-1-基)吡啶-2-胺作为原料,通过步骤4的合成方法,得到N-(3-(吡咯烷-1-基)吡啶-2-基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺。
1H NMR(400MHz,DMSO-d6)δ14.57(s,1H),7.83-7.72(m,3H),7.57(t,J=8.9Hz,1H),7.26-7.14(m,2H),3.53(s,4H),1.90-1.78(m,4H).
实施例31 N-(2-(吡咯烷-1-基)-3,5-二三氟甲基苯基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺的合成
使用3-氯-4-氟苯并[d]异噻唑-1,1-二氧化物和2-(吡咯烷-1-基)-3,5-二三氟甲基苯胺作为原料,通过步骤4的合成方法,得到N-(2-(吡咯烷-1-基)-3,5-二三氟甲基苯基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺。
1H NMR(400MHz,DMSO-d6)δ10.17(d,J=5.8Hz,1H),8.81(d,J=2.2Hz,1H),8.05-7.97(m,2H),7.95(d,J=2.2Hz,1H),7.84(ddd,J=10.6,6.5,2.5Hz,1H),3.28-3.20(m,4H),2.01-1.92(m,4H).
实施例32 N-(2-(环丙烷甲酰胺基甲基)苯基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺的合成
使用3-氯-4-氟苯并[d]异噻唑-1,1-二氧化物和2-(环丙烷甲酰胺基甲基)苯胺作为原料,通过步骤4的合成方法,得到N-(2-(环丙烷甲酰胺基甲基)苯基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺。
1H NMR(400MHz,DMSO-d6)δ10.48(d,J=4.7Hz,1H),8.77(t,J=5.9Hz,1H),7.99-7.88(m,2H),7.80-7.71(m,1H),7.55(dq,J=7.2,1.9,1.4Hz,1H),7.45-7.33(m,3H),4.29(d,J=5.8Hz,2H),1.54(tt,J=7.7,4.7Hz,1H),0.66-0.52(m,4H).
实施例33 N-(3-甲氧基苯基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺的合成
使用3-氯-4-氟苯并[d]异噻唑-1,1-二氧化物和3-甲氧基苯胺作为原料,通过步骤4的合成方法,得到N-(3-甲氧基苯基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺。
1H NMR(400MHz,DMSO-d6)δ9.82(s,1H),8.02-7.92(m,2H),7.78(ddd,J=10.3,6.0,2.9Hz,1H),7.43-7.34(m,3H),6.89(dt,J=6.0,2.9Hz,1H),3.80(s,3H).
实施例34 N-(2-(哌啶-1-基)苯基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺的合成
使用3-氯-4-氟苯并[d]异噻唑-1,1-二氧化物和2-(哌啶-1-基)苯胺作为原料,通过步骤4的合成方法,得到N-(2-(哌啶-1-基)苯基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺。
1H NMR(400MHz,DMSO-d6)δ10.21(d,J=7.2Hz,1H),8.45-8.39(m,1H),8.03-7.92(m,2H),7.85(ddd,J=10.9,7.1,2.0Hz,1H),7.45(dd,J=7.7,1.6Hz,1H),7.29(dtd,J=22.4,7.6,1.6Hz,2H),2.84(t,J=5.2Hz,4H),1.73(p,J=5.5Hz,4H),1.58(q,J=5.8Hz,2H).
实施例35 N-(3-三氟甲基苯基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺的合成
使用3-氯-4-氟苯并[d]异噻唑-1,1-二氧化物和3-三氟甲基苯胺作为原料,通过步骤4的合成方法,得到N-(3-三氟甲基苯基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺。
1H NMR(400MHz,DMSO-d6)δ10.14(s,1H),8.16(d,J=2.1Hz,1H),8.13-8.06(m,1H),8.04-7.94(m,2H),7.86-7.64(m,3H)
实施例36 N-(2-吗啉基苯基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺的合成
使用3-氯-4-氟苯并[d]异噻唑-1,1-二氧化物和2-吗啉基苯胺作为原料,通过步骤4的合成方法,得到N-(2-吗啉基苯基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺。
1H NMR(400MHz,DMSO-d6)δ10.16(d,J=8.7Hz,1H),8.43(dd,J=8.1,1.6Hz,1H),8.03-7.93(m,2H),7.85(ddd,J=10.8,7.1,1.9Hz,1H),7.51(dd,J=7.8,1.6Hz,1H),7.33(dtd,J=26.4,7.6,1.6Hz,2H),3.83-3.77(m,4H),2.92(d,J=9.1Hz,4H).
实施例37(R)-N-(2-(3-氟吡咯烷-1-基)苯基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺的合成
使用3-氯-4-氟苯并[d]异噻唑-1,1-二氧化物和(R)-2-(3-氟吡咯烷-1-基)苯胺作为原料,通过步骤4的合成方法,得到(R)-N-(2-(3-氟吡咯烷-1-基)苯基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺。
1H NMR(400MHz,DMSO-d6)δ10.02(d,J=6.5Hz,1H),7.95(dd,J=6.4,3.3Hz,2H),7.78(ddd,J=10.2,6.5,2.5Hz,1H),7.71(dd,J=7.9,1.6Hz,1H),7.26(td,J=7.8,1.6Hz,1H),7.12(dd,J=8.2,1.4Hz,1H),7.02(td,J=7.6,1.3Hz,1H),5.37(dq,J=54.8,3.0Hz,1H),3.64-3.31(m,3H),3.20(dt,J=8.9,6.0Hz,1H),2.27-2.09(m,2H)
实施例38(S)-N-(2-(3-氟吡咯烷-1-基)苯基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺的合成
使用3-氯-4-氟苯并[d]异噻唑-1,1-二氧化物和(S)-2-(3-氟吡咯烷-1-基)苯胺作为原料,通过步骤4的合成方法,得到(S)-N-(2-(3-氟吡咯烷-1-基)苯基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺。
1H NMR(400MHz,DMSO-d6)δ10.02(s,1H),7.95(dd,J=6.4,3.3Hz,2H),7.84-7.68(m,2H),7.26(ddd,J=8.6,7.4,1.6Hz,1H),7.12(dd,J=8.3,1.4Hz,1H),7.02(td,J=7.6,1.4Hz,1H),5.37(dq,J=54.7,3.4,2.9Hz,1H),3.62-3.35(m,3H),3.25-3.15(m,1H),2.28-2.08(m,2H)
实施例39 N-(2-(吡咯烷-1-基)-6-氟苯基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺的合成
使用3-氯-4-氟苯并[d]异噻唑-1,1-二氧化物和2-(吡咯烷-1-基)-6-氟苯胺作为原料,通过步骤4的合成方法,得到N-(2-(吡咯烷-1-基)-6-氟苯基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺。
1H NMR(400MHz,DMSO-d6)δ9.97(d,J=4.2Hz,1H),7.99-7.86(m,2H),7.73(ddd,J=10.0,8.0,1.1Hz,1H),7.19(td,J=8.3,6.7Hz,1H),6.56(dddd,J=8.2,5.1,3.9,1.2Hz,2H),3.33-3.25(m,4H),1.83-1.75(m,4H).
实施例40 N-(2-(吡咯烷-1-基)-3-氟苯基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺的合成
使用3-氯-4-氟苯并[d]异噻唑-1,1-二氧化物和2-(吡咯烷-1-基)-3-氟苯胺作为原料,通过步骤4的合成方法,得到N-(2-(吡咯烷-1-基)-3-氟苯基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺。
1H NMR(400MHz,DMSO-d6)δ10.47(s,1H),8.21(d,J=8.3Hz,1H),8.02-7.92(m,2H),7.81(ddd,J=10.8,7.3,1.8Hz,1H),7.40(td,J=8.4,5.9Hz,1H),7.14(dd,J=11.8,8.4Hz,1H),3.19(d,J=12.2Hz,4H),2.03-1.94(m,4H)
实施例41 N-(4-(吡咯烷-1-基)吡啶-3-基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺的合成
使用3-氯-4-氟苯并[d]异噻唑-1,1-二氧化物和4-(吡咯烷-1-基)吡啶-3-胺作为原料,通过步骤4的合成方法,得到N-(4-(吡咯烷-1-基)吡啶-3-基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺。
1H NMR(400MHz,DMSO-d6)δ8.01(d,J=5.9Hz,1H),7.90(s,1H),7.73(t,J=4.7Hz,3H),7.56(t,J=9.0Hz,1H),3.42(d,J=13.0Hz,4H),1.90-1.82(m,4H)
实施例42 N-(2-(4H-[1,2,4]三氮唑-4-基)苯基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺的合成
使用3-氯-4-氟苯并[d]异噻唑-1,1-二氧化物和2-(4H-[1,2,4]-三氮唑-4-基)苯胺作为原料,通过步骤4的合成方法,得到N-(2-(4H-[1,2,4]三氮唑-4-基)苯基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺。
1H NMR(400MHz,DMSO-d6)δ10.00(s,1H),8.79(s,2H),7.96(td,J=7.7,4.1Hz,1H),7.91(dd,J=7.6,1.0Hz,1H),7.84(dd,J=7.8,1.4Hz,1H),7.75(ddd,J=9.2,8.1,1.0Hz,1H),7.71-7.57(m,3H)
实施例43 N-(2-(1,3-噁嗪烷-2-酮)苯基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺的合成
使用3-氯-4-氟苯并[d]异噻唑-1,1-二氧化物和2-(1,3-噁嗪烷-2-酮)苯胺作为原料,通过步骤4的合成方法,得到N-(2-(1,3-噁嗪烷-2-酮)苯基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺。
1H NMR(400MHz,DMSO-d6)δ9.31(d,J=4.3Hz,1H),7.99-7.90(m,2H),7.87(dd,J=7.9,1.7Hz,1H),7.82-7.70(m,1H),7.57(dd,J=7.8,1.7Hz,1H),7.41(dtd,J=22.4,7.5,1.7Hz,2H),4.31(s,2H),3.84(s,2H),2.05(h,J=5.4Hz,2H).
实施例44 N-(2-(吡咯烷-2,5-二酮)苯基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺的合成
使用3-氯-4-氟苯并[d]异噻唑-1,1-二氧化物和2-(吡咯烷-2,5-二酮)苯胺作为原料,通过步骤4的合成方法,得到N-(2-(吡咯烷-2,5-二酮)苯基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺。
1H NMR(400MHz,DMSO-d6)δ9.35(d,J=5.6Hz,1H),7.95(dd,J=7.0,3.2Hz,2H),7.83(dd,J=8.1,1.5Hz,1H),7.75(ddd,J=9.4,7.0,2.1Hz,1H),7.59(td,J=7.7,1.6Hz,1H),7.49(td,J=7.7,1.5Hz,1H),7.39(dd,J=8.0,1.6Hz,1H),2.88-2.66(m,4H).
实施例45 N-(2-(3,3-二氟吡咯烷-1-基)苯基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺的合成
使用3-氯-4-氟苯并[d]异噻唑-1,1-二氧化物和(2-(3,3-二氟吡咯烷-1-基)苯胺作为原料,通过步骤4的合成方法,得到N-(2-(3,3-二氟吡咯烷-1-基)苯基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺。
1H NMR(400MHz,DMSO-d6)δ9.90-9.84(m,1H),8.03(dd,J=8.0,1.7Hz,1H),7.92(q,J=3.6,2.8Hz,2H),7.82-7.72(m,1H),7.34(dd,J=8.0,1.6Hz,1H),7.22(dtd,J=24.7,7.4,1.6Hz,2H),3.55(t,J=13.0Hz,2H),3.33(t,J=7.0Hz,2H),2.41(dt,J=14.7,7.0Hz,2H).
实施例46 N-(2-(吡咯烷-1-基)-3,5-二氟苯基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺的合成
使用3-氯-4-氟苯并[d]异噻唑-1,1-二氧化物和2-(吡咯烷-1-基)-3,5-二氟苯胺作为原料,通过步骤4的合成方法,得到N-(2-(吡咯烷-1-基)-3,5-二氟苯基)-4-氟苯并[d]异噻唑-1,1-二氧化物-3-胺。
1H NMR(400MHz,DMSO-d6)δ10.46(s,1H),8.04(d,J=8.2Hz,1H),7.99(s,2H),7.82(d,J=9.6Hz,1H),7.21(t,J=10.4Hz,1H),3.17(d,J=6.0Hz,4H),1.99(d,J=5.9Hz,4H).
实施例47化合物对786-O细胞系HIF-2α下游基因转录的影响
1.实验材料
人肾透明细胞癌细胞786-O(鼎国昌盛,CS0254),RPMI1640培养基(凯基生物),trypsin-EDTA(0.25%,Gibco),胎牛血清(Cellmax,SA211.02);Trizol试剂(Takara),反转录试剂盒(Takara),SYBR GREEN荧光定量试剂盒(翌圣生物)。
2.实验方法
荧光定量PCR
2.1细胞培养与给药
将786-O细胞使用含10%胎牛血清的RPMI1640培养基培养于皿中,生长至90%时,将其接种至12孔板。待细胞密度约60%,给药10μM的PT2385或10μM的化合物(DMSO终浓度1‰),培养后收样。
2.2 RNA提取
使用Trizol法抽提RNA,具体步骤如下:
a、样品匀浆:对于12孔板培养的贴壁细胞,移除培养基,用PBS清洗细胞。移除PBS,每孔加入Trizol试剂,吹吸混匀后转移至EP管,室温静置少许,待核蛋白复合物完全解离;
b、相分离:向细胞裂解液中加入氯仿,涡旋混匀并室温静置。4℃,12,000rpm离心10min。此时,样品分为三相,上层水相、中间相和下层酚-氯仿相。此时,RNA全部位于上层水相,将上层水相转移至新EP管中;
c、分离RNA:向EP管中加入异丙醇,涡旋混匀,4℃,12,000rpm离心10min收集RNA沉淀。弃上清,用75%乙醇清洗沉淀两次,4℃,12,000rpm离心8min,弃上清,打开EP管盖室温放置待乙醇挥发,干燥后的RNA为无色透明状。加入无菌水溶解RNA,测量浓度。
2.3反转录
表1反转录体系
2.4荧光定量PCR
表2荧光定量PCR体系
使用ΔΔC
使用qPCR检测化合物对786-O细胞系中HIF-2α下游基因VEGFA和NDRG1转录的影响。
如图1和2所示,本发明化合物能够显著增强VEGFA和NDRG1的转录,其中,化合物1、2、4、5、7、8、10、12、16、17、18、19、20、22、23、27、34、35、36、37、38效果明显,化合物1、5、8、10、16、17、18、19、34、35、36、37、38激动效果显著优于对照化合物。
实施例48化合物对HIF-2α下游基因EPO表达的影响
1.实验材料
人肝细胞癌Hep3B细胞(鼎国昌盛,CS0172),DMEM培养基(凯基生物),trypsin-EDTA(0.25%,Gibco),胎牛血清(Cellmax,SA211.02);酶联免疫吸附试剂盒(恒远生物)。
2.实验方法
酶联免疫吸附测定(enzyme-linked immunosorbent assay,ELISA);
a、将人肝细胞癌Hep3B细胞使用含10%胎牛血清的DMEM培养基培养于皿中,生长至90%时,将其接种至12孔板;
b、待细胞密度约60%,给药10μM的PT2385或10μM的其它化合物(DMSO终浓度1‰),缺氧培养;
c、取培养基,4℃,12,000rpm离心10min,取上清液冰上放置待用;
d、梯度稀释标准品,在酶标包被板上分别加入标准品或待测样品,37℃孵育并洗涤5次;
e、每孔加入酶标试剂50μL,37℃孵育并洗涤;
f、每孔按照ELISA显色剂试剂盒使用手册要求进行定量;
g、使用Excel绘制标准曲线回归方程,计算各样品的EPO浓度。使用GraphPadPrism绘图;数据处理用mean±SEM表示。
使用ELISA检测EPO蛋白的表达。
如图3所示,本发明化合物能够显著提高EPO的蛋白表达,其中,化合物1、2、3、5、6、7、8、9、10、12和14激动活性尤其明显,化合物1、2、5、8和10的激动效果优于对照化合物。
对照化合物:
尽管本发明的具体实施方式已经得到详细的描述,本领域技术人员将会理解,根据已经公开的所有教导,可以对那些细节进行各种修改和替换,这些改变均在本发明的保护范围之内。本发明的全部范围由所附权利要求及其任何等同物给出。
- 一种苯并噻唑醌类化合物及其制备方法和用途
- 一种取代的苯并异噻唑类化合物及其制备方法与用途
- 一种含有异羟肟酸的取代杂环类化合物及其制备方法和用途
- 一种乙酰化二苯并-1,3-二氧杂环辛类化合物的制备方法与作为抗菌药物的应用
- 一类5位环取代的具有苯甘氨醇类结构的2,4-二氨基嘧啶类化合物、其制备及用途
- 一种苯并氮杂环类化合物、其制备方法及用途
- 一种苯并氮杂环类化合物、其制备方法及用途