一种基于病理图像分析热激通路方法
文献发布时间:2024-04-18 19:58:53
技术领域
本发明属于图像处理技术领域,尤其涉及一种基于病理图像分析热激通路方法。
背景技术
基于病理图像分析热激通路对及疾病研究和医疗诊断有重要意义。目前研究表明54℃热刺激会激活促癌通路,从而促进食管鳞癌的发生和转移。然而,这些研究热激通路的发现是基因转录组数据的分析,具有相当的局限性。
发明内容
本发明提供一种基于病理图像分析热激通路方法,能够基于病理图像数据,提取影像组学特征,分析热激通路,以将病理组学特征分类,挖掘病理组学表型表达的潜在生物学意义。
本发明是这样实现的,一种基于病理图像分析热激通路方法,其特征在于,包括以下步骤:
步骤S1:获取组织病理图像数据和转录组测序数据,并划分训练集和测试集;
步骤S2:对组织病理图像数据进行预处理,得到预处理后的组织病理图像数据;
步骤S3:对预处理后的组织病理图像数据进行感兴趣区域ROI分割,提取并筛选病理组学特征,得到预后病理组学特征;构建病理组学标签,并评估病理组学标签的预测性能;
步骤S4:基于转录组测序数据,构建训练集和测试集的基因共表达模块,评估基因共表达模块在组间的保守性,保留高保守性的模块;
步骤S5:对每个保留下来的模块做基于训练集样本的基因集变异分析,以计算患者特定的GSVA评分;
步骤S6:进行Pearson相关性分析,分析各个保留模块的GSVA评分与病理组学标签之间的相关性,并筛选出与病理组学标签显著相关的模块;
步骤S7:对筛选出的模块中基因做通路富集分析,计算通路的GSVA评分,挖掘预后病理组学特征中与每一个特征显著相关的通路,选择相关性最高的通路,将特征进行分类,得到与热激通路相关的病理组学特征。
进一步的,在步骤S3中,对预处理后的组织病理图像数据进行感兴趣区域ROI分割,具体包括:利用Qupath软件在组织病理图像上手动分割感兴趣区域,随后,间隔两个月随机选取病人再次对其组织病理图像的ROI进行分割。
进一步的,在步骤S3中,提取并筛选病理组学特征,具体包括:
通过python从组织病理图像分割的ROI中提取病理组学特征;
计算组内相关系数ICC,来评估观察者内部之间特征提取的再现性,选取ICC大于等于0.85的特征;
计算每个所选特征的一致性指数C-index,来评估与患者总生存期的单变量相关性,选择C-index≥0.55特征,用zscore将特征进行归一化处理;
在训练集中,使用最小绝对收缩和选择算子LASSO-Cox回归算法,选择与预后最相关的病理组学特征,根据10倍交叉验证和最小准则的标准误差调整惩罚参数λ,筛选出系数不为0的与OS最相关的病理组学特征,得到预后病理组学特征。
进一步的,在步骤S3中,构建病理组学标签,并评估病理组学标签的预测性能,具体包括:
在训练集中,根据LASSO-Cox回归算法筛选出的病理组学特征值及其对应系数乘积的总和,构建病理组学标签,即病理组学预后分数:
pathomics score=β
其中,F
采用单因素Cox回归分析,计算C-index量化病理组学标签的性能,并在测试集中进行验证;根据病理组学预后分数的中位数作为截断值,将患者分为高、低风险组,评估截断值对患者生存状态的鉴别能力,并在测试队列中进行验证。
进一步的,在步骤S4中,具体包括:
用绝对中位差前5000且基因表达量不为0的基因,构建加权基因共表达网络;通过网络拓扑分析选择合适的软阈值,利用幂律分布,强化基因之间的强相关性,弱化基因之间的弱相关性;利用R语言中的“WGCNA”包,对训练集中的基因表达数据进行加权基因共表达网络分析,构建训练集和测试集的基因共表达模块;
对模块密度保存和模块连通性保存的加权汇总,得到统计量Zsummary,Zsummary用于评估基因模块在组间的保守性,作为训练集和测试集之间基因模块保存的度量;
训练集中保留测试集中Zsummary≥10的模块。
与现有技术相比,本发明的有益效果是:本发明所提出的一种基于病理图像分析热激通路方法及系统,可以量化大脑区域之间的间接连接并分析其拓扑属性,阐明大脑皮层中的复杂信息传递机制。在应用方面,本发明可以用于评估患者大脑连接的异常变化,为患者的诊断治疗提供指导意见。
附图说明
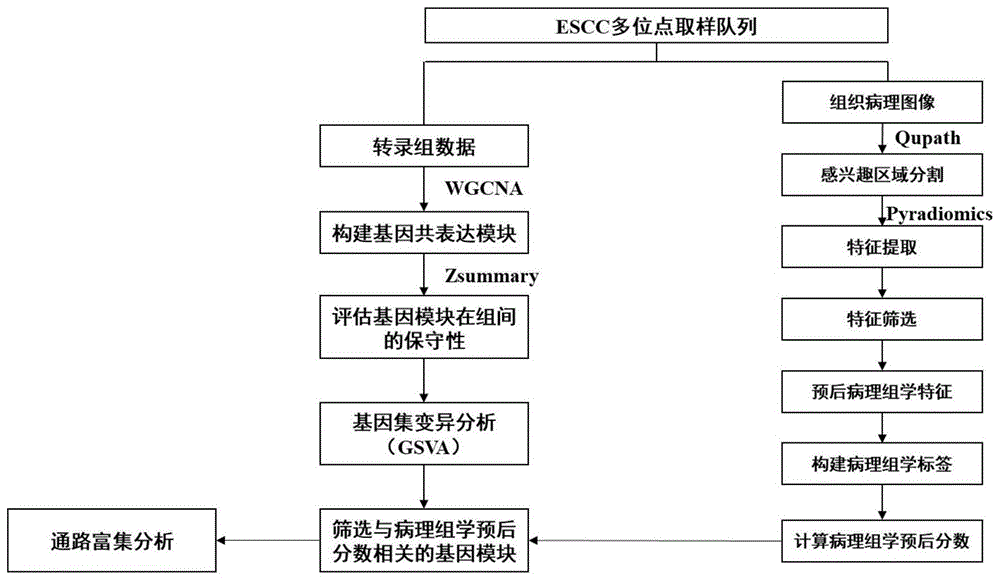
图1为本发明流程示意图;
图2为本发明勾画ROI以及提取特征示意图;
图3为本发明与病理组学显著相关的基因模块示意图;
图4为本发明预后病理组学特征显著相关的五条生物学通路。
具体实施方式
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合附图及实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
请参阅图1,本实施方式中,本发明公开一种基于病理图像分析热激通路方法,包括:
获取组织病理图像和转录组测序数据以及生存信息;
具体地,收集了三个数据集的HE染色的组织病理图像、转录组数据集以及生存信息分别是:
ESCC(Esophagealsquamouscellcarcinoma,食管鳞状细胞癌)多位点取样队列(N=39)作为训练集和测试集;
ESCC单位点取样队列(N=136)作为ESCC外部验证集;
以及癌症基因组图谱(Thecancergenomeatlas,TCGA)中的喉鳞状细胞癌(Laryngealsquamouscellcarcinoma,LSCC)(N=108)作为LSCC外部验证集。
对所述组织病理图像的获取具体包括:将ESCC多位点取样队列中39例食管鳞癌患者的10张组织微阵列芯片,采用常规流程用苏木素和伊红染料对组织芯片进行染色,用KF-PRO-005高分辨率全玻片扫描仪(KFBIO,宁波,中国)以40倍的光学放大率将其扫描后输入计算机;ESCC外部验证集有136张HE染色组织切片;LSCC外部验证集有108张HE染色的WSI;利用MATLABR2016a将三个数据集的组织病理图像转换为统一NII格式,用以特征提取。
使用R软件GenomicFeatures包将基因count值转换为FPKM值。
在一名有丰富阅片经验的病理医生指导下,利用Qupath软件,在组织病理图像上手动分割感兴趣区域。
病理组学特征的提取,通过python从图像分割的ROI中提取特征。
在ESCC多位点取样队列中,为了获得稳定且有预后价值的病理组学特征,采用了以下三个步骤进行筛选:
计算组内相关系数(Intraclasscorrelationcoefficient,ICC),来评估观察者内部之间特征提取的再现性;
计算每个所选特征的一致性指数(Concordanceindex,C-index)来评估与患者总生存期的单变量相关性;
在训练集中,使用最小绝对收缩和选择算子Cox(LeastabsoluteshrinkageandselectionoperatorCox,LASSO-Cox)回归算法,选择与预后最相关的病理组学特征;
病理组学标签的构建及评估,具体包括:
在训练集中,根据LASSO-Cox回归算法筛选出的病理组学特征值及其系数乘积的总和;
构建病理组学标签(Pathomicssignature,PS)也被称为病理组学预后分数;
构建基因共表达模块,具体包括:
利用R语言中“WGCNA”包,对训练集中的基因表达数据进行加权基因共表达网络分析;
保留测试集中高保守性(Zsummary≥10)的模块;
筛选与病理组学标签显著相关(P<0.01)的模块用于进一步分析。
请参阅图1-4,本实施方式中,本发明提供的以病理图像分析热激通路方法包括以下步骤:
S1、获取组织病理图像和转录组测序数据以及生存信息;
S2、对所述病理图像数据进行预处理,划分训练集和测试集;具体的,对病理图像数据进行预处理,划分训练集和测试集的过程如下:
(1)ESCC多位点取样队列(来自2014年至2016年间,经中国山西省肿瘤医院诊断并进行组织检查确诊为ESCC的39名患者,作为训练集和测试集;
(2)ESCC单位点取样队列(来自2017年5月至2018年7月间,经中国山西省肿瘤医院诊断并进行组织检查确诊为ESCC的136名患者)作为ESCC外部验证集,之前的研究中收集了155名ESCC患者,但由于其中19名患者HE染色的病理切片不符合本研究的要求(图像质量差,不能清晰显示感兴趣区域),因此剔除于本研究,剩余136名患者用于本研究中进行分析;
(3)108名来自癌症基因组图谱中的喉鳞状细胞癌患者作为LSCC外部验证集。由于本研究需要匹配的病理图像、转录组测序数据以及生存信息,因此只收集到了这108名患者的相关数据。通过GDCTCGA数据门户下载LSCC外部验证集的WSIs,而相匹配的转录组数据和临床生存信息则通过UCSCXena数据门户下载(https://xena.ucsc.edu/public-hubs/)。本研究经山西医科大学医学伦理委员会批准。从ESCC多位点取样队列的39名患者病例记录中,我们收集了这些患者的年龄、性别、肿瘤位置(中、下)、饮酒史、吸烟史等临床特征,肿瘤分化程度(G1-2,G2,G2-3,G3,其中G1-2划分为G2,G2-3划分为G3)、TNM分期、淋巴结转移信息等病理特征,以及总生存期和生存状态。总生存期是指初始病理诊断日期与死亡日期或随访结束日期之间的间隔。在最后一次随访时,仍然活着的患者或失去随访的患者被视为活着;
S3、组织图像的获取,感兴趣区域ROI的分割,具体的,勾画ROI的过程如下:
利用Qupath(版本0.2.3)软件在组织病理图像上手动分割感兴趣区域(Regionofinterest,ROI),随后,在ESCC多位点取样队列中,随机选取20名病人,间隔两个月,再次对其组织病理图像的ROI进行分割。
病理组学特征的提取、病理组学标签的构建及评估:
在ESCC多位点取样队列中,为了获得稳定且有预后价值的病理组学特征,采用了以下三个步骤进行筛选:
计算组内相关系数(Intraclasscorrelationcoefficient,ICC),来评估观察者内部之间特征提取的再现性。ICC>0.75的特征表示具有良好的再现性。在本研究中,选取了ICC大于等于0.85的特征。
计算每个所选特征的一致性指数(Concordanceindex,C-index)来评估与患者总生存期的单变量相关性,选择C-index≥0.55特征,用zscore将特征进行归一化处理。
在训练集中,使用最小绝对收缩和选择算子Cox(LeastabsoluteshrinkageandselectionoperatorCox,LASSO-Cox)回归算法,选择与预后最相关的病理组学特征。根据10倍交叉验证和最小准则的标准误差调整惩罚参数λ,筛选出系数不为0的与OS最相关的病理组学特征。
在训练集中,根据LASSO-Cox回归算法筛选出的病理组学特征值及其对应系数乘积的总和,构建病理组学标签(Pathomicssignature,PS)也被称为病理组学预后分数:
pathomics score=β
其中,F
为了评估病理组学标签的预测性能,采用单因素Cox回归分析,计算C-index量化病理组学标签的性能,并在测试集中进行验证。根据病理组学预后分数的中位数作为截断值,将患者分为高低风险组。评估截断值对患者生存状态的鉴别能力,并在测试队列中进行验证。
病理图像经过Qupath软件手动分割ROI后,使用Python软件提取了共945个病理组学特征。随后,从945个病理组学特征中,共筛选出ICC≥0.85的864特征;通过单因素预后分析,筛选出31个与预后相关的病理组学特征,用z-score对其进行归一化;最后,使用LASSO-Cox算法从31个特征中筛选与OS最相关的病理组学特征,当lambda.1se为0.001时,筛选出21个系数不为0的病理组学特征,这21个病理组学特征的系数就是后面的预后病理组学特征。
S4、构建基因共表达模块,具体包括:
利用R语言中的“WGCNA”包,对训练集中的基因表达数据进行加权基因共表达网络分析(Weightedgeneco-expressionnetworkanalysis,WGCNA);
用绝对中位差(Medianabsolutedeviation,MAD)前5000且基因表达量不为0的基因,构建加权基因共表达网络;
通过网络拓扑分析选择合适的软阈值,利用幂律分布,强化基因之间的强相关性,弱化基因之间的弱相关性,使得基因共表达网络中的基因之间的关系更符合无尺度网络分布;
基于基因表达矩阵和最佳软阈值,利用blockwiseModule函数构建基因共表达网络和模块划分,用sft$powerEstimate确定最佳软阈值β为9,在此基础上用blockwiseModule函数构建训练集和测试集的基因共表达模块;
在训练集中,得出了15个基因模块;在测试集中,得到了20个基因模块。
如图3所示,在训练集的15个模块中,有9个模块的Zsummary大于10,这些模块被认为是两个数据集之间高度保存的模块。9个模块中,有两个基因模块与病理组学标签显著相关,分别是图3中的上方圈出的模块一(r=0.32,P=6.03×10-4)和下方圈出的模块二(r=0.40,P=1.20×10-5)。
模块一中有1040个基因,模块二中有318个基因,Zsummary越大,表明该模块在训练集和测试集之间的保存程度越高。因此,选择图3中的模块一(Zsummary=32)为进一步研究的关键模块;
统计量Zsummary评估基因模块在组间的保守性,作为训练集和测试集之间基因模块保存的度量,Zsummary是模块密度保存(评估基因在测试集中是否保持高度连接)和模块连通性保存(评估基因在测试集是否保持连通性模式)的加权汇总。
其中,训练集中保留了测试集中高保守性(Zsummary≥10)的模块,丢弃了其他保守性较差的模块。
S5、对每个保留下来的基因模块做基于训练集样本的基因集变异分析(Genesetvariationanalysis,GSVA)以计算患者特定的GSVA评分。
S6、通过Pearson相关性分析,分析各个保留模块的GSVA评分与病理组学标签之间的相关性。筛选出与病理组学标签显著相关(P<0.01)的模块被用进一步分析。
S7、选择图3中的模块一为关键模块,随后对该模块中的基因做通路富集分析,计算通路的GSVA评分,挖掘了这21个预后病理组学特征中与每一个特征显著相关的五条通路,选择相关性最高的三条通路,将特征按照这三条通路中比例最高的通路进行分类,其中发现了3个病理组学特征和热激通路相关。
具体的,如图4所示,挖掘了这21个预后病理组学特征中与每一个特征显著相关的五条通路,选择相关性最高的三条通路(p<0.05),将特征按照这三条通路中比例最高的通路进行分类,将特征分为了三个分子类型:增殖、热激和代谢,揭示了病理组学特征潜在的生物学意义;
每条通路中不同病人GSVA评分的差异和不同病人的病理组学预后分数的差异,表明肿瘤个体间在基因组学和表型方面存在异质性;更重要的是,每条通路中每个病人不同肿瘤区域GSVA评分的差异和每个病人不同肿瘤区域病理组学预后分数的差异,表明同一个肿瘤内的不同区域在基因组学和表型方面也存在异质性。
以上仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
- 病理组织图像分析设备、病理组织图像分析方法和病理组织图像分析程序
- 病理组织图像分析设备、病理组织图像分析方法和病理组织图像分析程序