有利的抗HCV联合疗法
文献发布时间:2024-04-18 20:01:23
相关申请的交叉引用
本申请主张2021年6月17日提交的美国临时申请第63/212,047号的权益。该申请的全部内容在此引入作为参考。
技术领域
本发明是特定NS5B聚合酶抑制剂和特定NS5A抑制剂的药物组合,用于有利且协同的HCV治疗。
背景技术
丙型肝炎(HCV)是一种RNA单链病毒,是肝炎病毒属的成员。据估计,大多数肝病病例由HCV引起。HCV感染可导致肝硬化和肝癌,且如果任其发展,可能导致需要进行肝移植的肝衰竭。
RNA聚合酶是抗RNA单链病毒的药物开发的关键靶标。HCV非结构性蛋白NS5B RNA依赖性RNA聚合酶是负责启动和催化病毒RNA合成的关键酶。NS5B抑制剂存在两个主要亚类:核苷类似物和非核苷抑制剂(NNI)。核苷类似物被合成代谢为活性三磷酸盐,其作为聚合酶的替代底物。非核苷抑制剂(NNIs)与蛋白质上的变构区域结合。核苷或核苷酸抑制剂模拟天然的聚合酶底物和起到链终止剂的作用。它们抑制RNA转录的启动和/或新生RNA链的延长。
除了靶向RNA聚合酶,其他RNA病毒蛋白也可以被靶向用于治疗。作为治疗方法的额外靶标的HCV蛋白的例子有NS3/4A(一种丝氨酸蛋白酶)和NS5A(一种非结构性蛋白,其是HCV复制酶的重要组分且对细胞途径发挥一系列作用)。
2013年12月,首个核苷NS5B聚合酶抑制剂索非布韦(
是首个显示治疗特定类型HCV感染的安全性和有效性而无需共施用干扰素的药物。/>
许多固定剂量的药物组合已被批准用于治疗HCV。2014年,美国FDA批准
美国FDA还于2014年批准了AbbVie的VIEKIRA Pak
2015年7月,美国FDA分别批准Technivie
2017年8月,
与
HCV的其他经批准的疗法包括干扰素α-2b或聚乙二醇化的干扰素α-2b(
描述用于治疗黄病毒科(包括HCV)的核苷聚合酶抑制剂的美国专利和WO申请包括Idenix Pharmaceuticals(6,812,219;6,914,054;7,105,493;7,138,376;7,148,206;7,157,441;7,163,929;7,169,766;7,192,936;7,365,057;7,384,924;7,456,155;7,547,704;7,582,618;7,608,597;7,608,600;7,625,875;7,635,689;7,662,798;7,824,851;7,902,202;7,932,240;7,951,789;8,193,372;8,299,038;8,343,937;8,362,068;8,507,460;8,637,475;8,674,085;8,680,071;8,691,788,8,742,101,8,951,985;9,109,001;9,243,025;US2016/0002281;US2013/0064794;WO/2015/095305;WO/2015/081133;WO/2015/061683;WO/2013/177219;WO/2013/039920;WO/2014/137930;WO/2014/052638;WO/2012/154321);Merck(6,777,395;7,105,499;7,125,855;7,202,224;7,323,449;7,339,054;7,534,767;7,632,821;7,879,815;8,071,568;8,148,349;8,470,834;8,481,712;8,541,434;8,697,694;8,715,638,9,061,041;9,156,872和WO/2013/009737);Emory University(6,348,587;6,911,424;7,307,065;7,495,006;7,662,938;7,772,208;8,114,994;8,168,583;8,609,627;US 2014/0212382;和WO2014/1244430);Gilead Sciences/PharmassetInc.(7,842,672;7,973,013;8,008,264;8,012,941;8,012,942;8,318,682;8,324,179;8,415,308;8,455,451;8,563,530;8,841,275;8,853,171;8,871,785;8,877,733;8,889,159;8,906,880;8,912,321;8,957,045;8,957,046;9,045,520;9,085,573;9,090,642;和9,139,604)及(6,908,924;6,949,522;7,094,770;7,211,570;7,429,572;7,601,820;7,638,502;7,718,790;7,772,208;RE42,015;7,919,247;7,964,580;8,093,380;8,114,997;8,173,621;8,334,270;8,415,322;8,481,713;8,492,539;8,551,973;8,580,765;8,618,076;8,629,263;8,633,309;8,642,756;8,716,262;8,716,263;8,735,345;8,735,372;8,735,569;8,759,510和8,765,710);Hoffman La-Roche(6,660,721),(6,784,166;7,608,599,7,608,601和8,071,567);Alios BioPharma Inc.(8,895,723;8,877,731;8,871,737,8,846,896,8,772,474;8,980,865;9,012,427;US 2015/0105341;US2015/0011497;US2010/0249068;US2012/0070411;WO 2015/054465;WO 2014/209979;WO 2014/100505;WO2014/100498;WO 2013/142159;WO 2013/142157;WO 2013/096680;WO 2013/088155;WO2010/108135),Enanta Pharmaceuticals(US 8,575,119;8,846,638;9,085,599;WO 2013/044030;WO 2012/125900),Biota(7,268,119;7,285,658;7,713,941;8,119,607;8,415,309;8,501,699和8,802,840),Biocryst Pharmaceuticals(7,388,002;7,429,571;7,514,410;7,560,434;7,994,139;8,133,870;8,163,703;8,242,085和8,440,813),Alla Chem,LLC(8,889,701和WO 2015/053662),Inhibitex(8,759,318和WO/2012/092484),JanssenProducts(8,399,429;8,431,588,8,481,510,8,552,021,8,933,052;9,006,29和9,012,428);University of Georgia Foundation(6,348,587;7,307,065;7,662,938;8,168,583;8,673,926,8,816,074;8,921,384和8,946,244),RFS Pharma,LLC(8,895,531;8,859,595;8,815,829;8,609,627;7,560,550;US 2014/0066395;US2014/0235566;US2010/0279969;WO/2010/091386和WO 2012/158811)University College Cardiff ConsultantsLimited(WO/2014/076490,WO 2010/081082;WO/2008/062206),AchillionPharmaceuticals,Inc.(WO/2014/169278和WO 2014/169280),Cocrystal Pharma,Inc.(US9,173,893),Katholieke Universiteit Leuven(WO 2015/158913),Catabasis(WO 2013/090420)及Regents of the University of Minnesota(WO 2006/004637)提交的那些。
艾尔巴韦(Elbasvir)是FDA于2016年批准作为与格佐匹韦(grazoprevir)(一种NS3/4A蛋白酶抑制剂)的固定剂量组合的组分的HCV NS5A抑制剂。该组合(
Ruzasvir(MK-8408)是一种经临床评估用于治疗慢性HCV感染的口服泛基因型NS5A抑制剂。Ruzasvir对HCV NS5A(在广泛的HCV基因型和常见的临床变异体中)具有亚至低皮摩尔的亲和力,包括具有Y93H、Q30R、L31V和Y93C突变的HCV基因型1(Tong等,“Discovery of Ruzasvir(MK-8408):A Potent,Pan-Genotype HCV NS5A Inhibitor withOptimized Activity against Common Resistance-Associated Polymorphisms”,J.Med.Chem.2017,60,290-306)。转让给Merck and Company的WO 2014/110705和美国专利9,555,038号公开了ruzasvir、包含ruzasvir的药物组合物及其使用方法。
2015年,Merck,Sharp&Dohme发起了临床试验以确定格佐匹韦和乌普立布韦(uprifosbuvir)与艾尔巴韦或rusazvir组合对一系列HCV基因型的患者的疗效。这一种三药物方案在感染一系列HCV基因型的85-100%的患者中,在12周后获得了持续的病毒学反应(参见,例如,NCT02332707、NCT02332720,Lawitz,E.“Safety and efficacy of afixed-dose combination regimen of grazoprevir,ruzasvir and uprifosbuvir withor without ribavirin in participants with and without cirrhosis with chronichepatitis C virus genotype 1,2,or 3infection(C-CREST-1and C-CREST-2,part B):two randomized,phase 2,open-label trials”2017,Lancet Gastroenterol Hepatol,doi:10.1016/S2468-1253(17)30163-2)。格佐匹韦,三药物方案的一个组分,是NS3/4A蛋白酶抑制剂。这一种类的治疗剂可导致药物-药物相互作用和肝转氨酶水平升高。
2016年,默克启动了在不含NS3/4A抑制剂的两药物方案中ruzasvir和NS5B抑制剂乌普立布韦的二期开放标签临床试验。在每天研究剂量60mg ruzasvir和450mg乌普立布韦的情况下,该组合良好耐受,但观察到亚最佳效果,临床试验终止(C-BREEZE 1;NCT02759315)。在HCV GT1、GT2、GT4、GT5和GT6感染的参与者中,相同的组合但在更高的剂量(180mg ruzasvir、450mg乌普立布韦)下获得了超过90%的持续病毒学应答。然而,GT3感染的参与者中的反应仅为73.8%。此外,超过30%的试验参与者经历了与药物相关的不良事件(C-BREEZE 2;NCT02956629;Lawitz,E.“Efficacy and Safety of Two-Drug Direct-Acting Antiviral Agent Regimen Ruzasvir 180mg and Uprifosbuvir 450mg for12Weeks in Adults with Chronic Hepatitis C Virus Genotype 1,2,3,4,5,or 6”2019,J.Viral Hepat.26,9,1127-1138)。该第二,高剂量试验也被终止。
Atea Pharmaceuticals,Inc.已发现结构β-D-2’-去氧-2’-α-氟-2’-β-C-取代-2-修饰-N
在I期试验的多剂量组中,bemnifosbuvir在非肝硬化参与者使病毒负荷降低4.5log10IU/mL,和在肝硬化参与者中使病毒负荷降低4.6log10IU/mL。在肝硬化患者组中,其对包括难以治疗的GT3的一系列基因型同样有效(GT1b:4.0、4.0、4.5;GT2:5.0;GT3:4.8,5.2)(NCT 03219957;Berliba,E.et al.“Safety,Pharmacokinetics,and AntiviralActivity of AT-527,a Novel Purine Nucleotide Prodrug,in Hepatitis CVirus-Infected Subjects with or without Cirrhosis”2019,Antimicrob Agents Chemother,63(12):e01201-19)。
在bemnifosbuvir和第一代HCV NS5A抑制剂达卡他韦的联合试验中,所有10名受试者达到低于定量下限的HCV RNA。90%的受试者在治疗后12周出现持续的无法检测的病毒负荷(SVR12)。一名未经历SVR12的受试者存在一系列抗性相关变体(NS5A:R30Q,NS5B:L159F/A218S/C316N)(NCT04019717);Mungur,O.et al.“A combination of AT-527,apotent pan-genotypic guanosine nucleotide prodrug and daclatasvir was well-tolerated and effective in HCV-infected subjects”,2020,poster THU438 at TheInternational Liver Congress meeting)。但是,该试验终止。
仍然存在着对于开发安全、有效和良好耐受的抗HCV治疗的强烈医疗需求。潜在的药物抗性使得这一需求更加突出。HCV RNA聚合酶表现出高复制率,这造成在整个基因组中产生潜在抗性的单点和双点突变,并维持病毒准种。在用几乎所有单一疗法治疗时,在体外和体内均鉴别了抗性突变。
因此,本发明的目的是提供治疗和/或预防HCV感染的化合物、药物组合物、方法和剂型。
发明内容
本发明提供了化合物1(其为有效的泛基因NS5B聚合酶抑制剂)或其药学上可接受的盐(例如AT-527,bemnifosbuvir)与化合物2(其为NS5A抑制剂)或其药学上可接受的盐(ruzasvir,MK-8408)的高度协同组合用于治疗宿主(通常为人类)的丙型肝炎感染。
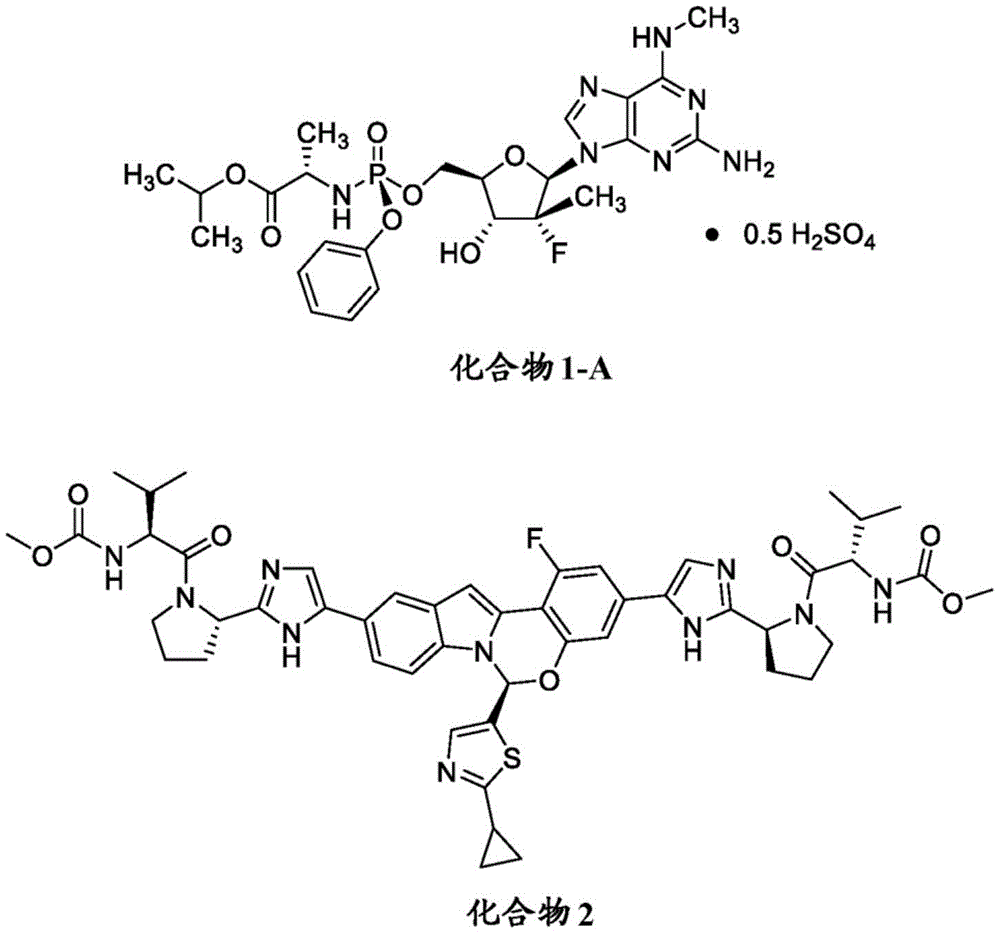
化合物1为异丙基(((2R,3R,4R,5R)-5-(2-氨基-6-(甲基氨基)-9H-嘌呤-9-基)-4-氟-3-羟基-4-甲基四氢呋喃-2-基)甲氧基)(苯氧基)磷酰基)-L-丙氨酸酯:
化合物1先前在美国专利号9,828,410;10,000,523;10,005,811;10,239,911;10,815,266;10,80,672;10,870,673;10,875,885;及转让给Atea Pharmaceuticals的PCT申请WO 2016/21276和WO 2019/200005中描述。
化合物1的半硫酸盐如下显示为化合物1-A:
美国专利号10,519,186;10,906,928;10,894,804;及转让给AteaPharmaceuticals的PCT申请WO 2018/144640和WO 2019/200005中公开了化合物1-A。
化合物2为ruzasvir(二甲基N,N’-([(6S)-6-(2-环丙基-1,3-噻唑-5-基)-1-氟-6H-吲哚并[1,2-c][1,3]苯并噁嗪-3,10-二基]双{1H-咪唑-5,2-二基-(2S)-吡咯烷-2,1-二基[(2S)-3-甲基-1-氧代丁烷-1,2-二基]}二氨基甲酸酯):
在一个实施方案中,化合物2是无定形固体。在又一实施方案中,化合物2是结晶固体。在一个实施方案中,化合物2作为其药学上可接受的盐施用。WO 2014/110705和美国专利号9,555,038中公开了化合物2。
先前ruzasvir与NS5B抑制剂乌普立布韦组合的临床试验因无效而终止。在此背景下,发现化合物1(例如AT-527)与ruzasvir的组合在多个剂量下测试时高度协同(参见实施例30和图34)。在每一组合剂量下,从实验观察的抗病毒活性减去预期的加性抗病毒保护,从而得到对于协同作用的正值、对于加性作用的零值或对于拮抗作用的负值。-50至50μM
以不同的作用机制共同作用的两种抗HCV药物的协同组合可作为两个或更多个单独的剂型或作为组合剂型系统地提供,例如口服。如果单独施用,药物的提供方式应使宿主受益于以协同生物学方式发挥作用的两种活性药物,例如,以实现重叠的药代动力学、血浆和/或AUC暴露的方式。为了有效治疗病毒,联合药物治疗在限制耐药性的出现方面尤其有利。
在一个非限制性实施方案中,化合物1以半硫酸盐提供。
在一个实施方案中,该固定剂量组合旨在于约或少于12周(例如约或少于10周、8周或6周或更短)内实现持续的病毒反应。除了有效治疗病毒外,联合药物治疗有助于限制耐药性的发生。
除非另有明确说明,否则本文所述剂型中活性化合物的重量为相对于化合物的游离形式或盐形式。例如,600mg化合物1-A等同于550mg化合物1。
在典型的实施方案中,化合物1的施用剂量为约300-1000mg(无论是否关于盐的重量),更典型地400或500至600或800mg,或500至750mg。在某些实施方案中,化合物1以约500至约1500mg的剂量施用。在一个实例中,550mg的化合物1以约600mg化合物1-A的剂量施用。在替代实施方案中,以约1200mg化合物1-A的剂量施用1100mg化合物1。
在典型的实施方案中,化合物2或其药学上可接受的盐以约20-500mg,更典型地为40-250mg的剂量施用,例如,包括但不限于剂型中至少60、70、75、100、125、150、180、200、225、250、270、300、350或400mg。在某些实施方案中,化合物2以包含至少约90、180、270或360mg的剂型施用。
在某些实施方案中,该组合包括550mg化合物1-A和约至少90、180、270或360mg化合物2或其药学上可接受的盐(例如180mg)。在某些实施方案中,该组合每天提供一次、两次或三次。
根据保健实施人员的建议,组合治疗剂可以一天施用一次、两次或三次或者更多次。在某些实施方案中,每天提供一次组合治疗剂。在其他实施方案中,组合治疗剂每天提供两次。在又一实施方案中,组合治疗剂一天提供三次。
在某些实施方案中,化合物1-A以600mg/天的固体剂量提供,化合物2以180mg/天在组合或单独的剂型中提供,两者一起可以每天给予1次、2次或3次。
在各个方面,化合物1或其药学上可接受的盐和化合物2或其药学上可接受的盐一起配制成单一剂型或以多个剂型提供(例如,两个或多个剂型,每一个具有两种活性物质,或其中一种剂型具有一种活性剂,另一种剂型具有另一种活性剂)。在替代实施方案中,化合物1或其药学上可接受的盐和化合物2或其药学上可接受的盐以单独的剂型提供,但其可以在宿主中协同地,例如协同性地发挥作用。例如,可以施用单独的剂型,使得存在重叠的AUC或其他药代动力学参数,这表明活性物质共同作用对抗病毒。
在本发明的一个方面,化合物1和化合物2以单独的药丸提供,并且在一天的整个过程中几乎在相同时间或同时施用。
化合物1(或其药学上可接受的盐,例如化合物1-A)和化合物2(或其药学上可接受的盐)的组合也可用于治疗相关病症,例如抗HCV抗体阳性和抗原阳性病症、基于病毒的慢性肝脏炎症、由晚期丙型肝炎导致的肝脏癌症(肝细胞癌(HCC))引起的肝癌、肝硬化、慢性或急性丙型肝炎、暴发性丙型肝炎、慢性持续性丙型肝炎和抗HCV基疲劳。
在某些实施方案中,化合物1或其药学上可接受的盐,例如化合物1-A,和化合物2或其药学上可接受的盐被施用最多24周、最多12周、最多10周、最多8周、最多6周或最多4周。在替代实施方案中,化合物1或其药学上可接受的盐(例如化合物1-A)和化合物2或其药学上可接受的盐被施用至少4周、至少6周、至少8周、至少10周、至少12周或至少24周。在某些实施方案中,化合物1或其药学上可接受的盐和化合物2或其药学上可接受的盐每天或每隔一天至少施用一次。在替代实施方案中,化合物1或其药学上可接受的盐和化合物2或其药学上可接受的盐每天施用两次。在替代实施方案中,化合物1或其药学上可接受的盐和化合物2或其药学上可接受的盐每天施用三次或更多次。
在某些实施方案中,患者是非肝硬化的。在某些实施方案中,患者为肝硬化的。在进一步实施方案中,肝硬化宿主具有代偿性肝硬化。在替代实施方案中,肝硬化宿主具有失代偿性肝硬化。在一个实施方案中,宿主具有Child-Pugh A肝硬化。在替代实施方案中,宿主具有Child-Pugh B或Child-Pugh C肝硬化。
上述组合也可用于治疗一系列HCV基因型。全球范围内已鉴定至少六种不同的HCV基因型,其每一种均具有多种亚型。基因型1-3在全球范围内普遍存在,而基因型4、5和6在地理上更为局限。基因型4在中东和非洲很常见。基因型5主要在南非发现。基因型6主要存在于东南亚。尽管在美国最常见的基因型是基因型1,但确定基因型和亚型可能有助于治疗类型和持续时间。例如,不同的基因型对不同药物的反应不同。最佳治疗时间因基因型感染而不同。在基因型内,亚型如基因型1a和基因型1b对治疗的反应也可能不同。一种基因型的感染并不排除不同基因型的随后感染。
在一个实施方案中,化合物1或其药学上可接受的盐(例如化合物1-A)和化合物2或其药学上可接受的盐的组合用于治疗HCV基因型1、HCV基因型2、HCV基因型3、HCV基因型4、HCV基因型5或HCV基因型6。在一个实施方案中,化合物1或其药学上可接受的盐和化合物2或其药学上可接受的盐用于治疗HCV基因型1a。在一个实施方案中,化合物1或其药学上可接受的盐和化合物2或其药学上可接受的盐用于治疗HCV基因型1b。在一个实施方案中,化合物1或其药学上可接受的盐和化合物2或其药学上可接受的盐用于治疗HCV基因型2a。在一个实施方案中,化合物1或其药学上可接受的盐和化合物2或其药学上可接受的盐用于治疗HCV基因型2b。在一个实施方案中,化合物1或其药学上可接受的盐和化合物2或其药学上可接受的盐用于治疗HCV基因型3a。在一个实施方案中,化合物1或其药学上可接受的盐和化合物2或其药学上可接受的盐用于治疗HCV基因型3b。在一个实施方案中,化合物1或其药学上可接受的盐和化合物2或其药学上可接受的盐用于治疗HCV基因型4a。在一个实施方案中,化合物1或其药学上可接受的盐和化合物2或其药学上可接受的盐用于治疗HCV基因型4d。在一个实施方案中,化合物1或其药学上可接受的盐和化合物2或其药学上可接受的盐用于治疗HCV基因型5a。在一个实施方案中,化合物1或其药学上可接受的盐和化合物2或其药学上可接受的盐用于治疗HCV基因型6a。在一个实施方案中,化合物1或其药学上可接受的盐和化合物2或其药学上可接受的盐用于治疗HCV基因型6b、6c、6d、6e、6f、6g、6h、6i、6j、6k、6l、6m、6n、6o、6p、6q、6r、6s、6t或6u。
本发明还包括特定的组合和剂型,其中化合物1-A可以是非晶或结晶盐的形式,并且独立地,化合物2可以是晶体或非晶体。
因此,本发明至少包括以下实施方案:
(a)化合物1或其药学上可接受的盐与化合物2或其药学上可接受的盐的有效抗HCV组合;
(b)化合物1或其药学上可接受的盐与化合物2或其药学上可接受的盐的组合的有效固体剂型;
(c)实施方案(a)或(b),其中化合物1是化合物1-A;
(d)实施方案(a)-(c),其中在组合中使用第三抗HCV有效药剂;
(e)实施方案(d),其中第三抗HCV有效药剂通过不同于化合物1、化合物1-A或化合物2的机制发挥作用;
(f)一种药物组合物,其包含在药学上可接受的赋形剂中实施方案(a)-(e)中任一项的组合。
(g)实施方案(f),其中所述组合为组合药物组合物的形式;
(h)实施方案(f),其中所述组合为每种活性剂的单独药物剂型,其以协同方式使用;
(i)(f)-(h)的药物剂型,其适合口服递送;
(j)(i)的剂型,其为丸剂、片剂或凝胶形式;
(k)(f)-(h)的药物剂型,其适合于肠胃外递送;
(l)(f)-(h)的药物剂型,其适用于静脉内递送;
(m)实施方案(a)-(l)中任一项的有效组合在制备用于治疗有需要的患者的丙型肝炎病毒感染的药物中的用途;
(n)一种制备旨在用于治疗需要的患者的丙型肝炎病毒感染的药物的方法,其特征在于在制备中使用实施方案(a)-(l)中任一项的有效组合;
(o)一种治疗丙型肝炎病毒感染的方法,包括向需要的患者施用实施方案(a)-(l)中任一项的有效组合;
(p)一种治疗丙型肝炎病毒感染的方法,包括向需要的患者施用实施方案(a)-(l)中任一项的有效组合;
(q)一种用于预防性治疗具有丙型肝炎病毒感染风险的患者的方法,包括向需要的患者施用实施方案(a)-(l)中任一项的有效组合;
(r)一种治疗与丙型肝炎病毒感染相关的病症的方法,所述病症选自基于病毒的慢性肝脏炎症、由晚期丙型肝炎导致的肝脏癌症(肝细胞癌(HCC))、肝硬化、慢性或急性丙型肝炎、暴发性丙型肝炎、慢性持续性丙型肝炎和抗HCV基疲劳,该方法包括对需要的患者施用实施方案(a)-(l)中任一项的有效组合;
(s)实施方案(m)-(r)中任一项,其中患者为肝硬化的。
(t)实施方案(m)-(r)中任一项,其中患者为非肝硬化的。
(u)实施方案(m)-(t)中任一项,其中HCV感染为基因型1。
(v)实施方案(m)-(t)中任一项,其中HCV感染为基因型2。
(w)实施方案(m)-(t)中任一项,其中HCV感染为基因型3。
(x)实施方案(m)-(t)中任一项,其中HCV感染为基因型4。
(y)实施方案(m)-(t)中任一项,其中HCV感染为基因型5。
(z)实施方案(m)-(t)中任一项,其中HCV感染为基因型6。
(aa)(a)-(l)的组合,用于治疗HCV或HCV相关病症。
在本发明中,化合物1-A可以以结晶形式在协同组合治疗中提供,或者可选地,结晶形式可以用于喷雾干燥制造程序。图1-15描述了由XRPD、DSC和TGA对化合物1-A物理形式的表征。化合物1-A可以结晶形式分离,这有助于合成和加工。
化合物1-A的药代动力学对组合治疗的成功是重要的。图16描述了化合物1-A的有利生物分布特性。相比于心脏,该化合物集中于肝脏(HCV感染的目标器官)。由于肝浓度高,有利于肝脏组合治疗。
同样,图17、18和21-23描述了化合物1-A和主要代谢物的药代动力学性质。尽管化合物1-A在8小时内发生代谢,但活性代谢物最多可存在24小时。在代谢物存在的24小时期间内,观察到HCV病毒RNA的抑制。在300mg的剂量下,化合物1-A代谢物的血药浓度高于HCVGT1b的EC
图19和图20显示了化合物1-A的泛基因型效力。该组合治疗中的泛基因型活性可防止抗性HCV突变体的发展。
图25-31描绘了施用化合物1的肝硬化宿主中的化合物1代谢物和HCV病毒RNA水平。图27突出显示,在宿主中,化合物1在肝硬化患者中与在非肝硬化患者同等有效。化合物1或其药学上可接受的盐与化合物2或其药学上可接受的盐的组合可用于治疗肝硬化和非肝硬化患者的HCV。包含化合物1或其药学上可接受的盐和化合物2或其药学上可接受的盐的药物组合物可被配制成口服剂型。图32描述了可以制备用于口服给药的片剂的示例过程。化合物1或其药学上可接受的盐和化合物2或其药学上可接受的盐组合在一起,过筛,混合并压片以提供作为口服剂型的组合。
附图说明
图1A是样品1-1(非晶化合物1)、1-2(结晶化合物1)和1-3(非晶化合物1-A)在稳定性研究之前XRPD衍射图的叠加图,其用于实施例2和实施例5中所述的表征目的。x轴为以度测量的2θ,y轴为以计数测量的强度。
图1B是非晶化合物1(样品1-1)的HPLC色谱图,用于确定实施例2中所述的纯度。样品的纯度为98.7%。x轴为以分钟计的时间,y轴为以计数测量的强度。
图2A是结晶化合物1(样品1-2)的HPLC色谱图,用于确定实施例2中所述的纯度。样品的纯度为99.11%。x轴为以分钟计的时间,y轴为以计数测量的强度。
图2B为用于实施例2中所述的表征目的在任何稳定性研究之前的结晶化合物1(样品1-2)的DSC和TGA图。x轴为以℃测量的温度,左侧y轴为以(W/g)测量的热流,右侧y轴为以百分比测量的重量。
图3是化合物1的X射线晶体学图像,显示了实施例2中所述的绝对立体化学。
图4A是样品1-1(非晶化合物1)、1-2(结晶化合物1)和1-3(非晶化合物1-A)在25℃和60%相对湿度下储存14天后的XRPD衍射图的叠加图,如实施例2和实施例5所述。x轴为以度计的2θ,y轴为以计数测量的强度。
图4B是样品1-4、1-5、1-6、1-7和1-9在25℃和60%相对湿度下储存7天后的XRPD衍射图的叠加图,如实施例4所述。x轴为以度计的2θ,y轴为以计数测量的强度。
图5A是样品1-4、1-6、1-7和1-9在25℃和60%相对湿度下储存14天后的XRPD衍射图的重叠图,如实施例4所述。x轴为以度计的2θ,y轴为以计数测量的强度。
图5B是如实施例5中所述的非晶化合物1-A(样品1-3)的XRPD图谱。x轴为以度计的2θ,y轴为以计数测量的强度。
图6A是非晶化合物1-A(样品1-3)的HPLC色谱图,用于确定实施例5中所述的纯度。样品的纯度为99.6%。x轴以分钟测量的时间,y轴以计数测量的强度。
图6B是用于实施例5中所述表征目的在任何稳定性研究之前,非晶化合物1-A(样品1-3)的DSC和TGA图。x轴为以℃测量的温度,左侧y轴为以(W/g)测量的热流,右侧y轴为以百分比测量的重量。
图7A是从化合物1-A的结晶(实施例6)鉴定的结晶样品(样品2-2、2-6和2-7)和不良结晶样品(样品2-3、2-4、2-5和2-8)的XRPD衍射图的重叠图。x轴为以度计的2θ,y轴为以计数测量的强度。
图7B是从化合物1-A的结晶(实施例6)中鉴定的非晶样品(样品2-9、2-10和2-11)的XRPD衍射图的重叠图。x轴为以度计的2θ,y轴为以计数测量的强度。
图8A是在25℃和60%相对湿度下储存6天后样品(样品2-2、2-3、2-4、2-5、2-6、2-7和2-8)的XRPD衍射图的重叠图(实施例6)。x轴为以度计的2θ,y轴为以计数测量的强度。
图8B是样品2-2的DSC和TGA图(实施例6)。x轴为以℃测量的温度,左侧y轴为以(W/g)测量的热流,右侧y轴为以百分比测量的重量。实施例2中给出了DSC和TGA收集的实验程序。
图9A是样品2-3的DSC和TGA图(实施例6)。x轴为以℃测量的温度,左侧y轴为以(W/g)测量的热流,右侧y轴为以百分比测量的重量。实施例2中给出了DSC和TGA收集的实验程序
图9B是样品2-4的DSC和TGA图(实施例6)。x轴为以℃测量的温度,左侧y轴为以(W/g)测量的热流,右侧y轴为以百分比测量的重量。实施例2中给出了DSC和TGA收集的实验程序
图10A是样品2-5的DSC和TGA图(实施例6)。x轴为以℃测量的温度,左侧y轴为以(W/g)测量的热流,右侧y轴为以百分比测量的重量。实施例2中给出了DSC和TGA收集的实验程序
图10B是样品2-6的DSC和TGA图(实施例6)。x轴为以℃测量的温度,左侧y轴为以(W/g)测量的热流,右侧y轴为以百分比测量的重量。实施例2中给出了DSC和TGA收集的实验程序
图11A是样品2-7的DSC和TGA图(实施例6)。x轴为以℃测量的温度,左侧y轴为以(W/g)测量的热流,右侧y轴为以百分比测量的重量。实施例2中给出了DSC和TGA收集的实验程序
图11B是样品2-8的DSC和TGA图(实施例6)。x轴为以℃测量的温度,左侧y轴为以(W/g)测量的热流,右侧y轴为以百分比测量的重量。实施例2中给出了DSC和TGA收集的实验程序
图12A是实施例7中讨论的非晶化合物1-B(样品3-12)的XRPD图谱。x轴为以度计的2θ,y轴为以计数测量的强度。无论使用何种溶剂,均未观察到丙二酸盐的结晶。
图12B是从化合物1与丙二酸盐的尝试结晶(实施例7)鉴定的非晶样品(样品3-6、3-10、3-11和3-12)的XRPD衍射图的重叠图。x轴为以度计的2θ,y轴为以计数测量的强度。
图13A是来自实施例7中所述的化合物1与丙二酸盐的尝试结晶的样品3-12的HPLC色谱图。样品纯度为99.2%。x轴为以分钟测量的时间,y轴以mAu为单位的强度。
图13B是如实施例8中所述使用LAG的结晶获得的固体样品(样品4-13、4-12、4-9、4-3和4-1)与化合物1(样品1-2)相比的XRPD衍射图的重叠图。所有XRDP匹配无额外峰的晶体酸反离子的图谱。x轴为以度计的2θ,y轴为以计数测量的强度。
图14A是如实施例10中所述使用乙酸乙酯作为结晶溶剂获得的样品(样品6-13、6-12、6-11、6-10、6-8、6-7、6-6、6-5、6-4和6-2)与结晶化合物1(样品1-2)相比的XRPD衍射图的重叠图。除样品6-2、样品6-4和样品6-5(其表现出轻微差异)外,发现XRPD图谱一般与化合物1图谱相匹配。x轴为以度计的2θ,y轴为以计数测量的强度。
图14B是如实施例9中所述的在MEK中二次溶解并加入反溶剂环己烷和扑酸后样品5-1的XRPD衍射图的重叠图。样品5-1(在扑酸中结晶)在成熟后为固体,但XRPD图谱与扑酸的图谱相匹配。
图15A是如实施例10中所述的使用乙酸乙酯作为结晶溶剂获得的样品(样品6-5、6-4和6-2)与结晶化合物1(样品1-2)相比的XRPD衍射图的重叠图。除样品6-2、样品6-4和样品6-5(其表现出轻微差异)外,XRPD图谱一般与化合物1图谱相匹配。x轴为以度计的2θ,y轴为以计数测量的强度,且标记结晶中使用的酸。
图15B是实施例14中描述的化合物1-A的XRPD图谱。x轴为以度计的2θ,y轴为以计数测量的强度。
图16A是大鼠、狗和猴的肝脏和心脏中的活性TP(代谢物1-6)浓度水平的图(实施例18)。x轴为对于每一物种以mg/kg计的剂量,y轴为以ng/g计的活性TP浓度。
图16B是在单次口服剂量的化合物1或化合物1-A后4小时测量的狗(n=2)的肝脏和心脏中的活性TP(代谢物1-6)浓度水平的图(实施例19)。x轴为每种化合物的剂量,单位为mg/kg,y轴为活性TP浓度,单位为ng/g。
图17是给予单一500mg/kg口服剂量的化合物1-A的大鼠中(实施例20)直至给药后72小时测量的化合物1和代谢物1-7的血浆分布图。x轴为以小时为单位的时间,y轴为以ng/mL为单位的血浆浓度。
图18是给予单一口服剂量的30mg、100mg或300mg化合物1-A的猴子中(实施例20)至给药后72小时测量的化合物1和代谢物1-7的血浆分布图。x轴为以小时为单位的时间,y轴为以ng/mL为单位的血浆浓度。
图19是索非布韦和化合物1针对HCV临床分离株的EC
图20是索非布韦和化合物1针对HCV基因型1a、1b、2a、3a、4a和5a的实验室株的EC
图21是如实施例24所述的在研究B部分的所有群组中施用单次口服剂量的化合物1-A后,化合物1的平均血浆浓度-时间分布图。在B部分的所有群组中化合物1被快速吸收和在约8小时内快速代谢。x轴是以小时为单位的时间,y轴是以ng/mL为单位的几何平均血浆浓度。
图22是如实施例24所述的在研究B部分的所有群组中施用单次口服剂量的化合物1-A后,代谢物1-7的平均血浆浓度-时间分布图。在B部分的所有群组中代谢物1-7均表现出持续的血浆浓度。x轴是以小时为单位的时间,y轴是以ng/mL为单位的几何平均血浆浓度。
图23A是如实施例24中所述的1b群组中登记的受试者的个体药代动力学/药效学分析。该图显示了血浆代谢物1-7暴露和HCV核糖核酸减少水平。虚线代表针对GT1b维持大于EC
图23B是实施例24中所述的1b群组中登记的受试者的个体药代动力学/药效学分析。该图显示了血浆代谢物1-7暴露和HCV RNA减少水平。虚线代表针对GT1b维持大于EC
图23C是实施例24中所述的1b群组中登记的受试者的个体药代动力学/药效学分析。该图显示了血浆代谢物1-7暴露和HCV RNA减少水平。虚线代表针对GT1b维持大于EC
图23D是如实施例24中所述的3b群组中登记的受试者的个体药代动力学/药效学分析。每张图表显示了血浆代谢物1-7的暴露和HCV RNA减少水平。虚线代表针对GT1b维持大于EC
图23E是如实施例24中所述的3b群组中登记的受试者的个体药代动力学/药效学分析。每张图表显示了血浆代谢物1-7的暴露和HCV RNA减少水平。虚线代表针对GT1b维持大于EC
图23F是如实施例24中所述的3b群组中登记的受试者的个体药代动力学/药效学分析。每张图表显示了血浆代谢物1-7的暴露和HCV RNA减少水平。虚线代表针对GT1b维持大于EC
图24是化合物1和索非布韦针对GT1、GT2、GT3和GT4 HCV感染患者的临床分离株的EC
图25是显示如实施例26和27中所述的在等同于92mg、275mg、368mg或550mg化合物1的单次剂量化合物1-A之后,非肝硬化的GT1b HCV感染受试者相对于基线的平均HCV RNA变化的图表。x轴为给药后的小时数,y轴为相对于基线的平均HCV RNA变化,单位为log
图26是显示如实施例26和27中所述的在化合物1-A的7天QD给药后,非肝硬化的GT1b HCV感染受试者中相对于基线的平均HCV RNA变化的图表。x轴为首次剂量后的天数,y轴为相对于基线的平均HCV RNA变化,单位为log
图27是比较如实施例26和27中所述的在化合物1-A的600mg/天QD剂量(相当于化合物1的550mg)之后,非肝硬化GT1 HCV感染受试者、非肝硬化GT3 HCV感染受试者和肝硬化GT1或GT3 HCV感染受试者中相对于基线的平均HCV RNA变化的图表。如图中所示,具有肝硬化的受试者表现出与非肝硬化受试者相似的平均HCV RNA变化。x轴为首次剂量后的天数,y轴为相对于基线的平均HCV RNA变化,单位为log
图28A是如实施例26和27中所述的在化合物1-A的600mg/天QD剂量(相当于550mg的化合物1)之后,非肝硬化GT1b HCV感染受试者的相对于基线的个体HCV RNA变化的图表。水平虚线(----)为定量极限(LOQ=15IU/mL),且50%的受试者获得HCV RNA 图28B是如实施例26和27中所述的在化合物1-A的600mg/天QD剂量(相当于550mg的化合物1)之后,非肝硬化性GT3 HCV感染受试者的相对于基线的个体HCV RNA变化的图表。水平虚线(----)是定量极限(LOQ=15IU/mL)。x轴为首次剂量后的天数,y轴为相对于基线的HCV RNA变化,单位为log 图28C为如实施例26和27中所述的在化合物1-A的600mg/天QD剂量(相当于550mg的化合物1)之后,肝硬化GT1或GT3 HCV感染受试者的相对于基线的个体HCV RNA变化的图表。水平虚线(----)是定量极限(LOQ=15IU/mL)。x轴为首次剂量后的天数,y轴为相对于基线的HCV RNA变化,单位为log 图29为GT1/GT3 HCV感染的肝硬化和非肝硬化受试者中代谢物1-7的平均血浆浓度-时间分布图。如实施例26和27所述,施用GT1感染的非肝硬化受试者给予等同于化合物1的138mg/d、275mg/d或550mg/d QD的化合物1-A,GT3感染的非肝硬化受试者给予600mg/dQD的化合物1-A(550mg/d的化合物1),GT1/GT3感染的肝硬化受试者给予600mg的化合物1-AQD(550mg/d的化合物1)。x轴是以小时为单位的时间,y轴是以ng/mL为单位的平均血浆浓度。 图30A为如实施例26和27中所述的非肝硬化GT1b HCV感染受试者在化合物1-A的600mg/天QD(相当于550mg的化合物1)之后的平均代谢物1-7血浆浓度(左y轴)和HCV RNA平均减少(右y轴)相对于时间的图。GT1b中化合物1的EC 图30B是如实施例26和27中所述的非肝硬化性GT3 HCV感染受试者在化合物1-A的600mg/天QD(相当于550mg的化合物1)后的平均代谢物1-7血浆浓度(左y轴)和HCV RNA平均减少(右y轴)相对于时间的图。GT3中化合物1的EC 图30C为如实施例26和27中所述的肝硬化GT1b HCV感染受试者在化合物1-A的600mg/天QD(相当于550mg的化合物1)后的平均代谢物1-7血浆浓度(左y轴)和平均HCV RNA减少(右y轴)相对于时间的图。GT1b中化合物1的EC 图30D是如实施例26和27中所述的肝硬化的GT3 HCV感染的受试者在化合物1-A的600mg/天QD(相当于550mg的化合物1)后的平均代谢物1-7血浆浓度(左y轴)和平均HCV RNA减少(右y轴)相对于时间的图。GT1b中化合物1的EC 图31是Emax模型,其中在第7天对非肝硬化GT1b HCV感染、非肝硬化GT3 HCV感染、肝硬化GT1b HCV和肝硬化GT3 HCV感染的受试者测量的HCV RNA减少相对于在化合物1-AQD给药后的代谢物1-7的AUC作图。如实施例26和27所述,非肝硬化GT1b HCV的受试者施用了相当于138mg/d、275mg/d或550mg/d化合物1QD 7天的化合物1-A的多个递增剂量。非肝硬化GT3受试者和肝硬化GT1/GT3受试者给予600mg化合物1-A(相当于550mg/d的化合物1)QD7天。显示了施用138mg/d、275mg/d或550mg/d的非肝硬化GT1b HCV的95%CI区间范围。该模型预测,大于或等于2000ng/mL x h的代谢物1-7暴露导致给药7天后至少4log的最大病毒负荷降低。所有受试者在550mg的化合物1剂量后能够达到大于2000ng/mL x h的代谢物1-7暴露,无论受试者是否表现出肝硬化。x轴为代谢物1-7的AUC,单位为ng/mL x h,y轴为第7天的HCV RNA减少,以log 图32是显示如实施例28中所述的化合物1-A的50mg和100mg片剂的制造工艺的流程图。在步骤1中,将微晶纤维素、化合物1-A、乳糖一水合物和交联羧甲基纤维素钠通过600μM筛网过滤。在步骤2中,来自步骤1的内容物加载到V型搅拌机中,并以25rpm混合5分钟。在步骤3中,硬脂酸镁通过600μM筛网过滤。在步骤4中,硬脂酸镁加载到包含步骤2的内容物(微晶纤维素、化合物1-A、乳糖一水合物和交联羧甲基纤维素钠)的V型搅拌机中,并以25rpm混合2分钟。然后将通用共混物分开用于产生50mg片剂和100mg片剂。为产生50mg片剂,将来自步骤4的共混物用6mm圆形标准凹口工具压制。为产生100mg片剂,将来自步骤4的共混物用8mm圆形标准凹口工具压制。片剂随后被包装到用PP帽感应密封和具有干燥剂的HDPE瓶中。 图33是NS5B聚合酶抑制剂化合物1-A和NS5A抑制剂化合物2。 图34是显示化合物1和化合物2的组合在何种浓度下是协同的、加性的或拮抗的三维组合表面图。如实施例30所述,基于两种化合物单独测试时的活性计算药物组合的效果。从实验测定的每一组合浓度的抗病毒活性中减去预期的加性抗病毒保护作用,得到正值(协同作用或增强作用)、负值(对抗作用)或零(加性)。在每个组合浓度下,组合测定的结果以三维形式呈现,产生了在加性平面之上(协同作用)或之下(拮抗作用)延伸的活性表面。化合物1和化合物2的组合在化合物1和化合物2两者的广泛浓度范围内是协同的。观察到化合物1的约0.156至约1和2.5至40或更高nM的协同。约0.001nM至约0.008nM的化合物2是观察到最大协同作用的化合物2的范围。 具体实施方式 本发明提供了特定NS5B聚合酶抑制剂和特定NS5A抑制剂的高活性的协同组合,其用于有利地治疗宿主(通常为人类)的丙型肝炎感染。 该组合治疗中使用的抗HCV化合物为:1)NS5B抑制剂异丙基((S)-(((2R,3R,4R,5R)-5-(2-氨基-6-(甲基氨基)-9H-嘌呤-9-基)-4-氟-3-羟基-4-甲基四氢呋喃-2-基)甲氧基)(苯氧基)磷酰基)-L-丙氨酸酯(化合物1)或其药学上可接受的盐;和2)NS5A抑制剂Ruzasvir(二甲基N,N’-([(6S)-6-(2-环丙基-1,3-噻唑-5-基)-1-氟-6H-吲哚并[1,2-c][1,3]苯并噁嗪-3,10-二基]双{1H-咪唑-5,2-二基-(2S)-吡咯烷-2,1-二基[(2S)-3-甲基-1-氧代丁烷-1,2-二基]})二氨基甲酸酯)(化合物2)或其药学上可接受的盐。在典型实施方案中,化合物1作为半硫酸盐衍生物(化合物1-A)施用。 /> 在一个实施方案中,药物的组合以固定剂量的剂型施用,例如丸剂或片剂。在替代实施方案中,两种化合物以根据标准药代动力学测量的需要的宿主以协同的方式获得两种化合物的益处的方式施用。 化合物1和化合物1-A 先前在美国专利号9,828,410;10,000,523;10,005,811;和10,239,911以及转让给Atea Pharmaceuticals的PCT申请WO 2016/21276和WO 2019/200005中描述了化合物1(异丙基((S)-(((2R,3R,4R,5R)-5-(2-氨基-6-(甲基氨基)-9H-嘌呤-9-基)-4-氟-3-羟基-4-甲基四氢呋喃-2-基)甲氧基)(苯氧基)磷酰基)-L-丙氨酸酯)。化合物1的合成在下文实施例1中描述。 化合物1-A先前在转让给Atea Pharmaceuticals的US 2018-0215776及PCT申请WO2018/144640和WO 2019/200005中公开。化合物1-A(异丙基((S)-(((2R,3R,4R,5R)-5-(2-氨基-6-(甲基氨基)-9H-嘌呤-9-基)-4-氟-3-羟基-4-甲基四氢呋喃-2-基)甲氧基)(苯氧基)磷酰基)-L-丙氨酸酯的半硫酸盐)的合成描述于下文实施例4中。在一个实施方案中,化合物1-A以其药学上可接受的组合物或固体剂型提供。在另一个实施方案中,化合物1-A是无定形固体。在一个实施方案中,化合物1-A是结晶固体。图1-15描绘了通过XRPD、DSC和TGA对化合物1-A物理形式的表征。 如上所述,化合物1-A已完成HCV感染患者的1b/2a期临床试验。该多部分研究评价了单剂量和多剂量化合物1-A在健康受试者、非肝硬化HCV感染患者和肝硬化HCV感染患者中的效果。化合物1-A在对所有测试的HCV感染群组施用时诱导了显著的抗病毒减少。化合物1-A在7天的过程中每天一次(QD)施用,且观察到强效的抗病毒活性。在给予600mg QD化合物1-A(相当于550mg化合物1)的非肝硬化HCV感染患者中,HCV GT1感染患者的平均最大HCV RNA减少为4.4log 除非另有说明,化合物1或其药学上可接受的盐,例如化合物1-A,以β-D-构型提供。在替代实施方案中,化合物1或其药学上可接受的盐,例如化合物1-A,可以以β-L-构型提供。化合物1或其药学上可接受的盐,例如化合物1-A,的氨基磷酸酯可以作为R或S手性磷衍生物或其混合物提供,包括外消旋或非对映体混合物。这些立体构型的所有组合为本文所述本发明的可选实施方案。 这些替代构型包括但不限于: /> 另外的替代构型包括 在一个实施方案中,在本文中本发明的任何方面,任何上述立体异构体或其药学上可接受的盐被用作化合物1或其药学上可接受的盐。在另一个实施方案中,上述立体异构体中的任何一种或其药学上可接受的盐在本文中本发明的任何方面用作化合物1-A。 在替代实施方案中,化合物1-A作为除化合物阐明中所描述的特定氨基磷酸酯之外的氨基磷酸酯的半硫酸盐提供。 在另一替代实施方案中,化合物1或其药学上可接受的盐作为除化合物阐明中所描述的特定氨基磷酸酯之外的氨基磷酸酯提供。本领域技术人员已知广泛的氨基磷酸酯,其可根据需要选择以提供本文所述的活性化合物。例如,化合物1或其药学上可接受的盐的氨基磷酸酯包括式A的化合物或其药学上可接受的盐: 其中: R R R R 在可选的非限制性实施方案中,本发明包括作为草酸盐(化合物1-B)、HCl盐(化合物1-C)或硫酸盐(化合物1-D)的化合物1。 化合物1和化合物1-A的代谢涉及5’-单磷酸盐的产生和随后N 或者,5’-单磷酸盐1-2可被代谢以生成嘌呤碱1-8。异丙基((S)-(((2R,3R,4R,5R)-5-(2-氨基-6-(甲基氨基)-9H-嘌呤-9-基)-4-氟-3-羟基-4-甲基四氢呋喃-2-基)甲氧基)(苯氧基)磷酰基)-L-丙氨酸酯的代谢途径如方案1所示: Atea Pharmaceuticals,Inc.在美国专利号9,828,410;10,000,523;10,005,811;10,239,911,10,815,266;10,870,672;10,870;673;10,870,885;10,519,186;10,906,928;10,894,804;及PCT申请号WO 2016/144918;WO 2018/048937;WO 2018/013937;和WO 2018/144640中公开了用于治疗HCV的β-D-2’-脱氧-2’-α-氟-2’-β-C-取代-2-修饰-N 化合物2 化合物2在转让给Merck and Company的WO 2014/110705和美国专利号9,555,038中公开。在一个实施方案中,化合物2作为其药学上可接受的盐施用。在一个实施方案中,使用固体形式的化合物2。在一个实施方案中,化合物2的固体形式为结晶固体。 Ruzasvir(二甲基N,N’-([(6S)-6-(2-环丙基-1,3-噻唑-5-基)-1-氟-6H-吲哚并[1,2-c][1,3]苯并噁嗪-3,10-二基]双{1H-咪唑-5,2-二基-(2S)-吡咯烷-2,1-二基[(2S)-3-甲基-1-氧代丁烷-1,2-二基]})二氨基甲酸酯,化合物2)的合成是本领域已知的。可用于合成化合物2的合成方法的非限制性实例包括实施例29中所示和在转让给Merck的WO2016/196932中报告的那些。 定义 本发明上下文中使用的术语“D-构型”指模拟糖部分的天然构型的主要构型,与非天然存在的核苷或“L”构型相反。术语“β”或“β异头体”用于指核苷类似物,其中核苷碱基被配置(布置)在核苷类似物中的呋喃糖部分的平面之上。 术语“共施用”和“共同施用”或组合治疗用于描述根据本发明的化合物1或其药学上可接受的盐与化合物2或其药学上可接受的盐组合的施用。在某些实施方案中,化合物1或其药学上可接受的盐(例如化合物1-A)和化合物2或其药学上可接受的盐与至少一种其他活性剂(例如在适当的情况下,至少一种另外的抗HCV剂)一起施用。共施用的时机最好由治疗患者的医学专家决定。有时,优选的是同时或至少以允许两种药物在被治疗患者中具有重叠的药理作用的方式施用。或者,选择用于组合治疗的药物可在不同时间施用于患者。当然,当存在多于一种病毒或其他感染或其他病症时,本发明的化合物可根据需要与其他药物组合以治疗该其他感染或病症。 本文中使用的术语“宿主”指HCV可在其中复制的单细胞或多细胞生物体,包括细胞系和动物,通常为人。术语“宿主”具体指感染的细胞、用HCV基因组的全部或部分转染的细胞以及动物,特别是灵长类动物(包括黑猩猩)和携带HCV基因组或其部分的人,其能够用本文所述组合治疗。在本发明的大多数动物应用中,宿主为人类患者,其包括但不限于具有重叠的药代动力学的剂量方案。然而,在某些情况下,本发明明确预期兽医应用(例如黑猩猩)。宿主可以是例如牛、马、鸟、狗、猫等,它能够寄宿病毒。 “药学上可接受的盐”是所公开化合物的衍生物,其中母体化合物被修饰为其无机和有机、酸或碱加成盐,而没有过度毒性。本发明化合物的盐可通过常规化学方法由母体化合物与碱性或酸性部分合成。通常,这类盐可通过使这些化合物的游离酸形式与化学计量量的适当碱(例如氢氧化钠、氢氧化钙、氢氧化镁或氢氧化钾、碳酸盐、碳酸氢盐等)反应,或通过使这些化合物的游离碱形式与化学计量量的适当酸反应来制备。这类反应通常在水中、有机溶剂中或两者的混合物中进行。一般地,在适用的情况下,典型的是非水性介质如醚、乙酸乙酯、乙醇、异丙醇或乙腈。本发明化合物的盐可任选以溶剂合物的形式提供。 药学上可接受的盐的实例包括但不限于碱性残基(例如胺)的无机或有机酸盐;酸性残基如羧酸的碱金属或有机盐;等等。药学上可接受的盐包括母体化合物的常规盐和季铵盐,例如由无过度毒性的无机酸或有机酸形成。例如,常规的酸式盐包括由无机酸如盐酸、氢溴酸、硫酸、氨基磺酸、磷酸、硝酸等衍生的那些;以及由有机酸如乙酸、丙酸、琥珀酸、乙醇酸、硬脂酸、乳酸、苹果酸、酒石酸、柠檬酸、抗坏血酸、扑酸、马来酸、顺丁烯二酸、羟基马来酸、苯乙酸、谷氨酸、苯甲酸、水杨酸、甲基丙烯酸、苯磺酸、对氨基苯磺酸、2-乙酰氧基安息香、富马酸、甲苯磺酸、甲磺酸、乙磺酸、苯磺酸、对氨基苯磺酸、2-乙酰氧基苯甲酸、富马酸、甲苯磺酸、甲烷磺酸、乙烷二磺酸、草酸、羟乙磺酸、HOOC-(CH 化合物可以以提供所需结果的任何摩尔比递送。例如,化合物可以提供小于摩尔当量的反离子,例如以半硫酸盐形式。或者,该化合物可以提供超过摩尔当量的反离子,例如以二硫酸盐的形式。化合物与反离子的摩尔比的非限制性例子包括1:0.25、1:0.5、1:1和1:2。 同位素取代 本发明包括化合物1或其药学上可接受的盐(例如化合物1-A)和化合物2或其药学上可接受的盐的组合,其中一种或两种化合物具有所需的同位素原子取代,其量高于同位素的天然丰度,即是富集的。同位素是具有相同原子序数但不同质量数的原子,即相同质子数但不同中子数。作为一般示例而非限制,氢的同位素,例如氘( 同位素(例如氘)取代可提供由更高的代谢稳定性导致的某些治疗优势,例如增加体内半衰期或降低剂量要求。在代谢降解位点处用氘取代氢可以降低该键处的代谢速率或消除该键处的代谢。在可能存在氢原子的化合物的任何位置处,氢原子可以是氢的任何同位素,包括氕( 术语“同位素标记的”类似物指的是作为“氘代类似物”、“ 治疗方法 如本文所用,治疗指以有效量将本发明的组合施用于宿主,例如感染或可能感染HCV病毒的人。在一个实施方案中,治疗方法包括将有效量的化合物1或其药学上可接受的盐和化合物2或其药学上可接受的盐施用于宿主,例如感染或可能感染HCV病毒的人。在另一个实施方案中,治疗方法包括将化合物1-A和化合物2施用于宿主,例如感染或可能感染HCV病毒的人。 术语“预防性的”或“预防的”,在使用时,指施用本发明的组合以预防或降低病毒病症发生的可能性。在替代实施方案中,本发明包括治疗性的和预防性的或预防的治疗。在一个实施方案中,该组合施用于已经暴露于丙型肝炎病毒感染并因此有感染风险的宿主。 本发明包括通过施用有效量的化合物1(例如1-A)和化合物2的协同组合来治疗丙型肝炎病毒的方法,包括药物抗性和多耐药形式的HCV及HCV感染的相关疾病状态、病症或并发症,包括肝硬化和相关肝毒性,以及HCV感染继发的其他病症,例如虚弱、食欲下降、体重减轻、乳房增大(尤其是男性)、皮疹(尤其是手掌上)、凝血困难、皮肤上的蜘蛛状血管、精神混乱、昏迷(脑病)、腹腔积液(腹水)、食管静脉曲张、门静脉高压、肾衰竭、脾脏增大、血细胞减少、贫血、血小板减少症、黄疸和肝细胞癌等。该方法包括向有需要的宿主(通常为人类)施用有效量的本文所述组合,任选地与至少一种另外的生物活性剂(例如,另外的抗HCV剂)组合,进一步任选地与药学上可接受的载体添加剂和/或赋形剂组合。在另一个实施方案中,该方法包括向有HCV感染风险的患者施用有效量的本发明组合。在另一个实施方案中,上述组合与药学上可接受的载体、添加剂或赋形剂一起使用,任选地与第三抗HCV剂组合。在另一个实施方案中,本发明的组合可在与肝炎相关的肝移植后施用于患者以保护新器官。 组合治疗和剂型也可用于治疗与HCV病毒暴露相关或因HCV暴露而发生的病症。例如,活性化合物可用于治疗HCV抗体阳性和HCV抗原阳性病症、基于病毒的慢性肝脏炎症、由晚期丙型肝炎导致的肝脏癌症(例如,肝细胞癌)、肝硬化、急性丙型肝炎、暴发性丙型肝炎、慢性持续性丙型肝炎和抗HCV基疲劳。该组合还可以预防性地使用以预防或限制抗HCV抗体或抗原阳性或已暴露于丙型肝炎的个体的临床疾病进展。 药物组合物和剂型 化合物1或其药学上可接受的盐和化合物2或其药学上可接受的盐的施用可采用任何所需的形式进行,包括但不限于口服、局部、肠胃外、肌内、静脉内、皮下、经皮(其可包括渗透增强剂)、口腔和栓剂施用,以及其它施用途径进行。在一个实施方案中,活性化合物或化合物组合以本领域众所周知的固体剂型提供,并在下文中进一步描述。肠溶包衣口服片剂也可用于增强化合物用于口服施用途径的生物利用度。最有效的剂型将取决于所选特定药剂的生物利用度/药代动力学以及患者的疾病严重程度。口服剂型是特别优选的,因为其易于施用且预期的有利患者依从性。 在某些实施方案中,根据本发明的药物组合物包含抗HCV病毒有效量的如本文所述的各自单独形式或组合形式的化合物1或其药学上可接受的盐和化合物2或其药学上可接受的盐,任选地与药学上可接受的载体、添加剂或赋形剂组合,进一步任选地与至少一种其他活性化合物组合或交替。 在一个实施方案中,该组合包括化合物1或其药学上可接受的盐(例如化合物1-A)和化合物2或其药学上可接受的盐在药学上可接受的载体中的固体剂型。该药物组合物可同时包含化合物1或其药学上可接受的盐和化合物2或其药学上可接受的盐,或者可选地,化合物可以在单独的剂型中,其施用方式为如根据标准药代动力学测量的,宿主以协同的方式获得两种化合物的益处。 本领域普通技术人员将认识到,治疗有效量将随着待治疗的感染或状况、其严重程度、待采用的治疗方案、所用药剂的药代动力学以及待治疗的患者或受试者(动物或人)而变化,并且该治疗量可由主治医生或专家确定。 化合物1或其药学上可接受的盐(例如化合物1-A)和化合物2或其药学上可接受的盐可与一种或多种药学上可接受的载体配制为一种或多种混合物。一般地,优选的是以可口服施用的形式施用一种或多种药物组合物,且特别是一种或多种固体剂型,例如丸剂或片剂。某些制剂可通过肠胃外、静脉内、肌内、局部、经皮、口腔、皮下、栓剂或其他途径(包括鼻内喷雾)施用。静脉内和肌内制剂通常在无菌盐水中施用。本领域普通技术人员可以对制剂进行改变以使其更易溶于水或其他溶媒,例如,这可以通过轻微的改变(盐制剂、酯化等)容易地实现,其完全在本领域技术人员的能力范围内。改变化合物1或其药学上可接受的盐(例如化合物1-A)和化合物2或其药学上可接受的盐)的施用途径和剂量方案以控制本发明化合物的药代动力学,从而在患者中获得最大的有益效果,也完全属于常规技术人员的技能范围。 在某些药物剂型中,化合物的前药形式,包括本发明化合物的酰化(乙酰化或其他)和醚(烷基和相关)衍生物、磷酸酯、硫代磷酰胺酯、氨基磷酸酯和各种盐形式,可用于实现所需的效果。本领域技术人员将认识到如何容易地将本发明的化合物改变为前药形式,以便于将活性化合物递送至宿主生物体或患者体内的目标部位。本领域技术人员还将利用前药形式的有利药代动力学参数(如适用)来将本发明化合物递送至宿主生物体或患者体内的目标部位,以使化合物的预期效果最大化。 本公开中提及的量通常指游离形式(即非盐、水合物或溶剂合物形式)。本文中通常描述的值代表游离形式当量,即,如同将施用游离形式的量。如果使用盐,则需要根据盐和游离形式之间的分子量比率计算该量。 根据本发明的治疗活性制剂中包含的化合物1或其药学上可接受的盐(例如化合物1-A)和化合物2或其药学上可接受的盐的量是实现根据本发明的期望结果的有效量,例如用于治疗HCV感染、降低HCV感染的可能性或者抑制、减少和/或消除HCV或其继发效应(包括继发HCV于发生的疾病状态、状况和/或并发症)。通常,本发明化合物在药物剂型中的治疗有效量可在例如每天约0.001mg/kg至约100mg/kg或更高的范围内。化合物1或化合物1-A可根据药物在患者体内的药代动力学以患者每天约0.1mg/kg至约15mg/kg范围的量施用。 在某些实施方案中,在剂型中提供600mg化合物1-A,即550mg化合物1。 在某些实施方案中,药物组合物的剂型包含单位剂型中的约1mg至约2000mg、约10mg至约1000mg、约100mg至约800mg、约200mg至约600mg、约300mg至约500mg或约400mg至约450mg的化合物1或等同量的化合物1-A,以及约1mg至约2000mg、约10mg至约1000mg、约100mg至约800mg的化合物2或等同量的化合物2的药学上可接受的盐。 在替代实施方案中,药物组合物的剂型包含单位剂型中的约500mg至约600mg、约550mg至约750mg、约600mg至约800mg或约1000mg至约1300mg的化合物1或等同量的化合物1-A,以及约100mg至约800mg、约150mg至约200mg、约250至约300mg或约350至约400mg的化合物2或等同量的化合物2的药学上可接受的盐。 在某些实施方案中,药物组合物为单位剂型中包含最多约10、约50、约100、约125、约150、约175、约200、约225、约250、约275、约300、约325、约350、约375、约400、约425、约450、约475、约500、约525、约550、约575、约600、约625、约650、约675、约700、约725、约750、约775、约800、约825、约850、约875、约900、约925、约950、约975或约1000mg或更多的化合物1或等同量的化合物1-A的剂型,例如固体剂型。 在某些实施方案中,药物组合物为包含最多约10、约50、约60、约100、约125、约150、约175、约200、约225、约250、约275、约300、约325、约350、约375、约400、约425、约450、约475、约500、约525、约550、约575、约600、约625、约650、约675、约700、约725、约750、约775、约800、约825、约850、约875、约900、约925、约950、约975或约1000mg或更多的化合物2或等同量的化合物2的药学上可接受的盐的剂型,例如固体剂型。 在替代实施方案中,药物组合物为包含最多约90、约180、约270或约360mg的化合物2或等同量的化合物2的药学上可接受的盐的剂型,例如固体剂型。 在一个实施方案中,每天一次向需要治疗HCV的宿主施用包含最高约800mg、最高约700mg、最高约600mg、最高约550mg、最高约500mg、最高约400mg、最高约300mg、最高约200mg或最高约100mg化合物1或等同量的化合物1-A和最高约360mg、最高约270mg、最高约180mg、最高约145mg、最高约130mg、最高约125mg、最高约110mg、最高约100mg、最高约90mg、最高约75mg、最高约70mg、最高约65mg、最高约60mg、最高约55mg、最高约50mg、最高约45mg、最高约40mg、最高约35mg、最高约30mg、最高约25mg、最高约20mg、最高约15mg、最高约10mg或最高约5mg化合物2或等同量的化合物2的药学上可接受的盐的固体剂型。 在一个实施方案中,每天一次向需要治疗HCV的宿主施用包含至少约100mg、至少约200mg、至少约300mg、至少约400mg、至少约500mg、至少约550mg、至少约600mg、至少约700mg、至少约750mg或至少约1100mg化合物1或等同量的化合物1-A和至少约5mg、至少约10mg、至少约15mg、至少约20mg、至少约25mg、至少约30mg、至少约35mg、至少约40mg、至少约45mg、至少约50mg、至少约55mg、至少约60mg、至少约65mg、至少约70mg、至少约75mg、至少约90mg、至少约100mg、至少约110mg,至少约125mg、至少约130mg、至少约145mg、至少约180mg、至少约270mg、至少约360mg、至少约400mg或至少约500mg的化合物2或等同量的化合物2的药学上可接受的盐的固体剂型。 在替代实施方案中,每天一次向需要治疗HCV的宿主施用包含最高约1100mg、最高约750mg或最高约550mg化合物1或等同量的化合物1-A和最高约360mg、最高约270mg、最高约180mg、最高约90mg化合物2或等同量的化合物2的药学上可接受的盐的固体剂型。 在替代实施方案中,每天两次向需要治疗HCV的宿主施用包含至少约500mg、至少约550mg、至少约750mg或至少约1100mg化合物1或等同量的化合物1-A和至少约90mg、至少约180mg、至少约270mg、至少约360mg化合物2或等同量的化合物2的药学上可接受的盐的固体剂型。 在一个实施方案中,本文所述化合物的组合以包含最高约600mg化合物1-A和最高约90、180、270或360mg化合物2或等同量的化合物2的药学上可接受的盐的单个片剂的形式施用。 在替代实施方案中,本文所述化合物的组合作为包含最高约600mg化合物1-A和最高约180mg化合物2或等同量的化合物2的药学上可接受的盐的单个片剂施用。 在替代实施方案中,本文所述化合物的组合作为包含最高约600mg化合物1-A和最高约270mg化合物2或等同量的化合物2的药学上可接受的盐的单个片剂施用。 在替代实施方案中,本文所述化合物的组合作为包含最高约600mg化合物1-A和最高约360mg化合物2或等同量的化合物2的药学上可接受的盐的单个片剂施用。 在替代实施方案中,本文所述化合物的组合作为包含最高约750mg化合物1-A和最高约180mg化合物2或等同量的化合物2的药学上可接受的盐的单个片剂施用。 在替代实施方案中,本文所述化合物的组合作为包含最高约750mg化合物1-A和最高约270mg化合物2或等同量的化合物2的药学上可接受的盐的单个片剂施用。 在替代实施方案中,本文所述化合物的组合作为包含最高约750mg化合物1-A和最高约360mg化合物2或等同量的化合物2的药学上可接受的盐的单个片剂施用。 在替代实施方案中,本文所述化合物的组合作为包含最高约1200mg化合物1-A和最高约180mg化合物2或等同量的化合物2的药学上可接受的盐的单个片剂施用。 在替代实施方案中,本文所述化合物的组合作为包含最高约1200mg化合物1-A和最高约270mg化合物2或等同量的化合物2的药学上可接受的盐的单个片剂施用。 在替代实施方案中,本文所述化合物的组合作为包含最高约1200mg化合物1-A和最高约360mg化合物2或等同量的化合物2的药学上可接受的盐的单个片剂施用。 或者,化合物1-A或等同量的化合物1的固体剂型可与含有化合物2或等同量的化合物2的药学上可接受的盐的单独固体剂型组合给药。该组合可根据医疗服务提供者的指示一天施用一次、两次、三次或最多四次。在一个实施方案中,化合物1-A或化合物1以与化合物2不同的单独时间表施用。例如,化合物1或等同量的化合物1-A可一天施用两次,而化合物2仅一天施用一次,反之亦然:化合物2可一天施用多次,而化合物1或等同量的化合物1-A仅一天施用一次。 在一个实施方案中,每天一次向需要治疗HCV的宿主施用包含最高约800mg、最高约700mg、最高约600mg、最高约500mg、最高约400mg、最高约300mg、最高约200mg或最高约100mg化合物1或等同量的化合物1-A的固体剂型每天施用一次,并且包含最高约145mg、最高约130mg,最高约125mg、最高约110mg、最高约100mg、最高约90mg、最高约75mg、最高约70mg、最高约65mg、最高约60mg、最高约55mg、最高约50mg、最高约45mg、最高约40mg、最高约35mg、最高约30mg、最高约25mg、最高约20mg、最高约15mg、最高约10mg或最高约5mg化合物2或等同量的化合物2的药学上可接受的盐的单独固体剂型。 在一个实施方案中,每天一次向需要治疗HCV的宿主施用包含至少约100mg、至少约200mg、至少约300mg、至少约400mg、至少约500mg、至少约600mg、至少约700mg或至少约800mg化合物1或等同量的化合物1-A的固体剂型,且每天一次施用包含至少约5mg、至少约10mg,至少约15mg、至少约20mg、至少约25mg、至少约30mg、至少约35mg、至少约40mg、至少约45mg、至少约50mg、至少约55mg、至少约60mg、至少约65mg、至少约70mg、至少约75mg、至少约80mg、至少约90mg、至少约100mg,至少约110mg、至少约125mg、至少约130mg或至少约145mg化合物2或等同量的化合物2的药学上可接受的盐的单独固体剂型。 在某些实施方案中,本文所述化合物的组合作为(两种活性剂的单独剂量或两种活性化合物的组合剂量)施用于需要的宿主: (1)单一剂型; (2)两个剂型; (3)三个或更多个剂型; (4)包含两个剂型的试剂盒; (5)包含三个或更多个剂型的试剂盒; 其中实施方案(1)-(5)中的每一个的剂型包含: (a)约500或550mg至约1100mg的化合物1-A和有效量的化合物2; (b)约100mg至约800mg的化合物1-A和有效量的化合物2; (c)约550至约650mg的化合物1-A和有效量的化合物2; (d)约500至约750mg的化合物1-A和有效量的化合物2; (e)约1000至约1300mg的化合物1-A和有效量的化合物2; (f)至少约600mg的化合物1-A和有效量的化合物2; (g)至少约750mg的化合物1-A和有效量的化合物2; (h)至少约1100mg的化合物1-A和有效量的化合物2; (i)实施方案(a)-(h)中任一项,其中存在约60mg至约500mg的化合物2; (j)实施方案(a)-(h)中任一项,其中存在约90mg至约360mg的化合物2; (k)实施方案(a)-(h)中任一项,其中存在约250mg至约300mg的化合物2; (l)实施方案(a)-(h)中任一项,其中存在约350mg至约400mg的化合物2; (m)实施方案(a)-(h)中任一项,其中存在至少约90mg的化合物2; (n)实施方案(a)-(h)中任一项,其中存在至少约180mg的化合物2; (o)实施方案(a)-(h)中任一项,其中存在至少约270mg的化合物2; (p)实施方案(a)-(h)中任一项,其中存在至少约360mg的化合物2; (q)实施方案(1)-(5)和(a)-(h)中任一项,其中该剂型每天施用一次; (r)实施方案(1)-(5)和(a)-(h)中任一项,其中该剂型每天施用两次; (s)实施方案(1)-(5)和(a)-(h)中任一项,其中该剂型每天施用三次或更多次; (t)实施方案(1)-(5)和(a)-(s)中任一项,其中该剂型是口服施用的; (u)实施方案(1)-(5)和(a)-(t)中任一项,其中所述宿主是人。 在替代实施方案中,可向需要的宿主提供一定量的化合物1,其导致化合物1的血浆浓度为约0.15nM至约1nM。在一个实施方案中,可向有需要的宿主提供一定量的化合物1,其导致化合物1的血浆浓度为约0.25至约40nM。在一个实施方案中,可向需要的宿主提供一定量的化合物1,其导致化合物1的血浆浓度为约40至约200nM。在一个实施方案中,可向需要的宿主提供一定量的化合物1,这导致化合物2的血浆浓度为约0.001nM至约0.008nM。 本发明组合的化合物通常口服施用,但也可肠胃外、局部或以栓剂形式施用,以及鼻内,作为鼻喷剂或如本文另外所述的施用。更一般地,这些化合物可以以一种或多种片剂、胶囊、注射剂、静脉内制剂、混悬剂、液体、乳剂、植入剂、颗粒、球、霜剂、软膏剂、栓剂、可吸入形式、透皮形式、口腔、舌下、局部、凝胶、粘膜等施用。 在某些实施方案中,该组合每天施用至少一次,最多24周。在某些实施方案中,组合每天施用至少一次,最多12周。在某些实施方案中,组合每天施用至少一次,最多10周。在某些实施方案中,组合每天施用至少一次,最多8周。在某些实施方案中,组合每天施用至少一次,最多6周。在某些实施方案中,组合每天施用至少一次,最多4周。在某些实施方案中,组合每天施用至少一次,持续至少4周。在某些实施方案中,组合每天施用至少一次,持续至少6周。在某些实施方案中,组合每天施用至少一次,持续至少8周。在某些实施方案中,组合每天施用至少一次,持续至少10周。在某些实施方案中,组合每天施用至少一次,持续至少12周。在某些实施方案中,组合每天施用至少一次,持续至少24周。在某些实施方案中,组合至少每隔一天施用,持续最多24周、12周、最多10周、最多8周、最多6周或最多4周。在某些实施方案中,组合至少每隔一天施用,持续至少4周、至少6周、至少8周、至少10周、至少12周或至少24周。 为本发明的目的,根据本发明的组合物的预防性或预防有效量落在与上述治疗有效量所示相同的浓度范围内,并且通常与治疗有效量相同。 为了制备根据本发明的药物组合物,治疗有效量的化合物1或其药学上可接受的盐(例如化合物1-A)和化合物2或其药学上可接受的盐可根据常规药物配混技术与药学上可接受的载体紧密混合以产生剂量。根据施用(例如,口服或肠胃外)所需的制剂的形式,载体可采取多种形式。在制备口服剂型的药物组合物时,可以使用任何常用的药物介质。因此,对于液体口服制剂,例如混悬剂、酏剂和溶液,可以使用合适的载体和添加剂,包括水、二醇、油、醇、调味剂、防腐剂、着色剂等。对于固体口服制剂如粉末、片剂、胶囊,以及对于固体制剂如栓剂,可使用合适的载体和添加剂,包括淀粉、糖载体如葡萄糖、歧管(manifold)、乳糖和相关载体、稀释剂、制粒剂、润滑剂、粘合剂、崩解剂等。如果需要,片剂或胶囊可以通过标准技术进行肠溶包衣或持续释放。使用这些剂型可能显著提高化合物在患者体内的生物利用度。 对于肠胃外制剂,载体通常包含无菌水或氯化钠水溶液,但也可包含其他成分,包括有助于分散的那些成分。当然,在使用无菌水并保持无菌的过程,组合物和载体也必须是无菌的。也可以制备可注射的混悬剂,在这种情况下,可以使用合适的液体载体、混悬剂等。 脂质体混悬剂(包括靶向病毒抗原的脂质体)也可通过常规方法制备以产生药学上可接受的载体。这可能适用于递送根据本发明的核苷化合物的游离核苷、酰基/烷基核苷或磷酸酯前药形式。 在根据本发明的典型实施方案中,药物组合物用于治疗、预防或延迟HCV感染或HCV的继发疾病状态、状况或并发症。 固体剂型 本发明的一个方面是活性化合物(单独或组合)或其药学上可接受的盐的固定剂型,任选地组合固定剂型。 一种或两种化合物可以以结晶或非结晶形式提供。在某些实施方案中,它们以单独或相同的口服剂型提供,例如囊形片、固体形式、喷雾干燥分散体、片剂、胶囊或其他形式的丸剂。 在一个实施方案中,固定剂量组合包括至少一种化合物或两种化合物(任一为药学上可接受的盐)的喷雾干燥固体分散体,并且该组合物适合于口服递送。在该实施方案的一个方面,固定剂量组合包括化合物1或其药学上可接受的盐和化合物2或其药学上可接受的盐,其中至少一种化合物处于喷雾干燥的固体分散体中。 在另一个实施方案中,固定剂量组合是至少一种化合物或其药学上可接受的盐的颗粒分层固体分散体,并且该组合物适合于口服递送。制备用于口服递送的固体剂型的示例性方法可以见图32。在该实施方案的一个方面,固定剂量组合是颗粒分层固体分散体,其包括化合物1或其药学上可接受的盐和化合物2或其药学上可接受的盐。在某些实施方案中,使用结晶化合物1-A制备喷雾干燥的分散体或颗粒分层的固体分散体组分。在替代实施方案中,化合物1或药学上可接受的盐(例如化合物1-A)或化合物2或药学上可接受的盐可以作为非晶化合物递送。 在其他实施方案中,固体分散体还包含至少一种选自共聚维酮、泊洛沙姆和HPMC-AS的赋形剂。在一个实施方案中,泊洛沙姆为泊洛沙姆407或可包括泊洛沙姆407的泊洛沙姆混合物。在一个实施方案中,HPMC-AS是HPMC-AS-L 在其他实施方案中,由化合物1或其药学上可接受的盐和化合物2或其药学上可接受的盐制备的固定剂量组合物还包含一种或多种以下赋形剂:磷酸甘油酯;磷脂酰胆碱;二棕榈酰磷脂酰胆碱(DPPC);二油烯基磷脂酰乙醇胺(DOPE);二油烯氧基丙基三乙基铵(DOTMA);二油酰磷脂酰胆碱;胆固醇;胆固醇酯;二酰基甘油;二酰基甘油琥珀酸酯;二磷脂酰甘油(DPPG);己烷癸醇;脂肪醇如聚乙二醇;聚氧乙烯-9-十二烷基醚;表面活性脂肪酸,如棕榈酸或油酸;脂肪酸;脂肪酸单甘油酯;脂肪酸二甘油酯;脂肪酸酰胺;脱水山梨醇三油酸酯( 在其他实施方案中,由化合物1或其药学上可接受的盐和化合物2或其药学上可接受的盐制备的固定剂量组合物还包含一种或多种以下表面活性剂:聚氧乙烯二醇、聚氧丙烯二醇、癸基葡萄糖苷、十二烷基葡萄糖苷、辛基葡萄糖苷、聚氧乙烯二醇辛基酚、TritonX-100、甘油烷基酯、十二酸甘油酯、椰油酰胺MEA、椰油酰胺DEA、十二烷基二甲基胺氧化物和泊洛沙姆。泊洛沙姆的例子包括泊洛沙姆188、237、338和407。这些泊洛沙姆的商品名为 还在其他实施方案中,由化合物1或其药学上可接受的盐和化合物2或其药学上可接受的盐制备的固定剂量组合物还包含一种或多种以下表面活性剂:聚乙酸乙烯酯、胆酸钠盐、二辛基磺基琥珀酸钠、十六烷基三甲基溴化铵、皂苷、糖酯、Triton X系列、脱水山梨醇三油酸酯、脱水山梨醇单油酸酯、聚氧乙烯(20)脱水山梨醇单月桂酸酯、聚氧乙烯(20)脱水山梨醇单油酸酯、油基聚氧乙烯(2)醚、硬脂基聚氧乙烯(2)醚、十二烷基聚氧乙烯(4)醚、氧化乙烯和氧化丙烯的嵌段共聚物、二乙二醇二油酸酯、四氢糠醇乙酸酯、油酸乙酯、肉豆蔻酸异丙酯、单油酸甘油酯、单硬脂酸甘油酯、单蓖麻油酸甘油酯、鲸蜡醇、硬脂醇、氯化十六烷基吡啶、苯扎氯铵、橄榄油、单月桂酸甘油酯、玉米油、棉籽油和葵花籽油。 在替代实施方案中,由化合物1或其药学上可接受的盐和化合物2或其药学上可接受的盐制备的固定剂量组合物通过包括溶剂或干法制粒,任选地随后压缩或压实、喷雾干燥、纳米混悬液加工、热熔挤出、挤出/滚圆、成型、滚圆、分层(例如,喷雾分层混悬液或溶液)等的方法制备。这类技术的实例包括使用适当的冲压机和模具直接压缩,例如其中冲压机和模具适配在适当的压片机上;使用合适的制粒设备(例如高剪切制粒机)的湿法制粒以形成待干燥成颗粒的湿颗粒;制粒,然后使用合适的冲压机和模具进行压缩,其中冲压机和模具适配在合适的压片机上;湿物料挤出以形成圆柱形挤出物,该挤出物在重力和磨损下被切割成所需长度或断裂成所需长度;挤出/滚圆,其中挤出物被滚圆成球形颗粒并通过滚圆而致密化;使用如常规锅或Wurster柱的技术将混悬液或溶液在惰性核心上喷雾成层;使用适配在压缩单元上的合适模具进行注射或压缩成型;等等。 示例性的崩解剂包括海藻酸、羧甲基纤维素钙、羧甲基纤维素钠、交联羧甲基纤维素钠(交联甲羧纤维素钠)、粉末状纤维素、壳聚糖、交联羧甲基纤维素钠、交聚维酮、瓜尔胶、低取代羟丙基纤维素、甲基纤维素、微晶纤维素、藻酸钠、淀粉乙醇酸钠、部分预胶化淀粉、预胶化淀粉、淀粉、羧甲基淀粉钠等,或其组合。 示例性润滑剂包括硬酯酸钙、硬酯酸镁、山嵛酸甘油酯、棕榈硬脂酸甘油酯、氢化蓖麻油、轻矿物油、十二烷基硫酸钠、十二烷基镁、硬酯基富马酸钠、硬脂酸、硬酯酸锌、二氧化硅、胶态二氧化硅、用二氧化硅处理的二甲基二氯硅烷、滑石或其组合。 本文所述的剂型核心可被包衣以形成包衣片剂。剂型核心可涂覆功能性或非功能性包衣,或功能性和非功能性包衣的组合。“功能性包衣”包括改变总组合物释放性质的片剂包衣,例如缓释或延迟释放包衣。“非功能性包衣”包括作为非功能性涂层的包衣,例如美容涂层。由于包衣的初始溶解、水化、穿孔等,非功能性包衣可能对活性剂的释放产生一些影响,但不会被视为与未包衣的组合物的重大偏离。非功能性包衣也可以掩盖包含活性药物成分的未包衣组合物的味道。包衣可包括光阻材料、光吸收材料或光阻材料和光吸收材料。 示例性聚甲基丙烯酸酯包括丙烯酸酯和甲基丙烯酸酯的共聚物,例如a.氨基甲基丙烯酸酯共聚物USP/NF,例如聚(甲基丙烯酸丁酯,(2-二甲基氨基乙基)甲基丙烯酸酯,甲基丙烯酸甲酯)1:2:1(例如,EUDRAGIT E 100、EUDRAGIT EPO和EUDRAGIT E 12.5;CASNo.24938-16-7);b.聚(甲基丙烯酸,丙烯酸乙酯)1:1(例如,EUDRAGIT L30 D-55、EUDRAGITL100-55、EASTACRYL 30D、KOLLICOAT MAE 30D和30DP;CAS No.25212-88-8);c.聚(甲基丙烯酸,甲基丙烯酸甲酯)1:1(例如,EUDRAGIT L 100、EUDRAGIT L 12.5和12.5P;也称为甲基丙烯酸共聚物,A型NF;CAS No.25806-15-1);d.聚(甲基丙烯酸,甲基丙烯酸甲酯)1:2(例如,EUDRAGIT S100、EUDRAGIT S12.5和12.5P;CAS No.25086-15-1);e.聚(丙烯酸甲酯,甲基丙烯酸甲酯,甲基丙烯酸)7:3:1(例如,Eudragit FS 30D;CAS No.26936-24-3);f.聚(丙烯酸乙酯,甲基甲基丙烯酸酯,甲基丙烯酸三甲铵乙基酯氯化物)1:2:0.2或1:2:0.1(例如,EUDRAGITS RL 100、RL PO、RL 30D、RL 12.5、RS100、RS PO、RS 30D或RS12.5;CASNo.33434-24-1);g.聚(丙烯酸乙酯,甲基丙烯酸甲酯)2:1(例如,EUDRAGIT NE 30D、Eudragit NE 40D、Eudragit NM 30D;CAS No.9010-88-2);等等,或其组合。 合适的烷基纤维素包括例如甲基纤维素、乙基纤维素等,或其组合。示例性水基乙基纤维素涂层包括AQUACOAT,进一步包含十二烷基硫酸钠和十六醇的30%分散体,可从FMC,Philadelphia,PA获得;SURELEASE,进一步包含稳定剂或其他涂层组分(例如油酸铵、癸二酸二丁酯、胶态无水二氧化硅、中链甘油三酯等)的25%分散体,可从Colorcon,WestPoint,PA获得;乙基纤维素,可从Aqualon或Dow Chemical Co(Ethocel),Midland,MI获得。本领域技术人员将理解,其他纤维素聚合物,包括其他烷基纤维素聚合物,可以替代部分或全部乙基纤维素。 可用于制备功能性包衣的其他合适材料包括乙酸羟丙甲纤维素琥珀酸酯(HPMCAS);乙酸纤维素邻苯二甲酸酯(CAP);聚乙酸乙烯邻苯二甲酸酯;中性或合成蜡、脂肪醇(例如十二烷醇、十四烷醇、硬脂醇、十六烷醇或特别地鲸蜡硬脂醇)、脂肪酸(包括脂肪酸酯)、脂肪酸甘油酯(单、二和三甘油酯)、氢化脂肪、碳氢化合物、普通蜡、硬脂酸、硬脂醇、具有烃主链的疏水和亲水材料或其组合。合适的蜡包括蜂蜡、糖蜡、蓖麻蜡、巴西棕榈蜡、微晶蜡、小烛树蜡和蜡样物质,例如在室温下通常为固体且熔点为约30℃至约100℃的材料,或其组合。 在其他实施方案中,功能性包衣可包括可消化的长链(例如,C8-C50,特别是C12-C40)、取代或未取代的烃,例如脂肪酸、脂肪醇、脂肪酸的甘油酯、矿物油和植物油、蜡或其组合。可以使用熔点在约25℃至约90℃之间的烃。具体地,可以使用长链烃材料、脂肪(脂族)醇。 包衣可任选地包含另外的药学上可接受的赋形剂,例如增塑剂、稳定剂、水溶性组分(例如成孔剂)、抗粘剂(例如滑石)、表面活性剂等,或其组合。 功能性包衣可包含影响功能性包衣的释放性能的释放调节剂。例如,释放调节剂可用作成孔剂或基质破裂剂。释放调节剂可以是有机的或无机的,且包括在使用环境中可从包衣中溶解、提取或浸提的材料。释放调节剂可包括一种或多种亲水性聚合物,包括纤维素醚和其他纤维素类,例如羟丙基甲基纤维素、羟丙基纤维素、羟乙基纤维素、甲基纤维素、醋酸纤维素邻苯二甲酸酯或醋酸羟丙基甲基纤维素邻苯二甲酸酯;聚维酮;聚乙烯醇;丙烯酸聚合物,例如胃溶解的Eudragit FS 30D、pH敏感的Eudragit L30D 55、L 100、S100或L100-55;或其组合。其他示例性释放调节剂包括聚维酮;糖(例如,乳糖等);金属硬脂酸盐;无机盐(例如磷酸氢钙、氯化钠等);聚乙二醇(例如聚乙二醇(PEG)1450等);糖醇(例如,山梨醇、甘露醇等);碱金属烷基硫酸盐(例如十二烷基硫酸钠);聚氧乙烯脱水山梨醇脂肪酸酯(例如,聚山梨醇酯);或其组合。示例性的基质破裂剂包括水不溶性有机或无机材料。有机聚合物,包括但不限于纤维素、纤维素醚如乙基纤维素、纤维素酯如乙酸纤维素、乙酸纤维素丁酸酯和乙酸纤维素丙酸酯;淀粉可以用作基质破坏剂。无机破裂剂的实例包括多种钙盐,例如磷酸一钙、磷酸二钙和磷酸三钙;二氧化硅和滑石。 包衣可任选地包含增塑剂以改善包衣的物理性能。例如,由于乙基纤维素具有相对高的玻璃转化温度,并且在正常包衣条件下不形成柔性膜,因此在将乙基纤维素用作包衣材料之前向乙基纤维素中加入增塑剂可能是有利的。通常,包衣溶液中包含的增塑剂的量基于聚合物的浓度,例如,根据聚合物,可为约1%至约200%,但最常见的是聚合物的约1重量%至约100重量%。然而,增塑剂的浓度可通过常规实验确定。 对于乙基纤维素和其他纤维素的增塑剂的实例包括如癸二酸二丁酯、邻苯二甲酸二乙酯、柠檬酸三乙酯、柠檬酸三丁酯、三醋精或其组合的增塑剂,尽管可能的是其他水不溶性增塑剂(例如乙酰化单酸甘油酯、邻苯二甲酸酯、蓖麻油等)也可以使用。 对于丙烯酸聚合物的增塑剂的实例包括柠檬酸酯,例如柠檬酸三乙酯NF、柠檬酸三丁酯、邻苯二甲酸二丁酯、1,2-丙二醇、聚乙二醇、丙二醇、邻苯二甲酸二乙酯、蓖麻油、三醋精或其组合,尽管可能的是其他增塑剂(例如乙酰化单酸甘油酯、邻苯二甲酸酯、蓖麻油等)也可以使用。 可以使用合适的方法将包衣材料施加到剂型核心的表面。可以使用如简单或复杂的凝聚、界面聚合、液体干燥、热和离子凝胶化、喷雾干燥、喷雾冷却、流化床涂布、锅包衣或静电沉积的工艺。 在某些实施方案中,在剂型核心和外部包衣之间使用任选的中间涂层。这种中间涂层可用于保护活性剂或核心亚单元的其他组分免受外部包衣中使用的材料的影响或提供其他性能。示例性中间涂层通常包括水溶性成膜聚合物。这种中间涂层可包括成膜聚合物,例如羟乙基纤维素、羟丙基纤维素、明胶、羟丙基甲基纤维素、聚乙二醇、聚氧化乙烯等,或其组合;和增塑剂。增塑剂可用于降低脆性和提高抗张强度和弹性。示例性的增塑剂包括聚乙二醇丙二醇和甘油。 组合与交替治疗 药物抗性有时通过编码病毒复制中使用的酶的基因的突变而发生。通过在组合治疗中添加另外的化合物,可以延长、增强或恢复组合治疗对HCV感染的疗效。这种进一步的组合治疗可以与另一种、且甚至可能两种或三种其他抗病毒化合物(诱导不同的突变或通过不同于主要组合的不同途径发挥作用)一起或交替施用。或者,组合的药代动力学、生物分布、半衰期或其他参数可通过这种组合治疗(如果认为是协调的,其可包括交替治疗)进行改变。 本发明已经提供了通过施用选择的NS5B抑制剂和NS5A抑制剂为HCV或与HCV感染相关的病症的治疗有利的组合治疗。通过加入第三、第四或甚至第五活性剂(共配制或单独提供)可以获得附加的治疗效果。 由于化合物1和化合物1-A是NS5B聚合酶抑制剂,且化合物2是NS5A抑制剂,因此将化合物1和化合物2与例如以下组合施用于宿主可能是有用的: (1)蛋白酶抑制剂,如NS3/4A蛋白酶抑制剂; (2)另一NS5A抑制剂; (3)另一NS5B聚合酶抑制剂; (4)NS5B非底物抑制剂; (5)可聚乙二醇化或以其他方式修饰的干扰素α-2a,和/或利巴韦林; (6)非基于底物的抑制剂; (7)解旋酶抑制剂; (8)反义寡脱氧核苷酸(S-ODN); (9)适体; (10)核酸酶抗性核酶; (11)iRNA,包括microRNA和SiRNA (12)针对病毒的抗体、部分抗体或域抗体,或 (13)诱导宿主抗体反应的病毒抗原或部分抗原。 可与本发明的组合进一步组合或交替施用的另外抗HCV剂的非限制性例子包括 (i)蛋白酶抑制剂,例如特拉匹韦( (ii)NS5A抑制剂,例如ACH-2928、ACH-3102、IDX-719、达卡他韦、ledispasvir、velpatasvir(Epclusa)、艾尔巴韦(MK-8742)、格佐匹韦(MK-5172)和奥比他韦(ABT-267); (iii)NS5B抑制剂,例如AZD-7295、克立咪唑、达萨布韦(Exviera)、ITX-5061、PPI-461、PPI-688、索非布韦( (iv)NS5B抑制剂,如ABT-333和MBX-700; (v)抗体如GS-6624; (vi)组合药物,如Harvoni(ledipasvir/索非布韦);Viekira Pak(奥比他韦/帕利瑞韦/利托那韦/达萨布韦);Viekirax(奥比他韦/帕利瑞韦/利托那韦);G/P(帕利瑞韦和glecaprevir);Technivie 如果施用该组合以治疗导致肝癌或肝硬化的晚期丙型肝炎病毒,在一个实施方案中,化合物可与通常用于治疗肝细胞癌(HCC)的另一种药物组合或交替施用,例如,如Andrew Zhu在“New Agents on the Horizon in Hepatocellular Carcinoma”Therapeutic Advances in Medical Oncology,V 5(1),January 2013,41-50中所述。用于组合治疗(其中宿主患有HCC或具有HCC的风险)的合适化合物的例子包括抗血管生成药、舒尼替尼、布立尼布、利尼法尼(linifanib)、雷莫芦单抗、贝伐单抗、西地尼布、帕唑帕尼、TSU-68、仑伐替尼、抗EGFR抗体、mTor抑制剂、MEK抑制剂和组蛋白脱乙酰酶抑制剂、卡培他滨、顺铂、卡铂、多柔比星、5-氟尿嘧啶、吉西他滨、伊立替康、奥沙利铂、拓扑替康和其他拓扑异构酶 一般方法 1 实施例中使用了以下缩写: BID:一天两次 DCM:二氯甲烷 EtOAc:乙酸乙酯 EtOH:乙醇 GT:基因型 HPLC:高压液相色谱 LD:负荷剂量 NaOH:氢氧化钠 Na MeOH:甲醇 Na NH PE:石油醚 硅胶(230-400目,吸附剂) t-BuMgCl:叔丁基氯化镁 THF:无水四氢呋喃(THF) TP:三磷酸盐 实施例1.化合物1和化合物1-A的合成 部分A:(2R,3R,4R,5R)-5-(2-氨基-6-(甲基氨基)-9H-嘌呤-9-基)-4-氟-2-(羟甲基)-4-甲基四氢呋喃-3-醇(1-7)的合成 在步骤1中,将化合物1-1溶解在DCM中,且在加入氯甲酸苄酯之前,将反应冷却至10℃,然后加入NEt 部分B:异丙基(羟基(苯氧基)磷酰基)-L-丙氨酸酯酯的二氢奎宁盐(1-12)的合成 苯基二氯磷酸酯(1-8,150g,1.0当量)加入1300mL乙酸异丙酯中。将溶液冷却至-10℃±5℃,然后添加苯甲醇(1-9,80.6克,1.05当量)和Et l-丙氨酸异丙酯盐酸盐(1-10,125g,1.05当量)和Et 过滤反应混合物,滤饼用20mL乙酸异丙酯洗涤。滤液用1N HCl、水和碳酸氢钠水溶液洗涤。分离的有机层用无水硫酸钠干燥,然后在40-50℃真空下浓缩至干,得到240g非对映体混合物(约1:1)形式的粗产物1-11。(浅黄色油;产率:89.6%mol/mol;HPLC纯度:83.4%(按面积);HPLC分析物:86.2%w/w)。产物包含约6%-7%残留苯甲醇。粗产物1-4直接用于下一步。 化合物1-11(135g,1.0当量,86.2%分析物)和奎宁(100g,1.0当量)加入650mL i-PrOH中。加入5%Pd/C(19.2克,60%水,按KF)后,在封闭系统中使用氢气袋在20-25℃进行氢化8小时。反应完成后,混合物通过Büchner漏斗过滤。在真空下浓缩滤液以除去溶剂。 向上述残留物中加入300mL TBME。在40℃至45℃真空下浓缩混合物以除去溶剂,然后用另外300mL MTBE重复该步骤。向其中加入600mL MTBE,混合物在40-45℃下搅拌1小时,然后在0-5℃下再搅拌1小时。过滤混合物,用100mL MTBE洗涤滤饼。将饼在45℃下无真空干燥16小时,得到152g异丙基(羟基(苯氧基)磷酰基)-L-丙氨酸酯二氢奎宁盐(1-12,白色固体;产率:69.5%mol/mol;HPLC纯度:97.91%)。 部分C:化合物1的合成 异丙基(羟基(苯氧基)磷酰基)-L-丙氨酸酯二氢奎宁盐(1-12,5.9克,1.5当量)、化合物1-7(2.0克,1.0当量)、DIPEA(0.83克,1.0当量)和HATU(3.65克,1.5当量)加入100毫升二氯甲烷中。将混合物加热至40℃并搅拌18小时。通过TLC和HPLC对反应进行监测。 反应完成后,将反应混合物冷却至室温,用1N盐酸(100mL×2)、水(100mL×2)和5%碳酸氢钠水溶液(15mL×1)洗涤。分离的有机相用2g无水硫酸钠干燥,过滤,并在40-45℃真空下浓缩以得到黄色油。 加入乙酸异丙酯(10mL)。搅拌后,混合物在真空下浓缩。然后,加入25mL乙酸异丙酯。将混合物加热至45℃以得到澄清溶液。在室温下搅拌2小时后,过滤固体沉淀并在45℃下真空干燥15小时,以得到2.0g粗化合物1(产率:53.8%mol/mol;HPLC纯度:93.1%(按面积)(含有3.7%的具有相反R 将粗化合物1(2.0g)和15mL乙酸异丙酯的混合物加热至80-85℃以提供溶液。将溶液冷却至20-25℃并搅拌1小时。过滤沉淀的固体,用乙酸异丙酯(1毫升)洗涤,并在50℃下真空干燥16小时,以得到1.7克化合物1(产率:45.7%mol/mol;HPLC纯度:98.99%)。 实施例2.非晶和结晶化合物1的表征 非晶化合物1和结晶化合物1最初通过XRPD、 化合物1的样品从乙酸乙酯/己烷再结晶,并用ORTEP绘制。化合物1的绝对结构通过单晶的再结晶得到确认。图3是化合物1的ORTEP图。晶体数据和测量数据见表3。基于X射线晶体学的化合物1的绝对立体化学如下所示: DSC数据在配备50位自动进样器的TA Instruments Q2000上收集。热容量的校准使用蓝宝石进行,能量和温度的校准使用经认证的铟进行。典型地,在针孔铝盘中,约3mg的每个样品以10℃/min的速度从25℃加热至200℃。在样品上保持50ml/min的干燥氮气吹扫。仪器控制软件为Advantage for Q Series v2.8.0.394和Thermal Advantage v5.5.3,数据使用Universal Analysis v4.5A进行分析。 TGA数据于配备16位自动进样器的TA Instruments Q500 TGA上收集。使用经认证的Alumel和Nickel对仪器进行温度校准。通常,将5-10mg的每个样品装载到预去皮的铝DSC盘上,并以10℃/min的速度从环境温度加热至350℃。对样品维持60ml/min的氮气吹扫。仪器控制软件为Advantage for Q Series v2.5.0.256和Thermal Advantage v5.5.3,数据使用Universal Analysis v4.5进行分析。 非晶化合物1(1-1): 1 结晶化合物1(1-2): 1 表1.结晶化合物1的峰列表 /> /> 表2.来自非晶化合物1和结晶化合物1的HPLC色谱图的相对保留时间 表3.化合物1的晶体和数据测量 /> 该初始表征后在25℃/60%相对湿度(RH)下储存14天,7天和14天后进行HPLC和XRPD分析。图4A为25℃/60%(RH)下14天后的XRPD。非晶化合物1(样品1-1)保持差的结晶,而结晶化合物1(样品1-2)的结晶度保持,但两种化合物在25℃/60%(RH)下14天后均稳定。 实施例3.草酸盐化合物1-B的形成 最初,化合物1的草酸盐(化合物1-B)通过将草酸盐与溶剂(5体积,100μL)混合并使任何溶液在室温下蒸发而形成。任何悬浮液成熟(室温-50℃)3小时,并获得结晶度。 表4显示了化合物1-B产生中使用的不同溶剂。除两种(环己烷和正庚烷)外,所有溶剂均提供了结晶产物。尽管化合物1-B具有高的结晶度和溶解度,但由于可能形成肾结石,草酸盐在临床开发中不可接受,因而研究了化合物1的其他盐。 表4.草酸盐化合物1-B的形成 实施例4.非晶化合物1的盐化合物 由于草酸盐化合物1-B(实施例3)因其可能形成肾结石而未在临床试验中继续进行,化合物1的非晶盐用表5中列出的反离子形成。将化合物1溶解于叔丁醇(20体积,6ml)中,并用酸反离子处理溶液(每个样品1当量,除样品1-9,其具有0.5当量的硫酸盐)。然后将样品冷冻,以通过冷冻干燥去除溶剂。样品1-4、1-5、1-6、1-7、1-8和1-9中的残留固体最初通过XRPD和HPLC进行分析。 表5.非晶盐形成详情 所有样品均获取 样品1-4,HCl(1:1)盐: 1 样品1-5,硫酸(1:1)盐: 1 样品1-6,富马酸(1:1)盐: 1 样品1-7,苯甲酸(1:1)盐: 1 样品1-8,琥珀酸(1:1)盐: 1 样品1-9,硫酸(0.5:1)盐: 1 然后将样品在25℃/60%相对湿度(RH)下储存14天,并在7天(图4B)和14天(图5A)后通过HPLC和XRPD进行分析。所有制备的盐保持无定形状态,观察结果见表6。在研究的过程中,发现单硫酸盐(样品1-5)和琥珀酸盐(样品1-8)物理上不稳定且易潮解或变成胶状。发现富马酸盐(样品1-6)和苯甲酸盐(样品1-7)为玻璃状固体。HCl盐(样品1-4)发现保持其物理外观。令人惊讶的是,半硫酸盐(样品1-9)也保持白色固体的物理外观,与单硫酸盐化合物(样品1-5)(其是粘性胶)相反。结果见表6。单HCl盐(样品1-4)和半硫酸盐(样品1-9)在25℃/60%相对湿度(RH)下储存2周后发现是物理和化学稳定的。尽管两种盐在两周内均稳定,但半硫酸盐优于HCl盐,因为HCl盐是吸湿的,从而与半硫酸盐相比,在长期储存或使用中较少可用。 表6.样品在25℃/60%RH下7天和14天后的稳定性 /> 实施例5.非晶化合物1-A的表征 非晶化合物1-A最初通过XRPD、 表7非晶化合物1-A的峰列表 表8.非晶化合物1-A的HPLC色谱图 非晶化合物1-A: 1 实施例6.非晶化合物1-A的结晶 由于半硫酸盐发现在14天稳定性研究后仍保持为固体,如表6所示,因此进行了使用11种不同溶剂研究结晶条件的初步试验。非晶化合物1-A在25℃下悬浮于5体积的溶剂中(样品2-1、2-2、2-3、2-4、2-5、2-6、2-7、2-8、2-9、2-10和2-11)。向非自由流动的那些样品(2-1、2-2、2-3、2-4、2-5、2-6、2-7、2-8和2-10)中加入额外的5体积溶剂。然后,样品在25-50℃(温度之间1℃/min,每个温度下4小时)下成熟6天,样品2-1除外,其观察到在1天后为澄清溶液,并允许在环境条件下蒸发。结果见表9。晶体图谱从异丁醇(样品2-1)、丙酮(样品2-2)、EtOAc(样品2-6)和iPrOAc(样品2-7)的结晶得到。从使用MEK(样品2-4)和MIBK(样品2-5)的结晶也鉴定了两个不良结晶的样品。图7A示出了XRPD图谱。 表9.化合物1-A的结晶条件 七个样品(样品2-2、2-3、2-4、2-5、2-6、2-7和2-8)通过DSC、TGA、 表10.结晶化合物1-A样品的表征 所有样品均获取 样品2-2: 1 样品2-3: 1 样品2-4: 1 样品2-5: 1 样品2-6: 1 样品2-7: 1 样品2-8: 1 实施例7.非晶丙二酸盐(化合物1-B)未能结晶 如实施例3所示,在确定化合物1的适当盐时,发现了结晶的草酸盐,但由于其可能导致肾结石,因此无法在临床试验中继续使用。因此,尝试使用与用于半硫酸盐的相同11种溶剂进行化学相关的丙二酸盐(化合物1-E)的结晶。将化合物1(12×50mg,样品3-1、3-2、3-3、3-4、3-5、3-6、3-7、3-8、3-9、3-10、3-11和3-12)溶解在叔丁醇(20体积)中,然后用1当量的丙二酸储备溶液(THF中1M)处理该溶液。然后冷冻样品以通过冷冻干燥去除溶剂。在室温下向样品3-1、3-2、3-3、3-4、3-5、3-6、3-7、3-8、3-9、3-10和3-11中加入相关溶剂(5体积)。任何得到的溶液允许在环境条件下蒸发,而胶或固体在25-50℃(温度之间1℃/min,每个温度4小时)下成熟5天。固体通过XRPD分析(图12B),但发现所有样品形成胶或是非晶的(图12B)。结果见表11。一个固体(非晶)样品(3-12)通过 样品3-12: 1 表11.非晶丙二酸盐化合物1-E的结晶条件 *在室温下蒸发 实施例8.使用液体辅助研磨(LAG)未能充分成盐 使用表12中的14种酸性反离子进行液体辅助研磨(LAG)研究以确定半硫酸盐以外的适当盐。 表12.LAG结晶中使用的反离子储备液 /> 化合物1(30mg)置于具有两个3毫米滚珠轴承的HPLC小瓶内。材料用溶剂(15μl乙醇,样品4-1、4-2、4-3、4-4、4-5、4-6、4-7、4-8、4-9、4-10、4-11、4-12、4-13和4-14)润湿,并加入1当量的酸反离子。然后,使用带有Automaxion适配器的Fritsch研磨系统以650rpm的速度将样品研磨2小时。研磨后的大部分样品被发现是透明胶,未进一步分析(表13)。观察到含有固体的那些通过XRPD分析,并且在所有情况下,发现获得的图谱与结晶酸反离子的图谱匹配,没有额外的峰(图13B)。 表13.化合物1的LAG的观察和XRPD结果 /> 实施例9.使用甲乙酮(MEK)未能获得充分的成盐 甲乙酮(MEK)随后被用作溶剂以研究半硫酸盐以外的适当盐。使用表12中的14种酸反离子,通过在室温下将化合物1(50mg)溶解在MEK(20vol)中进行研究。用1当量选定的反离子处理溶液(表12)。然后将样品以0.1℃/分钟的速度冷却至5℃,并在该温度下搅拌过夜。所有样品在环境条件下蒸发,并通过XRPD分析观察到的任何固体。除了使用硬脂酸的样品(样品4-12)和棕榈酸的样品(样品5-13)(其提供玻璃状溶剂)外,这种蒸发主要产生胶状物。这些固体通过XRPD确定是非晶的,而没有获得盐的结晶形式。结果见表14。 表14.化合物1溶解在MEK(20倍体积)中的结果 /> 酸添加前制备的储备液 *样品通过XRPD分析,给出了非晶图谱和来自酸反离子的峰 由于所有样品均为非晶的,所有样品在MEK(5vol)中重新溶解,并在室温下加入环己烷(20vol反溶剂),随后在25℃下搅拌1小时。然后,样品在50-5℃之间(温度之间1℃/min,每个温度4小时)成熟2天,之后循环改变为50-25℃再持续4天。样品成熟后用肉眼观察。结果见表15。成熟后,除5-1(用扑酸)外,所有样品均发现为胶状物。样品5-1(黄色固体)由XRPD分析,发现其图谱与已知形式的扑酸相匹配(图14B),因此未获得盐的结晶形式。 表15化合物1在MEK(5倍体积)和反溶剂中再溶解的结果 **通过XRPD分析的样品,其图谱匹配已知形式的扑酸(无额外峰) 实施例10.使用乙酸乙酯未能获得充分成盐 接下来乙酸乙酯被用于研究除半硫酸盐以外的合适的盐。利用表12中的14种酸反离子,通过在50℃下将化合物1(50mg)溶解在乙酸乙酯(20vol)中进行研究。溶液用1当量选定的反离子处理(表12)。然后将样品以0.1℃/分钟的速度冷却至5℃,并在该温度下搅拌4天。溶液在环境条件下蒸发,同时通过XRPD分析任何固体。使用乙酸乙酯结晶的结果见表16。与以MEK为溶剂的实施例8相反,观察到大多数样品在酸:化合物混合物冷却后为悬浮液(溶液在环境条件下蒸发的那些)。然而,一般发现XRPD衍射图与晶体化合物1相匹配。样品6-2、6-4和6-5具有一些细微差异(图14A和图15A)。未获得盐的结晶形式。 表16化合物1溶解在乙酸乙酯(20倍体积)中的结果 /> 实施例11.通过HPLC的化学纯度测定 实施例2和实施例4中的纯度分析在配备有二极管阵列检测器的Agilent HP1100系列系统上,并使用表17所示的方法利用ChemStation软件vB.04.03进行。 表17.测定化学纯度的HPLC方法 /> 实施例12.X射线粉末衍射(XRPD)技术 实施例2、3、4、5、6、7、8和9中的XRPD图谱是在PANalytical Empyrean衍射仪上使用Cu Kα辐射(45kV,40mA)以透射几何采集的。在入射光束上使用0.5°狭缝、4mm掩蔽和0.4rad Soller狭缝以及聚焦镜。放置在衍射束上的PIXcel 金属板的扫描模式使用角度(gonio)扫描轴,而Millipore板使用2θ扫描。使用硅粉(金属孔板)进行性能检查。数据收集的细节是角度范围2.5°至32.0°2θ,步长0.0130°2θ,总收集时间2.07分钟。 样品还使用Cu Kα辐射(40kV,40mA)、θ-2θ测角仪和V4发散及接收狭缝、Ge单色仪和Lynxeye检测器在Bruker D8衍射仪上采集。使用经认证的Corundum标准(NIST 1976)对仪器进行性能检查。用于数据采集的软件为Diffrac Plus XRD Commander v2.6.1,数据使用Diffrac Plus EVA v15.0.0.0进行分析和呈现。 样品在环境条件下作为平板样本使用接收的粉末运行。将样品轻缓装入切割成抛光的零背景(510)硅片的空腔中。在分析过程中,样品在其自身平面内旋转。数据收集的细节为角度范围2°至42°2θ,步长0.05°2θ,收集时间0.5s/步。 实施例13.非晶化合物1-A的合成 250mL的烧瓶中加入甲醇(151mL),并将溶液冷却至0-5℃。在10分钟内滴加浓硫酸溶液。向单独的烧瓶中加入化合物1(151g)和丙酮(910mL),并在25-30℃下于2.5小时内滴加硫酸/甲醇溶液。大量固体沉淀。溶液在25-30℃下搅拌12-15小时后,过滤混合物,用甲醇/丙酮(25mL/150mL)洗涤,并在55-60℃真空干燥以得到化合物1-A(121g,74%)。 化合物1-A的分析方法:使用Agilent 1100HPLC系统与Waters XTerra Phenyl 5μm 4.6*250mm柱以下列条件获得化合物1-A的纯度:1mL/min流速,在254nm下读取,30℃柱温,10μL注射量,30分钟运行时间。样品溶于ACN:水(90:10,v/v)。用于分离的梯度法如下所示。化合物1-A的Rt(min)约为12.0分钟。 实施例14.化合物1-A的表征 化合物1-A进一步通过肉眼、 表18.化合物1-A的附加表征数据概述 实施例15.化合物1和化合物1-A的溶解度 测试化合物1和化合物1-A在生物相关测试介质中的溶解度,包括模拟胃液(SGF)、禁食状态模拟胃液(FaSSIF)和进食状态胃液(FeSSIF)。化合物1的结果见表19,化合物1-A的结果见表20。样品在室温(20-25℃)下搅拌。化合物1-A在水中的溶解度在2小时时比化合物1高40倍以上,在24小时时高25倍以上。在SGF条件下,化合物1-A在24小时的溶解度为84.2mg/mL,而在同一时间点化合物1的溶解度为15.6mg/mL。化合物1-A在SGF条件下2小时比化合物1更易溶解,并且即使在48小时后仍足够稳定以进行测试,而化合物1未在48小时进行测试。 表19.化合物1溶解度测试结果 *样品看起来是澄清的,但溶解度仅为1.5mg/毫升。经进一步研究,发现搅拌棒上形成胶状膜。在标准制备过程中,化合物1活性药物成分在稀释剂(90%水/10%乙腈)中形成胶状球,其需要较长的超声处理时间才能完全溶解。 表20.化合物1-A溶解度测试结果 实施例16.化合物1-A的化学稳定性 通过监测有机纯度、水含量、 化合物1-A的纯度通过Shimadzu LC-20AD系统和Waters XTerra Phenyl,5μm,4.6x250mm柱以下列条件获得:1mL/min流速、254nm读数、35℃柱温和10μL注射体积。样品溶于乙腈-水(90:10)(v/v)。梯度方法如下所示。 化合物1-A(250mg)的水含量通过水滴定装置使用Karl Fischer滴定法测定。 结果见表21和表22。当化合物1-A在25℃和40℃下储存6个月时,降解速率最低。在3个月时,化合物1-A在25℃条件下的纯度为99.75%,在40℃条件下纯度为99.58%。6个月时,化合物1-A在25℃条件下仍为99.74%纯,在40℃条件下为99.30%纯。在25℃下,降解产物的百分比从第0天的0.03%增加到6个月后的0.08%。在40℃下,降解产物的百分比从0.03%增加到0.39%。在6个月的过程内,水的百分比在25℃下增加约0.6%,在40℃下增加约0.7%。 化合物1-A在1个月、2个月、3个月和6个月时通过 表21.化合物1-在25℃和40℃下6个月内的降解速率 表22.化合物1-A在降解研究中的表征 对化合物1-A进行了另外的化学稳定性研究,以确定杂质和水的含量。测试了三个条件:6个月期间的加速稳定性(40±2℃/75±5%相对湿度)、9个月期间的环境稳定性(25±2℃/60±5%相对湿度)和9个月期间的冰箱条件(5±3℃)下的稳定性。加速稳定性、环境稳定性和冰箱条件的结果分别见表23、表24和表25。根据这些研究的结果,化合物1-A化学上非常稳定。 在加速稳定性研究(表23)中,在测量化合物1-A的每个时间点(第1个月、第3个月和第6个月),化合物1-A的外观始终为白色固体,IR符合参考标准。六个月后,总相关物质1杂质仅为0.08%,未检测到相关物质2和异构体。 表23.化合物1-A的加速稳定性(40±2℃/75±5%相对湿度) N.D.:未检测到 在测量外观、IR、水和杂质水平的为期九个月的环境稳定性研究中,化合物1-A的外观始终为白色固体,IR始终与参考样品一致。结果(表24)强调了化合物1-A的化学稳定性。9个月后,样品中水的百分比仅为0.20%,总相关物质1杂质仅为0.02%。与加速稳定性研究类似,未检测到相关物质2和化合物1-A的任何异构体。 表24.化合物1-A的环境稳定性(25±2℃/60±5%相对湿度) /> 注:未检测到 冰箱条件下稳定性的测量结果见表25。甚至在9个月后仅检测到来自相关物质1的杂质和水。9个月后的含水量为0.32%,总相关物质1杂质仅为样品的0.01%。化合物1-A在冰箱条件下化学上非常稳定。 表25.化合物1-A在冰箱条件(5±3℃)下的稳定性 /> 注:未检测到 实施例17.单次口服化合物1-A后的血浆代谢物水平 将化合物1-A的单次口服剂量施用大鼠、狗和猴,并测量方案1中所示的某些代谢物的血浆水平。 化合物1-A向化合物1和代谢物1-7的转化见表26,代谢物1-8和代谢物1-2的结果见表27。在大鼠中,观察到低水平的化合物1暴露,但观察到高水平的代谢物1-7,即活性三磷酸盐(代谢物1-6)的核苷代谢物。在猴中,测量了化合物1的大致剂量比例的暴露。在狗中,测量了超比例的化合物1暴露,表明在肝脏中的首过代谢清除。在整个研究过程中,狗(高剂量组5/5)中观察到明显高于猴(高剂量组1/5)的呕吐。 表26.单次口服化合物1-A后化合物1和代谢物1-7的血浆水平 每物种每剂量3只雄性;*剂量制剂: 表27.单次口服剂量的化合物1-A后代谢物1-8和1-2的血浆水平 每物种每剂量3只雄性;*剂量制剂: 实施例18.化合物1-A口服剂量后活性三磷酸盐的组织暴露 化合物1-A口服剂量4小时后测量化合物1-A的活性三磷酸盐(代谢物1-6)的心脏和肝组织水平。在单剂量化合物1-A后4小时获取肝脏和心脏样品,快速冷冻、均质化并通过LC-MS/MS分析活性TP的细胞内水平。在大鼠、狗和猴子中测量组织水平,如图16A所示。在所有受试物种的肝脏中均检测到高水平的活性TP。由于首过肝代谢的饱和,狗的心脏中检测到相对较低的活性TP水平,并且在大鼠和猴的心脏中检测到无法定量的TP水平,表明活性TP的肝脏特异性形成。虽然未显示,但与化合物1的给药相比,化合物1-A给药改善了TP分布。 实施例19.化合物1和化合物1-A在狗中的药理学比较 对施用化合物1和化合物1-A的狗进行了头对头的比较。研究测量了在施用化合物1(25mg/kg)和化合物1-A(30mg/kg)后至4小时的化合物1和代谢物1-7(来自方案1)的血浆水平(表28),并且与化合物1-A相比,化合物1-A的代谢物1-7的AUC 表28.化合物1和化合物1-A施用后的血浆水平比较 /> a 肝脏/心脏比率三磷酸盐浓度表明,与化合物1相比,化合物1-A给药增加了三磷酸盐向肝脏的选择性递送,如表29所示。施用化合物1后,在心脏中测得的活性鸟嘌呤代谢物(1-6)的AUC 表29.化合物1和化合物1-A施用后肝脏和心脏暴露的比较 a b 与化合物1相比,当施用化合物1-A时,肝脏相对心脏的选择性增加的效果也显示在图16B中。将化合物1-A(30mg/kg)剂量后的活性三磷酸盐的心脏和肝脏组织水平与化合物1(25mg/kg)剂量后的活性三磷酸盐的组织水平进行比较。化合物1和化合物1-A的活性TP在肝脏中的浓度均高于心脏,但与化合物1相比,当施用化合物1-A时活性TP对于肝脏的选择性高于心脏。 实施例20.化合物1-A代谢物在大鼠和猴中的血浆分布 给予雄性SD大鼠和食蟹猴(每剂量组3只动物)单次口服剂量化合物1-A。通过LC-MS/MS分析用Dichlorvos处理的血液样品制备的等份血浆中化合物1和代谢物1-7(方案1中所示的化合物1-A的活性三磷酸盐的核苷代谢物)的浓度,并使用WinNonlin确定药代动力学参数。大鼠中单次500mg/kg剂量的结果见图17,猴中单次30、100或300mg/kg剂量的结果见图18。结果也总结在表30中。 代谢物1-7(化合物1-A的活性三磷酸盐(TP)的核苷代谢物)的高血浆水平表明形成了高的TP水平,即使在由于化合物1在大鼠血液中的短半衰期(<2分钟)而观察到母体核苷酸前药的血浆水平非常低的大鼠中也是如此。代谢物1-7的持续血浆水平反映了TP的长半衰期。 在猴中,化合物1的血浆暴露(AUC)大致与剂量成比例,而代谢物1-7的暴露略低于剂量-比例,尽管母体药物和活性TP的核苷类代谢物的AUC值持续增加直到测试的最高剂量(300mg/kg)。 在大鼠和猴中口服施用化合物1-A产生了代谢物1-7(化合物1-A的细胞内活性三磷酸盐的核苷代谢物)的高的剂量依赖性的血浆暴露;代谢物1-7暴露持续提高,直至测试的最高剂量,反映了这些物种中活性TP的大量形成。 表30.单次口服剂量化合物1-A后化合物1和1-7的血浆水平 剂量制剂: 实施例21.化合物1和化合物1-A的活性三磷酸对线粒体完整性的影响 将化合物1和化合物1-A的活性三磷酸盐(TP),代谢物1-6(方案1),通过人线粒体RNA聚合酶掺入的相对效率与索非布韦的活性TP和INX-189的活性TP的相对效率进行比较。化合物1和化合物1-A较少可能影响线粒体完整性,因为它们的活性三磷酸盐很少通过人线粒体RNA聚合酶掺入,其掺入效率类似于索非布韦的三磷酸盐掺入效率;INX-189的三磷酸盐的相对掺入效率高达55倍高。结果见表31。根据Arnold等(Sensitivity ofMitochondrial Transcription and Resistance of RNA Polymerase IIDependentNuclear Transcription to Antiviral Ribonucleotides.PLoS Pathog.,2012,8,e1003030)确定了人线粒体依赖性RNA聚合酶(POLRMT)对这些类似物的掺入。 表31.用人线粒体RNA聚合酶评价核苷酸类似物的动力学参数 *相对效率=(K 实施例22.化合物1对含NS5B序列的复制子的活性 使用一组复制子来确定化合物1和索非布韦的效力,该复制子包含源自6个实验室参考株(GT1a、1b、2a、2b、3a、4a和5a)(图19)和8个HCV患者血浆样品(GT1a、1b、2a、2b、3a-1、3a-2、4a和4d)(图20)的各种HCV基因型的NS5B序列。 化合物1对HCV的临床和实验室HCV株的效力高于索非布韦。化合物1在体外以EC 实施例23.化合物1-A在健康志愿者(部分A)和GT1-HCV感染患者(部分B)中的单次递增剂量(SAD)研究 在单次递增剂量(SAD)研究中测试了化合物1-A,以测量其在健康受试者中的安全性、耐受性和药代动力学(部分A)。部分A是一项随机、双盲、安慰剂对照的研究。部分A的健康受试者在禁食状态下接受单剂量化合物1-A或安慰剂。受试者从第-1天至第6天被限制在诊所内。 每一群组中的给药是交错进行的,使得在给药后48小时内,对2名受试者(1名活性药物:1名安慰剂)进行评估,然后再对群组的其余受试者给药。每一群组按递增顺序接受化合物1-A。顺序群组的给药基于对前一群组的可用安全性数据(至第5天)和血浆药代动力学数据(至第24小时)的审查进行。 在对这些数据进行令人满意的审查后,剂量递增。随着从先前的群组中得出的药代动力学和安全性数据,在群组3a-4a中评估的剂量以不超过100mg的增量进行调整。部分A中评估的总最大剂量不超过800mg。部分A的给药方案见表32。 表32.研究部分A化合物1-A施用的给药方案 *临床剂量以化合物1-A表示,括号内为化合物1碱的大致等同量 研究的部分A部分的健康志愿者为年龄在18岁至65岁之间的男性和女性受试者。在每个部分A群组中汇集了活性药物和安慰剂接受者,以保持研究的盲性。 化合物1-A也在单次递增剂量(SAD)研究中进行了测试,以测量其在GT1-HCV感染患者中的安全性、耐受性、药代动力学和抗病毒活性(部分B)。部分B的受试者在禁食状态下接受单一剂量的化合物1-A。从第-1天到第6天,患者被限制在诊所内。 部分B是在对部分A群组3a的安全性(至第5天)和血浆药代动力学(至第24小时)数据进行审查后开始的。在登记后续部分B群组前,对部分B第一群组(群组1b)的可用安全性数据(至第5天)和药代动力学数据(至第24小时)进行审查。仅在审查部分A中相应剂量的可用安全性和药代动力学数据以及部分B前一群组的可用安全性(至第5天)的审查后,才对后续部分B群组进行给药。 在对这些数据进行令人满意的审查后,HCV感染患者的剂量递增直至600mg。部分B的给药方案见表33。 表33.研究的部分B中化合物1-A的给药方案 *临床剂量以化合物1-A表示,括号内为化合物1碱的大致等同量。 感染HCV的患者为病毒负荷≥5log 部分A或部分B中均未记录任何严重不良事件,也未要求提前终止。所有不良反应的强度均为轻度至中度,且无明显的剂量相关分布,包括实验室参数、生命体征和ECG。 实施例24.化合物1-A的单次递增剂量(SAD)研究结果 化合物1和核苷代谢物1-7的药代动力学在单剂量化合物1-A后进行测量。在HCV感染患者中,600mg剂量化合物1-A后的代谢物1-7的C 表34.单剂量化合物1-A施用后在健康志愿者中化合物1和代谢物1-7的人体药代动力学 /> *基于24小时分布。 表35.化合物1-A施用后在HCV感染患者中化合物1和代谢物1-7的人体药代动力学 /> *基于24小时分布。 还计算了研究的部分A和部分B所有群组的化合物1和代谢物1-7的平均血浆浓度-时间分布。图21是单剂量化合物1-A后化合物1的平均血浆浓度,图22是单剂量化合物1-A后代谢物1-7的平均血浆浓度。如图21所示,化合物1在部分B的所有群组中被快速吸收和快速/广泛代谢。如图22所示,代谢物1-7是主要代谢物,并表现出持续的血浆浓度。化合物1的血浆暴露是剂量相关的,而代谢物1-7的暴露与剂量成比例。 对于部分B的HCV感染受试者,在施用化合物1-A之前、期间和之后进行HCV RNA定量测量。通过使用经验证的商业分析进行血浆HCV RNA测定。基线定义为第-1天和第1天(给药前)的平均值。单次300mg剂量化合物1-A(相当于270mg化合物1)对GT1b-HCV感染受试者产生显著的抗病毒活性。单次300mg剂量后的给药后24小时平均最大HCV RNA减少为1.7log 实施例25.针对HCV GT 1-4临床分离株,代谢物1-7的预测稳态谷水平超过化合物1EC 如图24所示,预测人体化合物1-A给药(600mg QD(550mg游离碱当量)和450mg QD(400mg游离碱当量))后代谢物1-7的稳态谷血浆水平(C 如图24所示,化合物1-A对所有测试的临床分离株的EC 如图24所示,化合物1-A在人体内以450mg QD(400mg游离碱当量)的剂量提供了约40ng/mL的稳态谷血浆浓度(C 因此,针对所有测试的临床分离株(甚至是难以治疗的GT3a),代理代谢物1-7的稳态血浆浓度几乎是EC95的两倍,这表明其性能优越。 相反,在所有测试的HCV临床分离株上,标准治疗核苷酸索非布韦的EC 化合物1-A 450mg稳态谷血浆浓度(C 通过三种方法预测600mg稳态谷血浆浓度(C 比较图24中功效和药代动力学稳态参数的数据清楚地表明,化合物1-A对于丙型肝炎的治疗具有意想不到的治疗重要性。事实上,化合物1-A施用后的预测稳态血浆水平预计比对于所有测试的基因型的EC 实施例26.评估化合物1-A的安全性/耐受性、药代动力学(PK)和抗病毒活性的三部分研究 对化合物1-A进行了三部分研究,以评估安全性/耐受性、药代动力学和抗病毒活性。这三个部分包括:1)在NC(非肝硬化的)GT1 HCV感染的丙患者中,每天一次(QD)施用最多600mg剂量化合物1-A(相当于550mg化合物1)的多个剂量,持续7天(部分C);2)600mg化合物1-A(相当于550mg化合物1)QD施用于NC GT3 HCV感染患者7天(部分D);和3)在患有GT1、GT2或GT3 HCV感染的Child-Pugh A(CPA)肝硬化患者群组中施用600mg化合物1-A(相当于550mg化合物1)QD 7天(部分E)。剂量以化合物1-A盐基的形式施用。游离碱化合物1当量通常在括号中给出。 部分C是随机、双盲、安慰剂对照的MAD研究,分为三个群组。受试者在禁食状态下给予150mg、300mg或600mg化合物1-A或安慰剂7天。只有在对数据进行令人满意的审查后,剂量才递增。部分D和部分E为开放标签研究,其中患者在禁食状态下接受600mg化合物1-A(相当于550mg化合物1)的剂量7天。 HCV感染患者未进行治疗,HCV RNA≥5log 部分A和部分B先前进行和在WO 2018/144640中描述。部分A和部分B为单次递增剂量(SAD)研究。在部分A中,健康受试者给予最多400mg化合物1-A(相当于367mg化合物1),在部分B中,GT1HCV感染受试者给予最多600mg化合物1-A(相当于550mg化合物1)。 实施例27.化合物1-A的研究结果 未报告任何严重不良事件、剂量限制毒性或提前停止。化合物1-A在最高测试剂量(600mg盐形式)持续7天耐受良好。观察到的唯一模式是,接受化合物1-A的受试者与接受安慰剂的受试者相比,大多数低级别脂质异常(胆固醇和甘油三酯提高)的发生率较高。然而,这一观察结果与显示在HCV感染受试者开始接受DAA治疗后伴随HCV清除的脂质的快速增加的先前公布的数据一致。另外,没有发现表明存在肝损伤。在接受化合物1-A的受试者中,ALT/AST值在治疗期间随时间下降。最后,在分析AE、实验室参数、ECG和生命体征时,不存在其他临床相关的剂量相关模式。 在部分B中,将相当于92mg、275mg、368mg或550mg化合物1的单剂量化合物1-A施用于分成给药群组(每群组n=3)的非肝硬化GT1b HCV感染受试者,以确定平均最大HCV RNA减少,结果见图25和表36。单剂量600mg化合物1-A(相当于550mg化合物1)施用于非肝硬化GT1b HCV感染受试者(n=3)导致平均最大HCV RNA减少2.3log 表36.单剂量化合物1-A后GT1b HCV患者中HCV RNA相对于基线的变化 /> 在部分C中,在非肝硬化GT1b HCV感染受试者(n=6)中,给药后7天观察到剂量相关的抗病毒活性,平均最大HCV RNA减少高达4.4log 在部分D中,在非肝硬化的GT3 HCV感染受试者(n=6)中观察到强效的抗病毒活性,平均最大HCV RNA减少为4.5log 部分E的CPA肝硬化HCV感染受试者中的抗病毒活性与非肝硬化GT1b和GT3群组相似。在部分E中,肝硬化HCV感染患者的平均最大HCV RNA减少为4.6log 部分C、部分D和部分E的平均最大HCV RNA变化见表37。图28A-28C是比较部分C的感染GT1 HCV的非肝硬化受试者、部分D的感染GT3 HCV的非肝硬化受试者和部分E的感染GT1/GT2/GT3 HCV的肝硬化受试者的平均最大减少的图。无论受试者是否感染GT1 HCV或GT3 HCV且无论受试者是否有肝硬化,受试者在给药7天后的平均最大减少相似。表37和表38总结了所有这些群组的抗病毒活性。在肝硬化受试者中观察到显著的早期病毒反应,在GT1和GT3受试者在最初24小时内分别导致2.4和2.2log 表37.部分B、部分C、部分D和部分E中的最大HCV RNA变化 *95%C.I. 表38.对于600mg化合物1-A的部分C、部分D和部分E中化合物1-A抗病毒活性总结 化合物1(化合物1-A的游离碱)被快速且良好地吸收,基于尿回收率,估计吸收的分数约为50%。在禁食状态下重复QD施用七天后,化合物1被快速吸收,随后快速代谢激活。 在部分C中每日施用7天后,化合物1显示出短的半衰期,并且不会随时间累积。化合物1的血浆暴露量略高于150mg至300mg之间的剂量比例,此后大部分为与剂量成比例。虽然代谢物1-7的血浆峰和总暴露量在150-300mg之间成剂量比例和300-600mg之间低于剂量-比例,但在研究的剂量范围内,代谢物1-7的谷水平大部分是成比例的。基于代谢物1-7谷水平,第三或第四剂量后基本达到稳态PK。代谢物1-7的形成在给药后约6小时达峰,代谢物1-7显示出长的半衰期(约13-30小时),这支持每天一次(QD)施用。在达到稳态后,长的半衰期导致所需的较高代谢物1-7谷值(50%-60%)。(活性三磷酸盐1-6在血浆中无法测量,因为其不离开细胞,因此血浆中可测量的1-7作为三磷酸盐1-6的替代并反映细胞内活性三磷酸盐)。 到第3天或第4天,代谢物1-7浓度在NC患者中达到稳态;在第5天,在肝硬化患者中达到稳态。总体而言,根据血浆暴露,轻度肝损害未显著影响化合物1-A的PK。未观察到食物对代谢物1-7的总暴露量和谷暴露量的影响。 图29是稳态下代谢物1-7的平均血浆浓度-时间分布图,比较了施用化合物1-A(相当于138mg/d QD的化合物1)的非肝硬化GT1 HCV感染的受试者、施用化合物1-A(相当于275mg/d QD的化合物1)的非肝硬化GT1 HCV感染的受试者、施用600mg的化合物1-A(相当于550mg的化合物1)的非肝硬化GT3 HCV感染的受试者和施用600mg的化合物1-A(相当于550mg的化合物1)的GT1或GT3 HCV感染的肝硬化受试者。使用LC-MS/MS测量代谢物1-7的血浆水平。 表39A、39B和39C显示了参与研究的受试者的平均PK结果。如表39A-39C和图29所示,非肝硬化和肝硬化受试者中的代谢物1-7的PK相似。 表39A.化合物1和代谢物1-7在第1天和稳态(SS)下的C 表39B.化合物1和代谢物1-7在第1天和稳态(SS)下的AUC和T #化合物1的AUC 表39C.化合物1和代谢物1-7在第1天和稳态(SS)下的C *C 图30A-30D是对GT1 HCV感染的非肝硬化受试者(图30A)、GT3 HCV感染的非肝硬化受试者(图30B)、GT1 HCV感染的肝硬化受试者(图30C)和GT3 HCV感染的肝硬化受试者(图30D)的PK/PD分析。左侧y轴为平均代谢物1-7浓度,右侧y轴为平均HCV RNA减少。水平虚线(-----)代表化合物1的EC 通过绘制代谢物1-7的AUC与HCV RNA减少的曲线生成的E 实施例28.化合物1-A的制剂描述和制造 化合物1-A片剂(50mg和100mg)的代表性非限制性批次配方见表40。片剂采用图32所示的直接压制工艺由通用共混物生产。 表40.50mg和100mg化合物1-A片剂的制剂 化合物1-A基于原样分析进行调整,同时调整微晶纤维素的百分比。将化合物1-A和赋形剂(微晶纤维素、乳糖一水合物和交联羧甲基纤维素钠)过筛,放入V型搅拌机(PKBlendmaster,0.5L碗)中,以25rpm混合5分钟。然后硬脂酸镁过筛、添加,并将共混物再混合2分钟。将通用共混物分开用于50mg和100mg片剂产生。然后,使用单冲研究压片机(KorschXP1)和重力粉末输送机以10片/分钟的速度对润滑的共混物进行压缩。使用圆形标准凹入6毫米工具和3.5kN力生产50mg片剂。100mg片剂使用8mm圆形标准凹口工具和3.9-4.2kN力生产。50mg和100mg片剂的规格见表41。 表41.化合物1-A的50mg和100mg片剂规格 如上所述生产的50mg和100mg片剂在三种条件下进行6个月的稳定性研究:5℃(冷藏)、25℃/60%相对湿度(环境)和40℃/75%相对湿度(加速)。50mg和100mg片剂在所有三种测试条件下均具有化学稳定性。 在冷藏条件(5℃)下,50mg和100mg片剂均保持白色固体,外观在T=0至T=6个月期间未发生变化。在为期6个月的研究中,无论是50mg片剂还是100mg片剂,均未报告任何大于0.05%的杂质。两种片剂6个月后的水含量也低于3.0%w/w。当片剂经历环境条件(25℃/60%相对湿度)时,报告了类似的结果;两种片剂在整个6个月内均未报告超过0.05%的杂质,且在6个月标记时水含量不超过3.0%w/w。当片剂处于加速条件(40℃/75%相对湿度)下时,50mg和100mg片剂的外观没有从白色圆形片剂发生改变。3个月后报告了一种杂质,但杂质仅为0.09%。6个月后报告了第二种杂质,但50mg和100mg片剂的总杂质百分比仅为0.21%。50mg片剂在6个月时的水含量为3.4%w/w,100mg片剂为3.2%w/w。 在单独的研究中,测量了50mg和100mg化合物1-A片剂在环境条件(25℃/60%相对湿度)下9个月的稳定性。50mg和100mg片剂的外观在9个月的过程中没有从白色圆形片剂发生改变。9个月后,50mg片剂中的杂质小于0.10%,100mg片剂中的杂质小于0.05%。9个月后,50mg片剂和100mg片剂的水含量分别仅为2.7%w/w和2.6%w/w。 实施例29:化合物2的合成 化合物2-9的合成 向250毫升圆底烧瓶中加入3-氯-5-氟苯酚(化合物2-1)和2-(2-溴-5-氯苯基)乙酸(化合物2-2,20.02克,80.0毫摩尔),与TfOH(91毫升)混合,并在氮气氛下加热至60℃。在该温度下搅拌16小时后,将混合物冷却至室温,并在20分钟内倒入已在冰/水浴中冷却的异丙醇(500mL)中。用水(125mL)(在10分钟内加入)稀释所得的浆液。在冰/水浴中老化30分钟后,过滤混合物,收集的固体用4:1异丙醇/水(50mL)洗涤。真空干燥固体,得到化合物2-3(20.14克,53.3毫摩尔,80%产率)。 将化合物2-3(2.03克,5.37毫摩尔)溶于2-甲基四氢呋喃(20.3毫升,10倍体积)中,并向该溶液中加入甲醇中的氨(11.51毫升7N,81毫摩尔,15当量)。)。所得溶液在室温下老化16小时,然后通过除去25mL溶剂进行浓缩和用甲苯(70mL)处理浆液。然后将所得溶液再蒸馏以进一步除去35mL溶剂,得到化合物2-5在20倍体积甲苯中的最终溶液。该溶液无需进一步纯化即可使用。 将化合物2-5(25克,163毫摩尔)、4-甲氧基苯胺(22.1克,180毫摩尔)和异丙醇(250毫升)装入配备空气冷凝器的1L圆底烧瓶中。将所得浆液加热至50℃并搅拌3.5小时,在此期间形成沉淀。将所得浆液冷却至0℃,老化1小时,过滤。用0℃的异丙醇(84mL)冲洗烧瓶和垫两次,固体在50℃的真空烘箱中干燥至恒重,得到化合物2-6。(39.0g,93%产率)。 将化合物2-6(7.54克,29.2毫摩尔)装入配备磁力搅拌棒、温度探头和氮气入口的3颈圆底烧瓶中,并加入化合物2-4在甲苯中的溶液(5.72重量%,174.8克,26.5毫摩尔)。将所得悬浮液在室温下搅拌直至固体溶解,并使用冰/水浴冷却所得溶液。加入TFA(2.45毫升,31.8毫摩尔),同时内部温度保持在5℃以下。将所得溶液在冰/水浴中搅拌16小时,随着其升温至室温。过滤所得浆液,用甲苯(27mL)洗涤烧瓶和垫,用碳酸氢钠水溶液(4wt%,54mL)和水(54mL)洗涤有机溶液。有机层真空浓缩至约25mL,用异丙醇(110mL)稀释,真空浓缩至总体积约50mL。将所得浆液加热至40℃,用水(10mL,30分钟内加入)稀释,在0℃老化1小时,过滤。用4:1异丙醇/水(25mL)洗涤烧瓶和垫,固体在50℃的真空烘箱中干燥至恒重,得到化合物2-7。(10.1g,74%产率) 向Pd(OAc) 在以下条件下使用SFC确定ee: 柱:ChiralCel OJ-3;4.6mm×150mm;3μm颗粒尺寸 温度:40℃ 压力:200巴 改性剂:添加25mM异丁基胺的IPA 流速:3.0毫升/分钟 条件:经5分钟1%改性剂/99%二氧化碳至40%改性剂/60%二氧化碳,在40%改性剂下保持1分钟 对于纯化的化合物2-9:(23mg,90%产率,91%ee)。 在45℃下将粗产物化合物2-9溶解于约50毫升甲苯和128毫升iPAC中。在45℃下分三份在2.5小时内加入(S)-樟脑磺酸(10.8克,46.4毫摩尔)。将其冷却至室温,并加入另外的(S)-樟脑磺酸(0.57克,2.4毫摩尔)。混合物在室温下老化16小时,然后过滤。固体用50ml1/2.5甲苯/乙酸异丙酯洗涤,然后用50ml乙酸异丙酯洗涤,并真空干燥,得到27.1g化合物2-9,为96%至>99%ee的樟脑磺酸盐。 化合物2的合成 在具有顶部搅拌器的500mL 3颈圆底烧瓶中,在氮气下加入化合物2-9((S)-CSA盐、10.0g或等同量的游离碱)、双(频哪醇合)二硼(8.50g)、乙酸钾(8.78g)和5-氯吲哚(0.46g)。加入脱气的2-Me-THF(130mL)和水(0.54mL)。在单独的容器中在氮气下加入乙酸钯(0.067g)和Xphos(2-二环己基膦基-2’,4’,6’-三异丙基联苯基)(0.293g)和脱气2-Me-THF(20mL),将混合物搅拌30分钟,然后向烧瓶加入化合物2-9。然后将混合物加热至75℃并在该温度下老化1小时或直至完全转化,然后冷却至室温。向混合物中加入水(30mL),分层。有机层用10%盐水(30mL)洗涤,然后用Cuno-3-碳(1.0g)处理约15小时。通过硅藻土垫过滤混合物以去除碳。将溶液真空浓缩至约35毫升混合物。加入种晶以引发结晶。混合物在室温下老化10分钟,然后缓慢加入乙腈(105mL)。过滤所得浆液,收集的固体用乙腈/2-Me-THF混合物(3:7,30mL)洗涤,然后在氮气流中干燥,得到化合物2-10。1HNMR(CDCl3,500MHz),8.23(s,1H),7.70(d,J=8.3Hz,1H),7.50(s,1H),7.31(d,J=9.6Hz,1H),7.29(d,J=7.1Hz,1H),7.23(d,J=8.3Hz,1H),7.18(d,J=3.3Hz,1H),7.10(s,1H),2.15-2.10(m,1H),1.39(s,6H),1.37(s,3H),1.37(s,3H),1.06-1.02(m,4H),1.01-0.95(m,4H)。 向高压容器中加入化合物2-10(10.0克,16.28毫摩尔,1.0当量)、化合物2-11(11.5克,2.15当量)、2-Me-THF(90mL)和K 将2-Me-THF(135mL)中的粗产物(14.66g)加入到三正丁基膦(2.32mL)的甲醇(19.4mL)溶液中。将混合物加热至70℃,然后加入溶于2-Me-THF(3.87mL)的(S)-扁桃酸(0.94g)溶液。在70℃老化数小时后,将反应混合物冷却至60℃,并加入另一份(S)-扁桃酸(3.06g)的2-Me-THF溶液(12.58mL)。该批次接种扁桃酸盐化合物2-12。在60℃下,在4小时内加入最后部分的2-Me-THF(22.25mL)中的S-扁桃酸(35.41g)。反应混合物在8小时内逐渐冷却至20℃,并在20℃下老化1小时。过滤浆液,用2-Me-THF(含2%重量的S)-扁桃酸)冲洗。收集的固体在60℃下干燥,得到固体的双扁桃酸盐化合物2-12(18.74g)。 将双扁桃酸盐化合物2-12(6克)与乙酸乙酯(48毫升)和水(25.7毫升)混合。在10分钟内向两相混合物中加入2M碳酸钾溶液(6mL,2.5当量),在此期间产生两相溶液。除去下层水层,有机层依次用8%盐水溶液(30mL)和水(2×30mL)洗涤。有机层通过蒸馏共沸干燥(最终溶液体积=30mL)。将庚烷(66mL)加入惰化烧瓶中。在2小时内向庚烷中加入含产物的乙酸乙酯流。在老化另外2小时后,过滤产物浆液,用庚烷(10.8mL)和乙酸乙酯(2mL)的混合物洗涤湿滤饼。固体在60℃真空干燥约15小时,得到化合物2作为游离碱。(4.40g,MS:M+H947.4047)。1H NMR(d6-DMSO,500MHz)δ(ppm)8.30(s,1H),8.22(br s,1H),8.10(br s,1H),8.00(s,1H),7.78(d,J=8.7Hz,1H),7.67-7.65(m,2H),7.52(br s,1H),7.38(s,1H),7.31-7.28(m,2H),7.19(d,J=3.2Hz,1H),5.16(t,J=7.4Hz,1H),5.14(t,J=7.4Hz,1H),4.15-4.11(m,2H),3.91-3.81(m,4H),3.55(s,6H),2.45-2.36(m,2H),2.26(m,1H),2.20-2.13(m,2H),2.13-2.06(m,2H),2.06-2.00(m,4H),0.99(m,2H),0.85-0.77(m,14H).13CNMR(d6-DMSO,126MHz)δ(ppm)175.20,171.21,171.15,158.61(d,J=251.0Hz),156.95,156.94,150.05,149.54(d,J=7.3Hz),148.88,141.37,133.88,133.00,131.11,130.68,129.14,128.78,125.35,121.40,120.38,118.15,117.01,114.18,111.04,110.75,107.30(d,J=23.4Hz),106.67(d,J=18.2Hz),102.94(d,J=8.6Hz),78.46,57.95,57.93,53.06,52.91,51.51,47.16,47.11,31.02,30.95,29.08,24.80,24.75,19.35,19.32,17.74,13.88,11.17,11.05。 实施例30:化合物1和化合物2的协同抗HCV活性 细胞培养-报告细胞系Huh-luc/neo-ET携带持续复制的13sgluc-ubi-neo/NS3-3'/ET复制子,其含有萤火虫荧光素酶基因-泛素-新霉素磷酸转移酶融合蛋白和EMCV·IRES驱动的NS3-5B HCV编码序列,该编码序列含有ET组织培养适应性突变(E1202G、T1208I和K1846T)。Huh-luc/neo-ET的原种培养物通过添加10%FCS、2mM谷氨酰胺、青霉素(100IU/ml)/链霉素(100μg/ml)和1X非必需氨基酸加1mg/ml G418的DMEM中培养物而扩增。将细胞以1:4分开,并在相同的培养基加250g/ml G418中传代两次。用胰蛋白酶处理细胞,并通过用台盼蓝染色计数,并以每孔7.5×103个细胞的细胞培养密度接种到96孔组织培养板中,和在37℃5%二氧化碳下孵育24小时。 化合物添加-孵育24小时后,移除培养基,用相同的培养基减去G418加稀释的测试化合物替换,一式三份。每个板中的六个孔仅接受介质作为无处理对照。细胞在37℃5%二氧化碳下再孵育72小时,然后通过荧光素酶终点检测抗HCV活性。重复板平行处理和孵育用于通过XTT染色评估细胞毒性。 细胞活力-在孵育72小时后,用四唑染料XTT(2,3-双(2-甲氧基-4-硝基-5-磺基苯基)-5-[(苯氨基)羰基]-2H-氢氧化四唑胺对处理的细胞的细胞培养单层进行染色,以评估在化合物存在下Huh-luc/neo-ET报道细胞系的细胞活力。细胞用四唑染料XTT染色。XTT-四唑被代谢活性的细胞的线粒体酶代谢成可溶甲臜产物,从而允许快速定量分析抗病毒测试物质对病毒诱导的细胞杀灭的抑制。XTT溶液以RPMl1640中1mg/ml的储备液每日制备。在PBS中以0.15mg/ml制备吩嗪甲氧基硫酸盐(PMS)溶液,并在-20℃下黑暗中储存。在使用前通过添加每ml XTT溶液40μl PMS即时制备XTT/PMS原液。向板的每个孔中加入50微升XTT/PMS,并将板在37℃下再孵育4小时。用粘合平板密封机将板密封,并轻轻摇动或倒置几次以混合可溶的甲臜产物,并用分Molecular Devices Vmax平板阅读仪在450/650nm下对板进行分光光度读数。 病毒复制的测量-根据制造商的说明(Perkin Elmer,Shelton,CT),使用britelite plus发光报告基因试剂盒在孵育72小时后通过荧光素酶活性测量复制子分析系统的HCV复制。简而言之,一小瓶britelite plus冻干基质溶解于10ml britele重构缓冲液中,并通过倒置轻轻混合。在室温下孵育5分钟后,将britelite plus试剂以每孔100μL的速度加入96个孔板中。平板用粘合剂膜密封,并在室温下孵育约10分钟以溶解细胞。将孔内容物转移至白色96孔板上,并在15分钟内使用Wallac 1450 Microbeta Trilux液体闪烁计数器测量发光。数据被导入定制的Microsoft Excel 2010电子表格中用于确定50%病毒抑制浓度(EC50)以进行单一浓度评估。 组合治疗分析 在上述抗HCV试验中,化合物2使用0.008nM的高测试浓度和四个连续的两倍稀释度与九个化合物1浓度组合进行评估。在抗病毒试验中,将50微升每种化合物以4倍(4x)所需浓度加入含细胞的96孔微量滴定板。向孔中加入50微升分析培养基,其中评价测试化合物作为单一化合物的抗病毒活性。 数据分析-原始数据从Softmax Pro收集并导入Prichard and ShipmanMacSynergy II软件模板(Prichard等1993.Antiviral Research 14:181-206)。药物组合的效果基于单独测试时两种化合物的活性进行计算。从实验确定的每一组合浓度下的抗病毒活性减去预期的加性抗病毒保护,得到正值(协同或增强)、负值(拮抗作用)或零(加性)。在每个组合浓度下,组合分析的结果以三维形式呈现,产生了在加性平面之上(协同)或之下(拮抗)延伸的活性的表面。计算表面的量,并表示为以95%置信区间计算的协同量(μM 在这些研究中,协同被定义为在95%置信区间下产生超过50μM 抗HCV组合治疗评估:评估化合物2与化合物1组合对在Huh-luc/neo-ET复制子细胞中HCV复制的抑制作用。以95%、99%和99.9%置信区间计算每种两药物组合在每种浓度下高于预期的病毒复制抑制百分比。以95%置信值获得的数据被三维绘制并计算协同量。下表42总结了95%置信度下组合的协同量。 表42:组合抗HCV活性评估 HCV复制子分析的结果表明,在组合抗病毒分析中使用的浓度范围内,化合物1在20和40nM下与化合物2在0.004和0.008nM下产生协同相互作用。在评估的浓度下未观察到拮抗或协同毒性相互作用。 本说明书已经参照本发明的实施方案进行了描述。然而,本领域普通技术人员理解,在不脱离如以下权利要求所述的本发明范围的情况下,可以进行各种修改和变化。因此,说明书应被视为说明性的而非限制性的,并且所有该等修改均旨在包括在本发明的范围内。
- 茶籽粕中综合提取茶籽皂素、茶籽多肽、茶籽多糖的方法
- 茶籽粕中综合提取茶籽皂素、茶籽多肽、茶籽多糖的方法