一种基于循环肿瘤DNA的突变基因检测方法及其应用
文献发布时间:2023-06-19 19:28:50
技术领域
本发明涉及生物检测技术领域,具体涉及一种基于循环肿瘤DNA的突变基因检测方法及其应用。
背景技术
MRD(Minimal Residual Disease)的概念源于白血病在诱导化疗达到完全缓解(或骨髓移植治疗)后,在患者体内依然残留着痕量白血病细胞的状态。转换到实体瘤的应用中,一般泛指在根治性治疗(如手术切除)后,传统影像学或实验室方法不能发现病灶,但通过液态活检技术可检出癌细胞来源分子的情况。实体瘤患者检出MRD,代表癌细胞在体内持续存在,癌症复发的风险较高。
检测循环肿瘤DNA(Circulating tumor DNA,ctDNA)在直接和实时检测MRD方面具有非常好的应用前景。肿瘤细胞死亡、破碎后,其DNA会被释放入血,形成ctDNA,因此,从理论上讲,可以通过探测血液中ctDNA的方法来捕捉影像学上不可见的微小残留病灶的踪迹,从而识别具有高度复发风险的患者,对评估疾病状态、判断疗效、预测复发、指导治疗具有重要的临床意义。MRD监测的时间点及检测灵敏度对于复发预测尤为重要。
近年来,基于NGS技术的ctDNA-MRD的临床数据和证据越来越多,实体瘤ctDNA-MRD检测的技术流派主要分为两大类:Tumor-informed assays(肿瘤知情分析)和Tumor-uninformed assays(肿瘤不知情分析)。Tumor-informed assays对原发肿瘤组织进行测序以鉴定患者的特异基因组变异图谱,然后设计引物定制panel进行个性化的ctDNA检测分析。Tumor-uninformed assays无需原发肿瘤组织,仅依赖于一组预先选定引物/探针设计与癌症类型相关的固定panel进行ctDNA检测分析。
固定化panel不需要根据每个患者进行定制,开发成本低,仅需要成熟的液相捕获平台,但是需要更多的数据量,并且可能产生假阴性;定制化panel开发成本高,周期长,存在引物设计的技术壁垒,但是测序成本低,易于发现有效位点,检测灵敏度和准确性更高。
发明内容
有鉴于此,本发明的目的在于提供一种基于循环肿瘤DNA的突变基因检测方法,能够有效提高肿瘤基因突变位点的灵敏度和特异性。
为解决上述技术问题,本发明提供了以下技术方案:
本发明提供了一种基于循环肿瘤DNA的突变基因检测方法,所述方法包括如下步骤:
对游离DNA进行单链建库后,利用特异性引物对所述文库进行PCR扩增,所得扩增产物经测序、数据分析后即可实现基因突变的检测;所述特异性引物为根据患者组织及血液基因组DNA进行全外显子测序信息获得初步的突变位点设计得到。
优选的,所述游离DNA由肿瘤患者术后血液中提取得到。
优选的,所述肿瘤包括肺癌、结直肠癌、乳腺癌、膀胱癌、食管癌。
优选的,所述PCR扩增为基于UMI的多重PCR扩增。
优选的,所述多重PCR包括两轮扩增,所述第一轮扩增的引物结构为overhang-UMI(NNWNNW)-GSP1/GSP2。
优选的,所述测序为二代测序,所述数据分析包括数据质控和分析。
本发明提供了一种基于循环肿瘤DNA的分子残留病灶检测试剂盒,所述试剂盒包括上述第一轮扩增的引物。
优选的,所述引物的设计原则包括:引物长度在15~25bp之间;引物GC含量在50%~75%之间;扩增子长度在80~120bp之间;引物3’端的碱基要严格配对。
本发明还提供了上述的检测方法在检测MRD中的应用。
本发明提供了一种基于循环肿瘤DNA的突变基因检测方法,采取定制化panel技术路线,采用单链文库构建的方法,将游离DNA进行了初步放大,然后基于UMI的多重PCR进行靶点富集,有效提升了检测灵敏度及特异性。经试验证实可知,本发明所述方法能够同时分析同一患者的多种基因突变结果,在突变等位基因比率(AF)上具有70%以上的灵敏度,90%以上的特异性,在MRD监控方面具有良好的应用前景。
附图说明
图1菁良基因2个ctDNA标准品及真实血浆游离DNA的片段分布对比。
图2为二轮多重PCR后2100峰图。
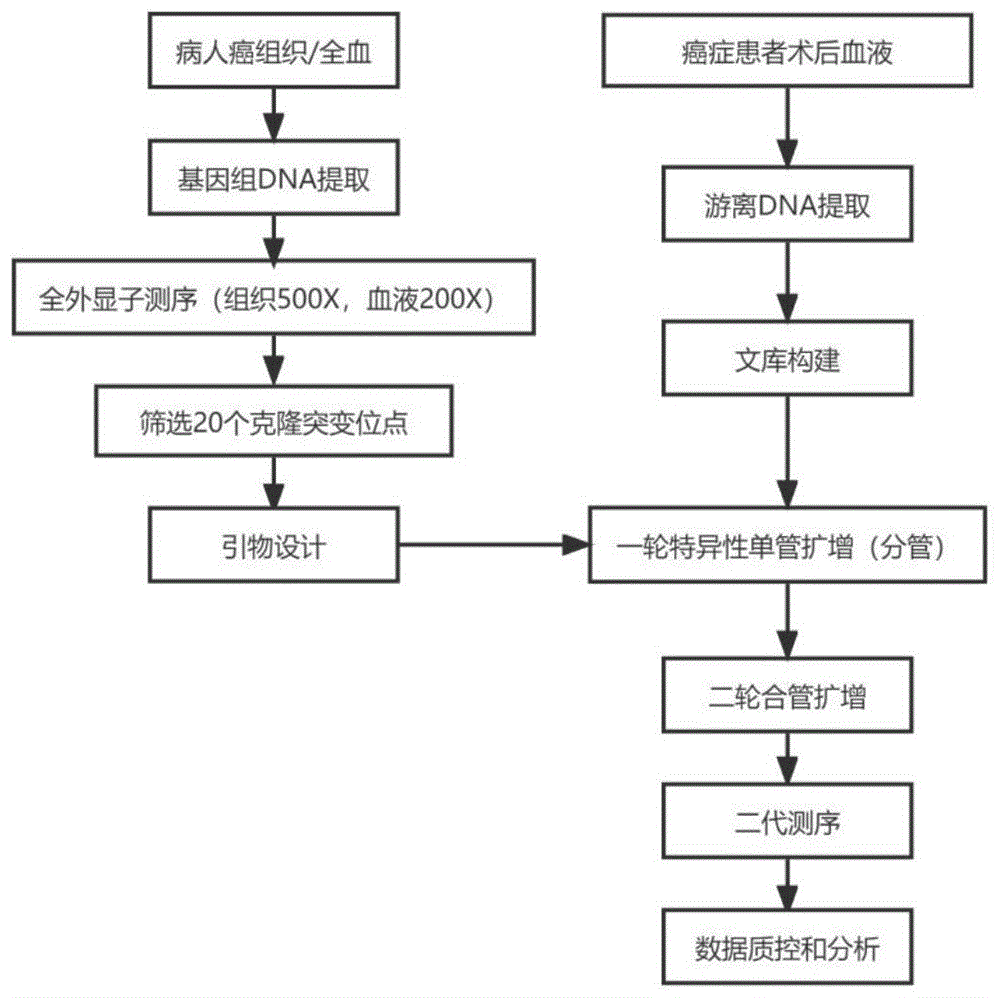
图3为本发明实验流程图。
具体实施方式
本发明提供了一种基于循环肿瘤DNA的突变基因检测方法,所述方法包括如下步骤:
对游离DNA进行单链建库后,利用特异性引物对所述文库进行PCR扩增,所得扩增产物经测序、数据分析后即可实现基因突变的检测;所述特异性引物为根据患者组织及血液基因组DNA进行全外显子测序信息获得初步的突变位点设计得到。
本发明中,所述游离DNA优选的由肿瘤患者术后血液中提取得到;所述肿瘤优选包括包括肺癌、结直肠癌、乳腺癌、膀胱癌、食管癌。本发明对所述游离DNA的提取方法并没有特殊限定,在本发明的具体实施例中,所述游离DNA优选的采用QIAamp CirculatingNucleicAcidKit进行提取。本发明中,所述全外显子测序优选的按照艾吉泰康AIExomeV2Plus试剂盒进行;所述组织基因组DNA测序平均深度优选为500X,所述血液样本测序平均深度优选为200X。
本发明中,所述PCR扩增优选为基于UMI的多重PCR扩增;所述多重PCR包括两轮扩增,所述第一轮扩增的引物结构优选为overhang-UMI(NNWNNW)-GSP1/GSP2。本发明中,所述引物设计含有UMI特殊结构,不同于常规的多重PCR的单管多靶区(高+低循环体系),引物间存在互相干扰产生非特异扩增,本发明采取单引物(低+高循环体系)扩增后组合的策略,在避免假阳性扩增风险的同时保证了真实的原始分子状态。本发明中,所述引物的设计原则优选的包括:引物长度在15~25bp之间;引物GC含量在50%~75%之间;扩增子长度在80~120bp之间;引物3’端的碱基要严格配对;退火温度60℃~66℃。
本发明中,所述测序优选为二代测序,所述二代测序的平台优选为IluminaNovaseq6000,所述数据分析优选的包括数据质控和分析。
本发明中,所述单链建库优选的按照ABclonal Scale ssDNA-seq Lib Prep Kit进行。本发明中,所述单链建库后的PCR扩增酶优选为2×KAPAHiFi HotStart ReadyMix,单链建库后的扩增引物优选为与后续多重PCR相匹配的通用引物。
本发明提供了一种基于循环肿瘤DNA的分子残留病灶检测试剂盒,所述试剂盒包括上述第一轮扩增的引物。
本发明还提供了上述的检测方法在检测MRD中的应用。
为使本发明的目的、技术方案和优点更加清楚明白,下面结合实施例对本发明进行详细的说明,但是不能把它们理解为对本发明保护范围的限定。
下述实施例中,如无特殊说明,均为常规方法。
下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
实施例1
为检测验证本发明的检测方法,以肺癌标准品作为样本进行以下实验
1.游离DNA标准品(菁良肺癌ctDNA标准品套装,货号GW-OCTM009),VAF分别为野生型、1%、0.1%、0.05%;具体的突变位点频率信息如下表1:
表1游离DNA标准品已知信息
2.引物设计
根据表1中6个突变位点的位置,在模板DNA的保守区内设计引物;引物长度在15~25bp之间;引物GC含量在50%~75%之间,Tm值最接近72℃;扩增子长度在80~120bp之间;避免引物内部出现二级结构,避免两条引物间互补,特别是3’端的互补;引物3’端的碱基要严格配对;退火温度60℃~66℃;
2.1单链建库扩增引物
引物序列如下:
ShortprimerF:GTTCAGACGTGTGCTCTTCCGATCT(SEQ ID
NO.1)
Short primer R:CCTACACGACGCTCTTCCGATCT(SEQ ID
NO.2)。
2.2多重PCR一轮特异性引物,5’端设计特殊overhang。每一对引物结构为overhang-UMI(NNWNNW)-GSP1/GSP2,其中,N为A、G、C、T中的任意一种,W为A或T。
引物序列如下表2:
表2引物序列
2.3多重PCR二轮扩增引物
引物序列如下:
Primer P5:
AATGATACGGCGACCACCGAGATCTACACGTTCAGAGTTCTACAGTCCGACGATC(SEQ IDNO.15);
Primer P7:
CAAGCAGAAGACGGCATACGAGATTCTCCAGCGTGACTGGAGTTCCTTGGCACCCGAGAATTCCA(SEQID NO.16);
3.文库构建试剂盒为ABclonal Scale ssDNA-seq Lib Prep Kit,PCR扩增酶为2×KAPAHiFi HotStart ReadyMix,扩增引物为与后续多重PCR相匹配的通用引物,具体试剂组分如下表3:
表3文库构建试剂
3.1热变性预处理
3.1.1取30ng 0.1%的cfDNA肺癌标准品至PCR管中,用Low-EDTATE稀释到总体积为15μL。
3.1.2待PCR仪器稳定到95℃后,将PCR管放入PCR仪中,进行95℃孵育2min后,立即将PCR管置于冰上进行冷却,静置2min。
3.2T7 Tailing&Ligation
3.2.1按照下表4配置T7 Tailing&Ligation预混液,需要在预处理前配制好,冰上放置时间不要超过20min。
表4 T7 Tailing&Ligation预混液
/>
3.2.2取25μLT7 Tailing&Ligation预混液加入到冰上放置的预处理DNA样本PCR管中(步骤3.1.2),使用移液器进行吹打混匀,然后瞬时离心使得反应液至管底。
3.2.3按照下表5设置程序,将PCR管置于PCR仪(热盖105℃)中,进行T7 Tailing&Ligation。
表5反应程序
3.3Second Strand Synthesis Reaction
3.3.1按照下表6体系配置Second Strand Synthesis Reaction预混液;
表6S econd Strand Synthesis Reaction预混液
3.3.2取46μL Second Strand Synthesis Reaction预混液加入到T7Tailing&Ligated mix(步骤3.2.3)中,使用移液器进行吹打混匀,然后瞬时离心使得反应液至管底;
3.3.3按照下表7设置程序,将PCR管置于PCR仪(热盖105℃)中,进行二链合成反应:
表7二链合成反应程序
3.3.4提前将DNA clean beads从2-8℃取出,静置平衡至室温,使用前涡旋或者震荡混匀;
3.3.5待Second Strand Synthesis Reaction结束后,在产物中加入105μL DNAcleanbeads(1.2X),吹打混匀;
3.3.6室温静置5min,然后转移至磁力架上5min,直至溶液变澄清,小心弃除上清;
3.3.7将离心管保持在磁力架上,加入200μL 80%乙醇静置30s,弃除全部上清;
3.3.8重复3.3.7,将磁珠用80%乙醇再洗1次,用10μL枪头将残留液体彻底吸干;
3.3.9干燥磁珠2-3min,待酒精挥发完全后,将PCR管移出磁力架,加入21μL Low-EDTATE,吹打混匀,然后室温静置2min;
3.3.10将PCR管放置到磁力架上室温静置,直到溶液变澄清,小心吸取20μL上清液至另一新的PCR管中备用。
3.4T5 Adapter Ligation
3.4.1按照下表8配制T5 Adapter Ligation反应体系,依次加入如下组分,使用移液器进行吹打混匀,然后瞬时离心使得反应液至管底。
表8 T5 Adapter Ligation反应体系
注:可以提前配制T5 Buffer II和T5 Adapter II的预混液,切不可以将T5Buffer II、T5 Adapter II和Ligase Mix预混,以免出现接头自连反应。
3.4.2按照下表9设置程序,将PCR管置于PCR仪(热盖加热功能关闭,或者热盖不要合上)中,进行连接反应。
表9连接反应程序
3.4.3提前将DNA clean beads从2-8℃取出,静置平衡至室温,使用前涡旋或者震荡混匀。
3.4.4连接反应结束后,加入40μL DNA cleanbeads(1.0X)到连接产物中,吹打混匀。
3.4.5室温静置5min,然后转移至磁力架上5min,直至溶液变澄清,小心弃除上清。
3.4.6将离心管保持在磁力架上,加入200μL 80%乙醇,静置30s,弃除全部上清。
3.4.7重复3.4.6,将磁珠用80%乙醇再次清洗一次,用10μL枪头将残留液体彻底吸干。
3.4.8干燥磁珠2-3min,将PCR管移出磁力架,加入21μL Low-EDTATE,吹打混匀,然后室温静置2min。
3.4.9将PCR管放置到磁力架上,室温静置直到溶液变澄清,小心吸取20μL上清液至另一新的PCR管中备用。
3.5PCR扩增
3.5.1按照下表10配制PCR反应体系:
表10 PCR反应体系
3.5.2使用移液器进行吹打混匀,然后瞬时离心使得反应液至管底,放置到PCR仪中。
3.5.3按照如下表11程序进行PCR反应
表11 PCR反应程序
3.5.4提前将DNA cleanbeads从2-8℃取出,静置平衡至室温,使用前涡旋或者震荡混匀。
3.5.5反应结束后,加入50μL DNA clean beads(1.0X)到PCR反应产物,吹打混匀。
3.5.6室温静置5min,然后转移至磁力架上5min,直至溶液变澄清,小心弃除上清。
3.5.7将离心管保持在磁力架上,加入200μL 80%乙醇,静置30s,弃除全部上清。
3.5.8重复3.5.7,将磁珠用80%乙醇再洗1次,用10μL枪头将残留液体彻底吸干。
3.5.9干燥磁珠2-3min,待酒精挥发完全后,加入21μL Low-EDTA TE,吹打混匀。
3.5.10室温静置2min,磁力架上1min,直到溶液变澄清,小心吸取20μL文库至另一的离心管中
3.6文库QC
3.6.1取1μL Qubit dsDNAHS Assay Kit定量。
3.6.2取1μL稀释到10ng/μL,进行Agilent 2100检测片段大小分布。
3.6.3文库(Lib DNA)置于-20℃保存或用于下一步实验。
4.单重位点特异性扩增
4.1使用表2中的6对带有UMI的特异性引物进行单管扩增,按照下表12反应体系配制反应液:
表12反应体系
4.2按照如下表13反应程序进行PCR扩增:
表13反应程序
4.3提前将DNA cleanbeads从2-8℃取出,静置平衡至室温,使用前涡旋或者震荡混匀;
4.4反应结束后,将6管反应产物混合为一管,转移至新的离心管中;
4.5在60μl混合物中加入90μl DNAcleanbeads(1.5×),吹打混匀;
4.6室温静置5min,然后转移至磁力架上5min,直至溶液变澄清,小心弃除上清;
4.7将离心管保持在磁力架上,加入200μL 80%乙醇,静置30s,弃除全部上清;
4.8重复上一步骤,将磁珠用80%乙醇再洗1次,用10μL枪头将残留液体彻底吸干;
4.9干燥磁珠2-3min,待酒精挥发完全后,加入19μLLow-EDTA TE,吹打混匀;
4.10室温静置2min,磁力架上1min,直到溶液变澄清,小心吸取16.8μL文库至新的PCR管中。
5.多重index扩增;
5.1按照如下表14反应体系配制反应液:
表14反应体系
5.2按照如下表15反应程序进行PCR扩增:
表15反应程序
5.3提前将DNA cleanbeads从2-8℃取出,静置平衡至室温,使用前涡旋或者5.4震荡混匀;
5.4反应结束后,在反应产物中加入40μl DNA cleanbeads(1×),吹打混匀;
5.5室温静置5min,然后转移至磁力架上5min,直至溶液变澄清,小心弃除上清;
5.6将离心管保持在磁力架上,加入200μL 80%乙醇,静置30s,弃除全部上清;
5.7重复上一步骤,将磁珠用80%乙醇再洗1次,用10μL枪头将残留液体彻底吸干;
5.8干燥磁珠2-3min,待酒精挥发完全后,加入20μL Low-EDTA TE,吹打混匀;
5.9室温静置2min,磁力架上1min,直到溶液变澄清,小心吸取18μL文库至新的PCR管中。
6文库QC
6.1取1μL Qubit dsDNAHS AssayKit定量。
6.2取1μL进行Agilent 2100检测片段大小分布,结果如图2。
6.3将文库进行上机测序,对测序所得原始数据进行QC质控和处理:Trim,Mapping,blast,UMI聚类去重后得到突变频率结果如表16。
表16测序结果
/>
/>
/>
/>
*predict_vaf为0的位点,突变频率≥0.0002为假阳性,假阳率=假阳位点*/所有位点=6.94%=5/72;
*predict_vaf为0.001的位点,突变频率<0.0002为假阴性,假阴率=假阴位点*/所有位点=15.3%=11/72。
根据图2可以看出,利用本发明所述方法对标准品实际检出大小与标准品的理论扩增子的大小一致,且无其他杂峰。表明本发明所述方法的特异性和方法学可靠稳定性良好。
将表16中利用本发明所述检测方法检测到的VAF与标准品已知的VAF(表1)进行比较,评估本发明所述方法对SNV突变位点的检测能力。利用本发明表16中标准品6个位点12次重复统计数据,计算得到本发明检测方法在突变等位基因比率(AF)上具有84.7%的灵敏度(61/72),表17中预期突变0.1%的位点,检出阳性用灰色填充),93.1%的特异性(1-5/72),表17中预期突变为0的位点,检出假阳性用灰色填充)。
其中,本发明中突变位点的阳性判断标准为:以突变频率为0.02%为阈值,大于该值为阳性,否则阴性。
具体判断结果如下表17:
表17突变位点阳性判断结果
实施例2
经过WES对每个患者筛选出10~20个肿瘤突变位点,在这些位点上进行本发明的多重PCR扩增,以患者血液白细胞gDNA检测值为背景去除克隆性造血干扰,检测手术前肺癌患者血浆样本的ctDNA,测试本发明通过ctDNA检测肺癌阳性的性能。
一、临床样本
采集10例手术前肺癌患者的10mL外周血,以及手术中获得的组织标本用于突变检测,所有患者经病理学确诊。
二、样本处理
外周血样本经离心分离血浆后,提取游离DNA样本,样本溶解在35μL体积的无核酸酶水中。提取的样本使用Qubit定量,浓度在0.5-5ng之间,取30ng用于后续实验。
三、实验流程
1.从患者组织及血液样本中提取基因组DNA(组织提取采用QIAamp DNA FFPEKit;血液提取采用试剂盒QIAamp DNA Blood mini Kit)
2.从患者手术后血液样本中提取游离DNA(QIAamp Circulating NucleicAcidKit)
3.全外显子测序(艾吉泰康AIExomeV2 Plus建库试剂盒),组织样本测序平均深度500X,血液样本测序平均深度200X,测序仪为Illumina NovaSeq 6000
4.根据测序数据结果,进行位点筛选及引物设计
5.后续实验流程见实施例1中3.1~6.3
6.患者临床信息如下表18:
表18患者临床信息
ctDNA检测阳性的判定标准为:每位患者选出的所有突变位点中,突变频率大于0.02%判定为阳性位点;每位患者至少2个阳性位点,则其在样本水平为阳性。
10例临床样本测序实验数据如表19-28所示(位点突变频率,是由平均有效测序深度大于100000X的条件下计算得出;带*的是白细胞检测阳性的位点,该位点若ctDNA检出阳性,则不计入总阳性位点数):
表19患者1临床样本测序实验数据
白细胞阳性位点:0
ctDNA阳性位点:0
表20患者2临床样本测序实验数据
白细胞阳性位点:0
ctDNA阳性位点:0
表21患者3临床样本测序实验数据
白细胞阳性位点:1
ctDNA阳性位点:13
表22患者4临床样本测序实验数据
白细胞阳性位点:0
ctDNA阳性位点:2
表23患者5临床样本测序实验数据
/>
白细胞阳性位点:1
ctDNA阳性位点:2
表24患者6临床样本测序实验数据
/>
白细胞阳性位点:0
ctDNA阳性位点:19
表25患者7临床样本测序实验数据
/>
白细胞阳性位点:1
ctDNA阳性位点:2
表26患者8临床样本测序实验数据
/>
白细胞阳性位点:1
ctDNA阳性位点:11
表27患者9临床样本测序实验数据
白细胞阳性位点:1
ctDNA阳性位点:1
表28患者10临床样本测序实验数据
白细胞阳性位点:2
ctDNA阳性位点:5
根据以上结果可以看出,同一患者进行白细胞、ctDNA检测,白细胞阳性的患者有1例(患者10),特异性达到了90%(9/10);除去白细胞阳性的位点,10例患者中,ctDNA检测出阳性位点大于2的患者有7例(除了患者1、2、9之外的患者),检测灵敏度70%(7/10)。
以上所述仅为本发明的实施例,并非因此限制本发明的专利范围,凡是利用本发明说明书内容所作的等效结构或等效流程变换,或直接或间接运用在其他相关的技术领域,均同理包括在本发明的专利保护范围内。
- 一种检测血浆游离DNA中肺癌相关突变基因的试剂盒及其应用
- 基于C2c2的肿瘤相关突变基因的gRNA、检测方法、检测试剂盒
- 一种肿瘤标志物循环肿瘤DNA检测芯片及试剂盒
- 一种单链分子标签接头及单链DNA建库方法及其在检测循环肿瘤DNA中应用
- 一种比率式聚集诱导ECL/EC传感平台、其制备方法及其检测循环肿瘤DNA的应用