一种电化学介导乙烯基苯胺与醇合成2,3-二烷氧基取代吲哚啉化合物、合成方法及应用
文献发布时间:2023-06-19 19:28:50
技术领域
本发明涉及吲哚啉化合物合成技术领域,尤其涉及一种电化学介导乙烯基苯胺与醇合成2,3-二烷氧基取代吲哚啉化合物、合成方法及应用。
背景技术
吲哚啉骨架广泛存在于天然产物和药物分子中,具有较好的药理活性,吲哚啉骨架由于其特殊的化学和生物性质是各种天然产物中的优势结构,被认为是与药物有关的一大类化合物中的“核心骨架”,2,3-二取代吲哚啉化合物,在合成和药物化学中都有重要的应用,许多天然产物和药物的结构中都包含了这种杂环骨架(Med.Res.Rev.1995,15,3;J.Med.Chem.2014,57,1488;J.Am.Chem.Soc.2015,137,4666.),例如,氟喹唑啉是一种以吲哚啉为核心骨架的天然抗真菌生物碱,对P388血癌细胞腺具有生物毒性(Chin.J.Chem.,2014,32,757-770);毛壳素,一种由内生毛壳菌产生的细胞毒性吲哚啉类生物碱,对癌细胞SW1116具有较强的毒性(Org.Lett.,2006,8,5709-5712.);此外,吲哚啉也是药物合成的关键中间体(J.Comb.Chem.2009,11,303);
自合成化学发展以来,吲哚啉衍生物的功能化和合成引起了有机化学家的极大兴趣,但是现有的合成方式多局限于取代吲哚骨架的直接去芳构化,因此,开拓更多的吲哚啉骨架的合成方法是非常有必要的。
发明内容
本发明的目的在于提供一种电化学介导乙烯基苯胺与醇合成2,3-二烷氧基取代吲哚啉化合物、合成方法及应用,以解决现有技术中缺乏2,3-二烷氧基取代吲哚啉化合物合成路径的问题。
为实现上述目的,本发明提供了一种2,3-二烷氧基取代吲哚啉化合物,结构如式(I)所示:
其中,R为烷基,R
本发明还公开一种电化学介导乙烯基苯胺与醇合成2,3-二烷氧基取代吲哚啉化合物的合成方法,应用于制备如前所述的2,3-二烷氧基取代吲哚啉化合物,
包括如下步骤:
分别将醇类、乙烯基苯胺、电解质加入到反应容器中,并加入溶剂溶解;
随后用碳棒作阳极、镍片作阴极,恒电流,预设温度条件下进行搅拌反应;
反应完成后,用乙酸乙酯萃取混合物;
萃取获得的有机层用无水硫酸钠干燥,减压旋干溶剂,并通过柱色谱纯化残余物,得到2,3-二烷氧基取代吲哚啉化合物。
其中,醇类、乙烯基苯胺、电解质的用量比为2:0.3:0.09,其中,乙烯基苯胺以及电解质均以毫摩尔计,醇类以毫升计。
其中,所述醇类为甲醇、乙醇、烯丙醇、环丙基甲醇中的至少一种。
其中,所述溶剂为乙腈、甲醇、乙腈与甲醇混合液中的至少一种。
其中,所述恒电流为10~20毫安。
其中,所述预设温度为40℃~80℃。
其中,所述搅拌反应的时间为1-3小时。
其中,所述电解质为四丁基六氟膦酸铵,四乙基六氟膦酸铵,四丁基四氟硼酸铵中的至少一种。
本发明还公开如前所述的2,3-二烷氧基取代吲哚啉化合物在制备抗肿瘤药物中的应用。
本发明的一种电化学介导乙烯基苯胺与醇合成2,3-二烷氧基取代吲哚啉化合物、合成方法及应用,以乙烯基苯胺为原料,醇类作为亲核试剂,在电化学的条件下,通过电氧化环化合成了一系列2,3-二烷氧基吲哚啉化合物,并通过体外抗肿瘤活性筛选,发现合成的部分吲哚啉化合物具有良好的抗肿瘤活性。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
图1是本发明一种2,3-二烷氧基取代吲哚啉化合物提供的合成工艺示意图。
图2是本发明一种电化学介导乙烯基苯胺与醇合成2,3-二烷氧基取代吲哚啉化合物的合成方法的步骤示意图。
具体实施方式
下面详细描述本发明的实施例,所述实施例的示例在附图中示出,其中自始至终相同或类似的标号表示相同或类似的元件或具有相同或类似功能的元件。下面通过参考附图描述的实施例是示例性的,旨在用于解释本发明,而不能理解为对本发明的限制。
请参阅图1,本发明提供了一种2,3-二烷氧基取代吲哚啉化合物,结构如式(I)所示:
其中,R为烷基,R
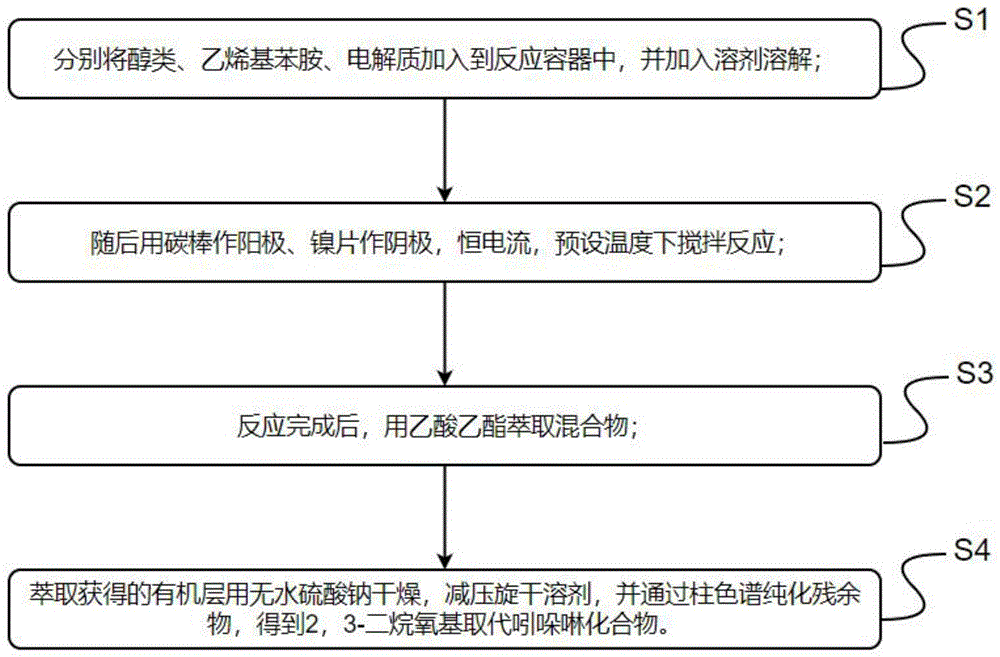
请参阅图2,本发明还公开一种电化学介导乙烯基苯胺与醇合成2,3-二烷氧基取代吲哚啉化合物的合成方法,应用于制备如前所述的2,3-二烷氧基取代吲哚啉化合物,
包括如下步骤:
S1:分别将醇类、乙烯基苯胺、电解质加入到反应容器中,并加入溶剂溶解;
S2:随后用碳棒作阳极、镍片作阴极,恒电流,预设温度条件下搅拌反应1-3小时;
S3:反应完成后,用乙酸乙酯萃取混合物;
S4:萃取获得的有机层用无水硫酸钠干燥,减压旋干溶剂,并通过柱色谱纯化残余物,得到2,3-二烷氧基取代吲哚啉化合物。
进一步的,醇类、乙烯基苯胺、电解质的用量比为2:0.3:0.09,其中,乙烯基苯胺以及电解质均以毫摩尔计,醇类以毫升计。
进一步的,所述醇类为甲醇、乙醇、烯丙醇、环丙基甲醇中的一种。
进一步的,所述溶剂为乙腈、甲醇、乙腈与甲醇混合液中的一种。
进一步的,所述电解质为四丁基六氟膦酸铵,四乙基六氟膦酸铵,四丁基四氟硼酸铵中的一种。
进一步的,所述恒电流为10~20毫安。
进一步的,所述预设温度为40℃~80℃。
进一步的,所述搅拌反应的时间为1-3小时。
本发明还公开如前所述的2,3-二烷氧基取代吲哚啉化合物在制备抗肿瘤药物中的应用。
具体实施步骤为:分别将2毫升醇、0.3毫摩尔乙烯基苯胺、0.09毫摩尔电解质(四丁基六氟膦酸铵,四乙基六氟膦酸铵,四丁基四氟硼酸铵等)加到10毫升三颈瓶中,加入4毫升溶剂(乙腈,甲醇,乙腈与甲醇混合液等)溶解,用碳棒作阳极,镍片作阴极,在10-20毫安恒电流,40-80摄氏度条件下进行搅拌反应1-3小时,薄层层析色谱法监测反应进程,反应完成后,用乙酸乙酯(3×5毫升)萃取混合物。有机层用无水硫酸钠干燥,减压旋干溶剂,并通过柱色谱(硅胶,乙酸乙酯/石油醚=1:20-40洗脱)纯化残余物,得到目标产物。
实施例1:
(2,3-二甲氧基-3-甲基吲哚啉-1-基)苯基甲酮(4a)的制备和表征:
分别将2毫升甲醇、0.3毫摩尔N-(2-(1-丙烯-2-基))苯基)苯甲酰胺、0.09毫摩尔四丁基六氟膦酸铵加到10毫升三颈瓶中,加入4毫升乙腈溶解,用碳棒作阳极,镍片作阴极,在10毫安恒电流,80摄氏度条件下进行搅拌反应1.5小时,薄层层析色谱法监测反应进程,反应完成后,用乙酸乙酯(3×5毫升)萃取混合物。有机层用无水硫酸钠干燥,减压旋干溶剂,并通过柱色谱(硅胶,乙酸乙酯/石油醚=1:25洗脱)纯化残余物,得到目标产物4a,表征如下:
yellow oil(66%,58.9mg).
实施例2:
(2,3-二甲氧基-3-甲基吲哚-1-基)(邻甲苯基)甲酮(4b)的制备和表征:
分别将2毫升甲醇、0.3毫摩尔2-甲基-N-(2-(1-丙烯-2-基))苯基)苯甲酰胺、0.09毫摩尔四丁基四氟硼酸铵加到10毫升三颈瓶中,加入4毫升乙腈溶解,用碳棒作阳极,镍片作阴极,在15毫安恒电流,60摄氏度条件下进行搅拌反应1.5小时,薄层层析色谱法监测反应进程,反应完成后,用乙酸乙酯(3×5毫升)萃取混合物。有机层用无水硫酸钠干燥,减压旋干溶剂,并通过柱色谱(硅胶,乙酸乙酯/石油醚=1:25洗脱)纯化残余物,得到目标产物4b,表征如下:
yellow oil(52%,48.6mg).
实施例3:
(2,3-二甲氧基-3-甲基吲哚-1-基)(间甲苯基)甲酮(4c)的制备和表征:
分别将2毫升甲醇、0.3毫摩尔3-甲基-N-(2-(1-丙烯-2-基))苯基)苯甲酰胺、0.09毫摩尔四乙基六氟膦酸铵加到10毫升三颈瓶中,加入4毫升乙腈与甲醇混合液(v:v=3:1)溶解,用碳棒作阳极,镍片作阴极,在20毫安恒电流,40摄氏度条件下进行搅拌反应2小时,薄层层析色谱法监测反应进程,反应完成后,用乙酸乙酯(3×5毫升)萃取混合物。有机层用无水硫酸钠干燥,减压旋干溶剂,并通过柱色谱(硅胶,乙酸乙酯/石油醚=1:25洗脱)纯化残余物,得到目标产物4c。
yellow oil(55%,51.4mg).
实施例4:
(2,3-二甲氧基-3-甲基吲哚-1-基)(对甲苯基)甲酮(4d)的制备和表征:
分别将2毫升甲醇、0.3毫摩尔4-甲基-N-(2-(1-丙烯-2-基))苯基)苯甲酰胺、0.09毫摩尔四乙基六氟膦酸铵加到10毫升三颈瓶中,加入4毫升乙腈溶解,用碳棒作阳极,镍片作阴极,在10毫安恒电流,80摄氏度条件下进行搅拌反应1.5小时,薄层层析色谱法监测反应进程,反应完成后,用乙酸乙酯(3×5毫升)萃取混合物。有机层用无水硫酸钠干燥,减压旋干溶剂,并通过柱色谱(硅胶,乙酸乙酯/石油醚=1:25洗脱)纯化残余物,得到目标产物4d。
yellow oil(54%,50.4mg).
实施例5:
(4-氯苯基)(2,3-二甲氧基-3-甲基吲哚-1-基)甲酮(4e)的制备和表征:
分别将2毫升甲醇、0.3毫摩尔4–氯-N-(2-(1-丙烯-2-基))苯基)苯甲酰胺、0.09毫摩尔四丁基六氟膦酸铵加到10毫升三颈瓶中,加入4毫升甲醇溶解,用碳棒作阳极,镍片作阴极,在20毫安恒电流,60摄氏度条件下进行搅拌反应3小时,薄层层析色谱法监测反应进程,反应完成后,用乙酸乙酯(3×5毫升)萃取混合物。有机层用无水硫酸钠干燥,减压旋干溶剂,并通过柱色谱(硅胶,乙酸乙酯/石油醚=1:25洗脱)纯化残余物,得到目标产物4e。
yellow oil(58%,57.7mg).
实施例6:
(2,3-二甲氧基-3-甲基吲哚-1-基)(萘-2-基)甲酮(4f)的表征和制备:
分别将2毫升甲醇、0.3毫摩尔N-(2-(1-丙烯-2-基))苯基)-2-萘甲酰胺、0.09毫摩尔四乙基六氟膦酸铵加到10毫升三颈瓶中,加入4毫升乙腈溶解,用碳棒作阳极,镍片作阴极,在10毫安恒电流,40摄氏度条件下进行搅拌反应1小时,薄层层析色谱法监测反应进程,反应完成后,用乙酸乙酯(3×5毫升)萃取混合物。有机层用无水硫酸钠干燥,减压旋干溶剂,并通过柱色谱(硅胶,乙酸乙酯/石油醚=1:20洗脱)纯化残余物,得到目标产物4f。
yellow oil(52%,54.2mg).
实施例7:
(2,3-二甲氧基-3-甲基吲哚-1-基)(噻吩-2-基)甲酮(4g)的表征和制备。
分别将2毫升甲醇、0.3毫摩尔N-(2-(1-丙烯-2-基))苯基)噻吩-2-甲酰胺、0.09毫摩尔四丁基六氟膦酸铵加到10毫升三颈瓶中,加入4毫升乙腈溶解,用碳棒作阳极,镍片作阴极,在20毫安恒电流,70摄氏度条件下进行搅拌反应1.5小时,薄层层析色谱法监测反应进程,反应完成后,用乙酸乙酯(3×5毫升)萃取混合物。有机层用无水硫酸钠干燥,减压旋干溶剂,并通过柱色谱(硅胶,乙酸乙酯/石油醚=1:30洗脱)纯化残余物,得到目标产物4g。
yellow oil(52%,47.3mg).
实施例8:
1-(2,3-二甲氧基-3-甲基吲哚-1-基)乙烷-1-酮(4h)的表征和制备:
分别将2毫升甲醇、0.3毫摩尔N-(2-(1-丙烯-2-基))苯基)乙酰胺、0.09毫摩尔四乙基六氟膦酸铵加到10毫升三颈瓶中,加入4毫升甲醇溶解,用碳棒作阳极,镍片作阴极,在20毫安恒电流,70摄氏度条件下进行搅拌反应1.5小时,薄层层析色谱法监测反应进程,反应完成后,用乙酸乙酯(3×5毫升)萃取混合物。有机层用无水硫酸钠干燥,减压旋干溶剂,并通过柱色谱(硅胶,乙酸乙酯/石油醚=1:30洗脱)纯化残余物,得到目标产物4h。
yellow oil(59%,41.6mg).
实施例9:
1-(2,3-二甲氧基-3-甲基吲哚-1-基)-2-苯乙烷-1-酮(4i)的制备与表征:
分别将2毫升甲醇、0.3毫摩尔2-苯基-N-(2-(1-丙烯-2-基))苯基)乙酰胺、0.09毫摩尔四丁基六氟膦酸铵加到10毫升三颈瓶中,加入4毫升乙腈溶解,用碳棒作阳极,镍片作阴极,在20毫安恒电流,80摄氏度条件下进行搅拌反应3小时,薄层层析色谱法监测反应进程,反应完成后,用乙酸乙酯(3×5毫升)萃取混合物。有机层用无水硫酸钠干燥,减压旋干溶剂,并通过柱色谱(硅胶,乙酸乙酯/石油醚=1:25洗脱)纯化残余物,得到目标产物4i。
yellow oil(72%,67.3mg).
实施例10:
(5-环丙基-2,3-二甲氧基-3-甲基吲哚-1-基)(苯基)甲酮(4j)的制备和表征:
分别将2毫升甲醇、0.3毫摩尔N-(4-环丙基-2-(丙-1-烯-2-基))苯基)苯甲酰胺、0.09毫摩尔四丁基四氟硼酸铵加到10毫升三颈瓶中,加入4毫升乙腈溶解,用碳棒作阳极,镍片作阴极,在10毫安恒电流,60摄氏度条件下进行搅拌反应1小时,薄层层析色谱法监测反应进程,反应完成后,用乙酸乙酯(3×5毫升)萃取混合物。有机层用无水硫酸钠干燥,减压旋干溶剂,并通过柱色谱(硅胶,乙酸乙酯/石油醚=1:25洗脱)纯化残余物,得到目标产物4j。
yellow oil(51%,51.6mg).
实施例11:
(5-溴-2,3-二甲氧基-3-甲基吲哚-1-基)(苯基)甲酮(4k)制备和表征:
分别将2毫升甲醇、0.3毫摩尔N-(4-溴-2-(丙-1-烯-2-基))苯基)苯甲酰胺、0.09毫摩尔四丁基六氟膦酸铵加到10毫升三颈瓶中,加入4毫升乙腈溶解,用碳棒作阳极,镍片作阴极,在20毫安恒电流,60摄氏度条件下进行搅拌反应2.5小时,薄层层析色谱法监测反应进程,反应完成后,用乙酸乙酯(3×5毫升)萃取混合物。有机层用无水硫酸钠干燥,减压旋干溶剂,并通过柱色谱(硅胶,乙酸乙酯/石油醚=1:20洗脱)纯化残余物,得到目标产物4k。
yellow oil(56%,63.2mg).
实施例12:
(2,3-二甲氧基-3-苯基吲哚-1-基)(苯基)甲酮(4l)制备和表征:
分别将2毫升甲醇、0.3毫摩尔N-(2-(1-苯基乙烯基)苯基)苯甲酰胺、0.09毫摩尔四丁基六氟膦酸铵加到10毫升三颈瓶中,加入4毫升乙腈溶解,用碳棒作阳极,镍片作阴极,在20毫安恒电流,55摄氏度条件下进行搅拌反应1小时,薄层层析色谱法监测反应进程,反应完成后,用乙酸乙酯(3×5毫升)萃取混合物。有机层用无水硫酸钠干燥,减压旋干溶剂,并通过柱色谱(硅胶,乙酸乙酯/石油醚=1:20洗脱)纯化残余物,得到目标产物4l。
yellow oil(57%,61.5mg).
实施例13:
1-(2,3-二乙氧基-3-甲基吲哚-1-基)-2-苯乙烷-1-酮(4m)制备和表征:
分别将2毫升乙醇、0.3毫摩尔2-苯基-N-(2-(1-丙烯-2-基))苯基)乙酰胺、0.09毫摩尔四丁基四氟硼酸铵加到10毫升三颈瓶中,加入4毫升乙腈溶解,用碳棒作阳极,镍片作阴极,在15毫安恒电流,80摄氏度条件下进行搅拌反应2.5小时,薄层层析色谱法监测反应进程,反应完成后,用乙酸乙酯(3×5毫升)萃取混合物。有机层用无水硫酸钠干燥,减压旋干溶剂,并通过柱色谱(硅胶,乙酸乙酯/石油醚=1:40洗脱)纯化残余物,得到目标产物4m。
yellow oil(70%,71.3mg).
实施例14:
1-(2,3-二烯丙基-3-甲基吲哚-1-基)-2-苯乙烷-1-酮(4n)制备和表征:
分别将2毫升烯丙醇、0.3毫摩尔2-苯基-N-(2-(1-丙烯-2-基))苯基)乙酰胺、0.09毫摩尔四丁基六氟膦酸铵加到10毫升三颈瓶中,加入4毫升乙腈溶解,用碳棒作阳极,镍片作阴极,在10毫安恒电流,70摄氏度条件下进行搅拌反应3小时,薄层层析色谱法监测反应进程,反应完成后,用乙酸乙酯(3×5毫升)萃取混合物。有机层用无水硫酸钠干燥,减压旋干溶剂,并通过柱色谱(硅胶,乙酸乙酯/石油醚=1:25洗脱)纯化残余物,得到目标产物4n。
yellow oil(50%,54.5mg).
实施例15:
1-(2,3-二环丙基甲氧基-3-甲基吲哚-1-基)-2-苯乙烷-1-酮(4o)的制备和表征:
分别将2毫升环丙基甲醇、0.3毫摩尔2-苯基-N-(2-(1-丙烯-2-基))苯基)乙酰胺、0.09毫摩尔四丁基四氟硼酸铵加到10毫升三颈瓶中,加入4毫升乙腈溶解,用碳棒作阳极,镍片作阴极,在20毫安恒电流,60摄氏度条件下进行搅拌反应1.5小时,薄层层析色谱法监测反应进程,反应完成后,用乙酸乙酯(3×5毫升)萃取混合物。有机层用无水硫酸钠干燥,减压旋干溶剂,并通过柱色谱(硅胶,乙酸乙酯/石油醚=1:20洗脱)纯化残余物,得到目标产物4o。
yellow oil(52%,61.1mg).
化合物药理活性研究:
本申请利用MTT筛选了化合物对4株癌细胞株(MGC-803,MIA PaCa-2,MDA-MB-231和HeLa)的体外抑制活性,结果如表1所示:
表1
实验结果表明大部分化合物对肿瘤细胞株具有良好的抑制活性,其中化合物4o对肿瘤细胞株的抑制效果最好。如表1所示,化合物4o对MGC-803,MIAPaCa-2和HeLa的IC50值分别为17.2±1.0,11.6±0.6和14.8±1.3μM,在温和的电化学条件下,通过乙烯基苯胺与醇反应,得到了一系列2,3-二烷氧基吲哚啉化合物,利用MTT法对所合成化合物的抗肿瘤活性进行了研究,实验结果表明大部分化合物对肿瘤细胞株具有良好的抑制活性,其中化合物4o对肿瘤细胞株的抑制效果最好。
本发明的一种电化学介导乙烯基苯胺与醇合成2,3-二烷氧基取代吲哚啉化合物、合成方法及应用,以乙烯基苯胺为原料,醇类作为亲核试剂,在电化学的条件下,通过电氧化环化合成了一系列2,3-二烷氧基吲哚啉化合物,并通过体外抗肿瘤活性筛选,发现合成的部分吲哚啉化合物具有良好的抗肿瘤活性。
以上所揭露的仅为本发明一种较佳实施例而已,当然不能以此来限定本发明之权利范围,本领域普通技术人员可以理解实现上述实施例的全部或部分流程,并依本发明权利要求所作的等同变化,仍属于发明所涵盖的范围。