一种RNA建库的方法
文献发布时间:2023-06-19 09:26:02
技术领域
本发明涉及基因测序技术领域,具体涉及一种RNA建库的方法。
背景技术
高通量测序技术简写为NGS,可以对数百万条DNA分子序列同时测定;其测序流程包括样本的提取,文库构建,测序及结果分析,文库构建包括RNA文库构建和DNA文库构建。传统的RNA文库构建是mRNA富集后通过高温和Mg
另外,传统的一种Tn5转座酶量只针对特定的RNA的投入量,转座酶对模板投入量的依赖性较强。
文献“RNA sequencing by direct tagmentation of RNA/DNA hybrids”(DiLin,Fu Yusi,Sun Yue,et al.Proc Natl Acad Sci USA.2020,117(6):2886-2893.)公开了一种新的基于Tn5转座酶的转录组测序快速建库方法,与已有的其它方法相比,大大简化了建库的过程;同时对样品的用量还很小(10ng-100pg),不仅可以用于高质量的单细胞转录组测序,而且对新型冠状病毒(2019-nCoV)等样本的测序质量和速度可能有一定的提升,但是其对样品量范围仍有一定的局限,使用时影响到效率。
因此,有必要研究一种对样品用量范围更广的RNA快速测序建库方法,通过降低转座酶对模板投入的依赖性,克服建库周期长,步骤复杂,容易出错的缺陷,在满足快速灵活建库的同时,扩大样品量的适用范围。
发明内容
针对现有技术的缺陷,本发明提供一种RNA建库的方法,具备灵活及快速的特点,适用于更广的样本用量范围。
为了实现上述目的,本发明提出一种RNA建库的方法,包括以下步骤:
1.投入total RNA合成First cDNA,形成杂交链;
2.用固定有Tn5酶的磁珠在对First cDNA杂交链进行片段化;所述磁珠为链霉素亲和素磁珠;
3.将片段化的杂交链缺口补齐,经扩增得到文库。
进一步地,所述total RNA起始投入量为10ng-1ug。
进一步地,所述固定有Tn5酶的磁珠的制备方法如下:先将突变型Tn5转座酶与带有生物素标记的双链接头复合组装,形成转座复合体;然后将转座复合体与链霉亲和素磁珠进行组装,最后形成Tn5转座酶磁珠复合体。
进一步地,所述步骤1为:反应中加入缓冲液5X First Strand Buffer,Betaine,Oligo(dT)
进一步地,所述步骤1还可以选择通过Poly(A)mRNA Capture Module富集mRNA进行first cDNA合成,通过2X Oligo(dT)25Capture Beads中polyT与mRNA的polyA结合捕获mRNA,去除其他RNA;然后在体系中加入4X First cDNA Synthesis Buffer反应液与mRNA结合,通过第一链合成酶混合物合成RNA/cDNA杂交链。
进一步地,上述两种所述1st cDNA合成方法中所述第一链合成酶混合物包括逆转录酶和RNA inhibitor,所述逆转录酶为ABScript III逆转录酶或ABScript II逆转录酶。
进一步地,所述步骤2片段化反应体系包含打断缓冲液2X tagment buffer,所述2X tagment buffer组分包括20mM Tris-HCI,10mM MgCl
进一步地,所述步骤2中终止Tn5转座复合体磁珠进行打断的终止反应液为6XTermination Buffer。
进一步地,所述GAP延伸酶与2X PCR mix一起完成GAP的补齐,最后使用2X PCRmix完成最终的文库的富集;
进一步地,所述GAP延伸酶为ABScript III逆转录酶或等温扩增酶;所述等温扩增酶为Bst3.0 DNA Polymerase或Bst2.0 WarmStart DNA polymerase;所述2X PCR mix为ABclonal,RK20698。
与现有技术相比,本发明的有益效果为:
1)本发明提供的RNA建库方法打破了传统的一种Tn5转座酶量或酶浓度只针对一种特定的RNA的投入量的建库方法,降低转座酶对模板投入的依赖性,使得起始材料RNA的投入量比较灵活,可以根据自己需求投入10ng到1ug不同量的RNA;
2)本发明实验步骤简单,用时短,可以有效的降低成本。
附图说明
为了更清楚地说明本发明实施例的技术方案,下面将对实施例中所需要使用的附图作简单地介绍,应当理解,以下附图仅示出了本发明的某些实施例,因此不应被看作是对范围的限定,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他相关的附图。
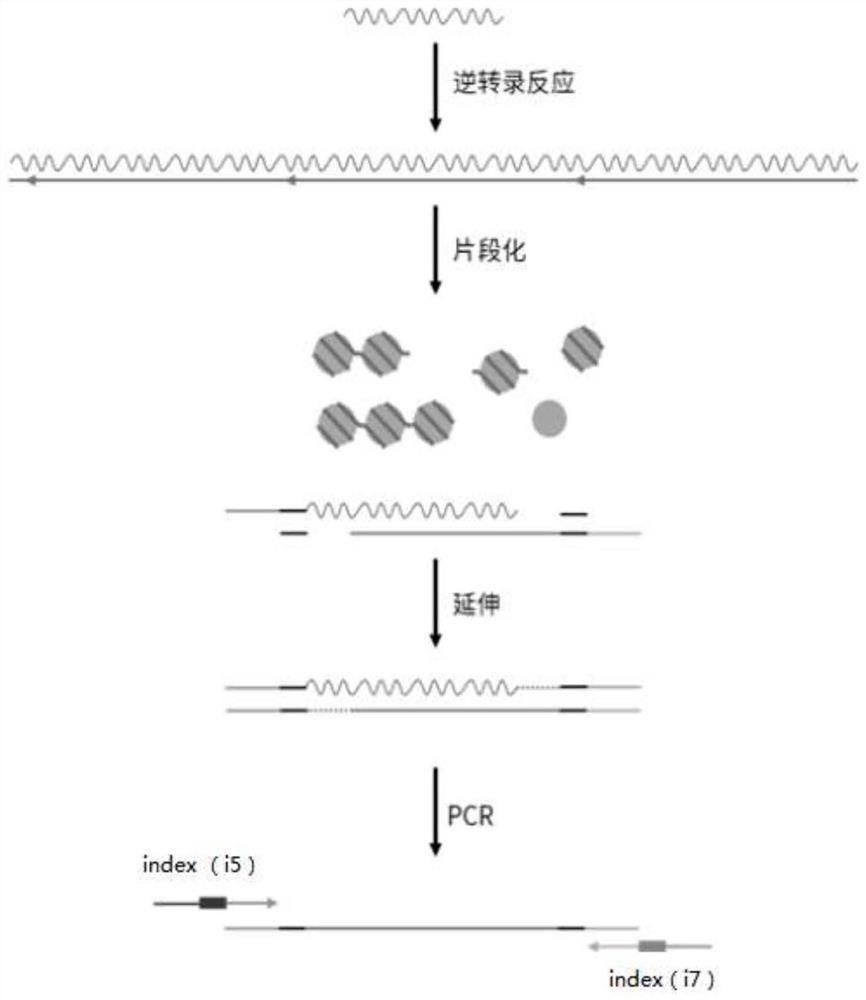
图1是本发明RNA建库的方法流程图;
图2为本发明Tn5转座酶磁珠复合体制备流程图;
图3本发明10ng total RNA用Oligo(dT)
图4为本发明100ng total RNA用Oligo(dT)
图5为本发明1ug total RNA用Oligo(dT)
图6为本发明10ng total RNA用polyA法捕获mRNA后进行first cDNA合成构建的文库;
图7为本发明100ng total RNA用polyA法捕获mRNA后进行first cDNA合成构建的文库;
图8为本发明1ug total RNA用polyA法捕获mRNA后进行first cDNA合成构建的文库;
图9为相同投入量的不同物种的total RNA用Oligo(dT)
图10为相同投入量的不同物种的total RNA用polyA捕获mRNA进行first cDNA合成构建的文库。
具体实施方式
以下实例用于说明本发明,但不限制本发明的范围。在不背离本发明精神和实质的前提下,对本发明的方法、步骤或条件所作的修改或替换,均属于本发明的范围。
本发明主要采用了一种新型的Tn5酶的形式,即将结合了特定接头的Tn5酶通过一定方法固定于链霉亲和素磁珠上,形成Tn5酶磁珠复合体,并将这种磁珠应用于RNA建库当中,所述建库的方法流程示意图如图1所示。
实施案例1:转座复合体与磁珠组装
步骤1:DNA linker引物制备
DNA linker引物包含转座酶识别的mosaic end sequences以及一段与磁珠相连接的序列,具体序列如下:
Biotin-Tn5 ME-A:5'-NNNNNNNNNN(5-100bp)-TCGTC GGCAG CGTCA GATGT GTATAAGAGA CAG-3' 5'-Biotin
Biotin-Tn5 ME-B:5'-NNNNNNNNNN(5-100bp)-GTCTC GTGGG CTCGG AGATG TGTATAAGAG ACAG-3' 5'-Biotin Tn5 MErev:5’-CTGTCTCTTATACACATCT-3’ 5’-磷酸化修饰
步骤2:转座酶复合体的组装
DNA linker退火buffer,转座酶组装buffer,Tn5转座酶由武汉爱博泰克生物科技有限公司生产的One-step DNA Lib prep kit试剂盒提供。
2.1 DNA linker组装
将DNA linker序列溶于1xTE buffer中,调整浓度为100uM,将Biotin-Tn5ME-A序列/Biotin-Tn5 ME-B序列与Tn5 MErev序列已1:1的摩尔比进行DNA linker组装。将配制好DNA linker与退火buffer混匀,放到PCR仪上进行组装,组装程序如下:
95℃,2min;0.1℃/S降温;22℃,5min,12℃保持。
2.2 DNA linker与转座酶转座复合体组装
配制反应体系如下:
配制好的体系放置于PCR仪上30℃,1h。组装结束后用Tn5 storage buffer稀释0.25倍备用。
步骤3:转座复合体与磁珠组装
将组装好的转座复合体与磁珠进行孵育,制备包含转座酶复合体的磁珠;其中磁珠为赛默飞的M-270磁珠,将链霉素亲和磁珠使用wash buffer清洗两遍,重新溶于初始磁珠相同体积的wash buffer中,添加转座酶复合体进行室温孵育,孵育0.5-1h。其中磁珠与转座复合体的组装体积比可以根据需要进行调整,磁珠:转座酶复合体比例为10/1与1/1之间,本实验中磁珠:转座酶复合体比例为2/1,本实验中孵育完成后,置于磁力架上,washbuffer清洗两遍,后储存于保存buffer中。
实施例2同一物种不同total RNA投入量建库
一、利用Oligo(dT)
以total RNA起始投入,分别投入10ng,100ng,1ug mouse total RNA直接进行first cDNA的合成。
1.first cDNA合成
1.1取出Total RNA样本,冰上融解,将10ng~1ug的样本加Nuclease-free Water稀释至8μL,冰上放置备用;
1.2取出5X First Strand Buffer,Oligo(dT)
*First Strand Synthesis Enzyme Mix成分包括逆转录酶和RNA inhibitor,逆转录酶为ABScript III逆转录酶(ABclonal RK20408)或ABScript II逆转录酶(ABclonalRK21400)或其他市售的能够合成全长first cDNA的逆转录酶。
1.3使用移液器吹打混匀,瞬时离心,将体系置于PCR仪(75℃热盖),反应条件如下:25℃10min,42℃30min,70℃10min。
2.First cDNA tagmentation
2.1分别将2X Tagment buffer和10X Tagment Beads从冰箱拿出,恢复至室温,按照下表配制反应体系:
*2X Tagment buffer包括20mM Tris-HCI PH7.5,10mM MgCl
2.2使用移液器上下吸打,充分混匀,立即放到PCR仪上进行打断反应,反应如下(热盖温度75℃):55℃10min,12℃hold。
2.3反应结束,立即取出PCR管,加入10μL 6X Termination Buffer(选自ABclonalRK20205),涡旋混匀或使用移液器吹打混匀,置于PCR仪(热盖温度75℃),反应如下:37℃5min,12℃hold。
2.4待反应结束后,将PCR管直接置于磁力架上,待溶液澄清后,移除上清液(注:移除上清时请注意不要碰到磁珠)。
2.5将PCR管保持在磁力架上,加入200μL的Beads Washing Buffer漂洗磁珠,孵育30s,弃除上清。
2.6重复步骤2.5一次。
2.7保持PCR管在磁力架上,用10μL移液器移除残留的Beads Washing Buffer,微量残留不影响PCR反应。
3.延伸和PCR
3.1按照下表配制延伸和PCR体系,并吹打混匀:
*2X PCR Mix选择ABclonal,RK20698。
*Gap Extension Enzymes选自ABScript III逆转录酶(ABclonal,RK20408)或者ABScript II逆转录酶(ABclonal,RK21400)或者其他等温扩增没Bst3.0 DNA Polymerase(NEB,M0374)或Bst2.0 WarmStart DNA polymerase(NEB,M0538)等。
*N5XX Primer和N7XX Primer均来自Dual DNA Adapter 96 Kit for One-stepDNA Lib Prep(Cat.RK20290)。
3.2将配制好的体系(步骤3.1)按照50μL/样加入到步骤2.7中的PCR管中,移液器吹打或者振荡重悬磁珠,瞬时离心后置于PCR仪中进行PCR反应(热盖温度105℃)。
*:PCR循环数按照下表进行选择:
3.3提前将Agencourt AMPure XP beads从2-8℃取出,静置平衡至室温,使用前涡旋或者振荡混匀。
3.4 PCR反应结束,直接在PCR反应管中加入50μL Agencourt AMPure beads(1X)枪头吹打混匀;。
注:不需要弃去反应管中的反应磁珠。
3.5室温静置5min,然后将PCR管转移至磁力架上~5min,待溶液澄清后,弃除上清。
3.6将离心管保持在磁力架上,加入200μL 80%乙醇,静置30s,弃除全部上清;
3.7重复步骤3.5一次,将PCR管保持在磁力架上,用10μL移液器移除管底残留的乙醇。
3.8干燥磁珠2-3min,待酒精挥发完全后(磁珠颜色从光亮褐色变为磨砂褐色),加入22μL Low-EDTA TE,吹打混匀。
3.9室温静置2min,磁力架上1min,直到溶液变澄清,小心吸取20μL上清至另一离心管中;
建库产量:
整个RNA建库过程可以实现2小时内完成。
每个样本随机取一个进行Agilent 2100分析,如图3-5所示,其中图3为10ngtotal RNA用Oligo(dT
二、polyA法捕获mRNA后进行first cDNA合成
分别投入10ng,100ng,1ug mouse total RNA,用来自ABclonal公司的polyA mRNApurification module(RK20341)富集mRNA后进行first cDNA的合成,具体步骤如下:
1.mRNA的富集
1)将RNA取出冰上溶解,取10ng~1μg total RNA溶于Nuclease-free Water中至总体积50μL,冰上放置备用。
2)将2X Oligo(dT)
3)将混合物放入PCR仪中进行孵育(热盖温度≥75℃):65℃5min;25℃5min。
1.1孵育结束后,取出离心管置于磁力架上约2min,至溶液变澄清,弃上清。
1.2加入200μL Washing Buffer吹打混匀,置于磁力架上,至溶液变澄清,弃上清。
1.3加入15μL Nuclease-free Water,吹打混匀后PCR仪上孵育(热盖温度105℃),80℃2min。
1.4反应结束后立即置于磁力架上,溶液澄清后,取13μL上清置于新的PCR管中,进入下一个反应。
2.First cDNA的合成
*4X First cDNA Synthesis Buffer用来自ABclonal公司的BK0005。
2.1提前将4X First cDNA Synthesis Buffer从-20℃取出融解,冰上按照下表配制反应体系:
2.2使用移液器吹打混匀,瞬时离心,将体系置于PCR仪(75℃热盖)
3.First cDNA tagmentation
3.1分别将2X Tagment buffer和10X Tagment Beads从冰箱拿出,恢复至室温,按照下表配制反应体系:
3.2使用移液器上下吸打,充分混匀,立即放到PCR仪上进行打断反应(热盖温度75℃)。
3.3反应结束,立即取出PCR管,加入10μL 6X Termination Buffer*,涡旋混匀或使用移液器吹打混匀,置于PCR仪(热盖温度75℃):
3.4待反应结束后,将PCR管直接置于磁力架上,待溶液澄清后,移除上清液。
注:移除上清时请注意不要碰到磁珠。
3.5将PCR管保持在磁力架上,加入200μL的Beads Washing Buffer漂洗磁珠,孵育30s,弃除上清。
3.6重复步骤3.5一次。
3.7保持PCR管在磁力架上,用10μL移液器移除残留的Beads Washing Buffer,微量残留不影响PCR反应。
4.延伸和PCR
4.1按照下表配制延伸和PCR体系,并吹打混匀
*N5XX Primer和N7XX Primer均来自Dual DNA Adapter 96 Kit for One-stepDNA Lib Prep(Cat.RK20290)。
4.2将配制好的体系(步骤3.1)按照50μL/样加入到步骤2.7中的PCR管中,移液器吹打或者振荡重悬磁珠,瞬时离心后置于PCR仪中进行PCR反应(热盖温度105℃):
*:PCR循环数按照下表进行选择:
4.3提前将Agencourt AMPure XP beads从2-8℃取出,静置平衡至室温,使用前涡旋或者振荡混匀。
4.4PCR反应结束,直接在PCR反应管中加入50μL Agencourt AMPure beads(1X)枪头吹打混匀;
注:不需要弃去反应管中的反应磁珠。
4.5室温静置5min,然后将PCR管转移至磁力架上~5min,待溶液澄清后,弃除上清。
4.6将离心管保持在磁力架上,加入200μL 80%乙醇,静置30s,弃除全部上清。
4.7重复步骤3.5一次,将PCR管保持在磁力架上,用10μL移液器移除管底残留的乙醇。
4.8干燥磁珠2-3min,待酒精挥发完全后(磁珠颜色从光亮褐色变为磨砂褐色),加入22μL Low-EDTA TE,吹打混匀。
4.9室温静置2min,磁力架上1min,直到溶液变澄清,小心吸取20μL上清至另一离心管中;
建库产量:
整个RNA建库过程可以实现2小时内完成。
每个样本随机取一个进行Agilent 2100分析,如图6-8所示,其中图6为10ngtotal RNA用polyA法捕获mRNA后进行first cDNA合成构建的文库,图7为100ng total RNA用polyA法捕获mRNA后进行first cDNA合成构建的文库,图8为1ug total RNA用polyA法捕获mRNA后进行first cDNA合成构建的文库,文库大小分布均匀,均在200~1000bp左右。
测序数据:Total RNA进入1st cDNA合成与传统的polyA捕获mRNA法进入firstcDNA合成数据比较如下:
结果分析:使用相同物种,投入不同total RNA量建库,用Oligo(dT)
实施案例3.相同投入量的不同物种的total RNA建库
一、利用Oligo(dT)
以total RNA起始投入,分别投入相同量的human,rat,mouse total RNA直接进行first cDNA的合成。
1.first cDNA合成
1.1取出Total RNA样本,冰上融解,将10ng~1ug的样本加Nuclease-free Water稀释至8μL,冰上放置备用。
1.2取出5X First Strand Buffer,Oligo(dT)
1.3使用移液器吹打混匀,瞬时离心,将体系置于PCR仪(75℃热盖)。
2.First cDNA tagmentation
2.1分别将2X Tagment buffer和10X Tagment Beads从冰箱拿出,恢复至室温,按照下表配制反应体系:
2.2使用移液器上下吸打,充分混匀,立即放到PCR仪上进行打断反应(热盖温度75℃):55℃10min,12℃hold。
2.3反应结束,立即取出PCR管,加入10μL 6X Termination Buffer*,涡旋混匀或使用移液器吹打混匀,置于PCR仪(热盖温度75℃):37℃5min,12℃hold。
2.4待反应结束后,将PCR管直接置于磁力架上,待溶液澄清后,移除上清液。
注:移除上清时请注意不要碰到磁珠。
2.5将PCR管保持在磁力架上,加入200μL的Beads Washing Buffer漂洗磁珠,孵育30s,弃除上清。
2.6重复步骤2.5一次。
2.7保持PCR管在磁力架上,用10μL移液器移除残留的Beads Washing Buffer,微量残留不影响PCR反应。
3.延伸和PCR
3.1按照下表配制延伸和PCR体系,并吹打混匀
*N5XX Primer和N7XX Primer均来自Dual DNA Adapter 96 Kit for One-stepDNA Lib Prep(Cat.RK20290)。
3.2将配制好的体系(步骤3.1)按照50μL/样加入到步骤2.7中的PCR管中,移液器吹打或者振荡重悬磁珠,瞬时离心后置于PCR仪中进行PCR反应(热盖温度105℃):
*:PCR循环数按照下表进行选择:
3.3提前将Agencourt AMPure XP beads从2-8℃取出,静置平衡至室温,使用前涡旋或者振荡混匀。
3.4PCR反应结束,直接在PCR反应管中加入50μL Agencourt AMPure beads(1X)枪头吹打混匀;
注:不需要弃去反应管中的反应磁珠。
3.5室温静置5min,然后将PCR管转移至磁力架上~5min,待溶液澄清后,弃除上清。
3.6将离心管保持在磁力架上,加入200μL 80%乙醇,静置30s,弃除全部上清。
3.7重复步骤3.5一次,将PCR管保持在磁力架上,用10μL移液器移除管底残留的乙醇。
3.8干燥磁珠2-3min,待酒精挥发完全后(磁珠颜色从光亮褐色变为磨砂褐色),加入22μL Low-EDTA TE,吹打混匀。
3.9室温静置2min,磁力架上1min,直到溶液变澄清,小心吸取20μL上清至另一离心管中;
建库产量:
每个样本随机取一个进行Caliper分析,如图9所示,其中A为1ug human totalRNA用Oligo(dT)
二、polyA法捕获mRNA后进行first cDNA合成
分别投入10ng,100ng,1ug mouse total RNA,用来自ABclonal公司的polyA mRNApurification module(RK20341)富集mRNA后进行first cDNA的合成,具体步骤如下:
1.mRNA的富集
1.1将RNA取出冰上溶解,取10ng~1μg total RNA溶于Nuclease-free Water中至总体积50μL,冰上放置备用。
1.2将2X Oligo(dT)
1.3将混合物放入PCR仪中进行孵育(热盖温度≥75℃):65℃5min,25℃5min。
1.4孵育结束后,取出离心管置于磁力架上约2min,至溶液变澄清,弃上清。
1.5加入200μL Washing Buffer吹打混匀,置于磁力架上,至溶液变澄清,弃上清。
1.6加入15μL Nuclease-free Water,吹打混匀后PCR仪上孵育(热盖温度105℃):80℃2min。
1.7反应结束后立即置于磁力架上,溶液澄清后,取13μL上清置于新的PCR管中,进入下一个反应。
2.First cDNA的合成
2.1提前将4X First cDNA Synthesis Buffer从-20℃取出融解,冰上按照下表配制反应体系:
2.2使用移液器吹打混匀,瞬时离心,将体系置于PCR仪(75℃热盖):25℃10min,42℃30min,70℃10min。
3.First cDNA tagmentation
3.1分别将2X Tagment buffer和10X Tagment Beads从冰箱拿出,恢复至室温,按照下表配制反应体系:
3.2使用移液器上下吸打,充分混匀,立即放到PCR仪上进行打断反应(热盖温度75℃):55℃10min,12℃hold。
3.3反应结束,立即取出PCR管,加入10μL 6X Termination Buffer,涡旋混匀或使用移液器吹打混匀,置于PCR仪(热盖温度75℃):37℃5min,12℃hold。
3.4待反应结束后,将PCR管直接置于磁力架上,待溶液澄清后,移除上清液。注:移除上清时请注意不要碰到磁珠。
3.5将PCR管保持在磁力架上,加入200μL的Beads Washing Buffer漂洗磁珠,孵育30s,弃除上清。
3.6重复步骤3.5一次。
3.7保持PCR管在磁力架上,用10μL移液器移除残留的Beads Washing Buffer,微量残留不影响PCR反应。
4.延伸和PCR
4.1按照下表配制延伸和PCR体系,并吹打混匀:
*N5XX Primer和N7XX Primer均来自Dual DNA Adapter 96 Kit for One-stepDNA Lib Prep(Cat.RK20290)。
4.2将配制好的体系(步骤3.1)按照50μL/样加入到步骤3.7中的PCR管中,移液器吹打或者振荡重悬磁珠,瞬时离心后置于PCR仪中进行PCR反应(热盖温度105℃):
*:PCR循环数按照下表进行选择:
4.3提前将Agencourt AMPure XP beads从2-8℃取出,静置平衡至室温,使用前涡旋或者振荡混匀。
4.4 PCR反应结束,直接在PCR反应管中加入50μL Agencourt AMPure beads(1X)枪头吹打混匀。
注:不需要弃去反应管中的反应磁珠。
4.5室温静置5min,然后将PCR管转移至磁力架上~5min,待溶液澄清后,弃除上清。
4.6将离心管保持在磁力架上,加入200μL 80%乙醇,静置30s,弃除全部上清。
4.7重复步骤3.5一次,将PCR管保持在磁力架上,用10μL移液器移除管底残留的乙醇。
4.8干燥磁珠2-3min,待酒精挥发完全后(磁珠颜色从光亮褐色变为磨砂褐色),加入22μL Low-EDTA TE,吹打混匀。
4.9室温静置2min,磁力架上1min,直到溶液变澄清,小心吸取20μL上清至另一离心管中;建库结果:
每个样本随机取一个进行Caliper分析,如图10所示,其中A为1ug human totalRNA用polyA法捕获mRNA后进行first cDNA合成构建的文库,B为1ug rat total RNA用polyA法捕获mRNA后进行first cDNA合成构建的文库,C为1ug mouse total RNA用polyA法捕获mRNA后进行first cDNA合成构建的文库,D为100ng human total RNA用polyA法捕获mRNA后进行first cDNA合成构建的文库,E为100ng rat total RNA用polyA法捕获mRNA后进行first cDNA合成构建的文库,F为100ng mouse total RNA用polyA法捕获mRNA后进行first cDNA合成构建的文库,文库大小分布均匀,均在200~1000bp左右。
结果分析:相同total RNA投入量的不同物种建库测试,用Oligo(dT)
因此本发明提供的RNA建库起始材料RNA的投入量比较灵活,打破了传统的一种Tn5转座酶量只针对特定的RNA的投入量的建库方法,降低转座酶对模板投入的依赖性,可以根据自己需求投入10ng到1ug不同量的RNA,在满足快速灵活建库的同时,扩大样品量的适用范围。另外,本发明提供的RNA建库过程操作简单,仅需要2个小时完成,克服建库周期长、步骤复杂、容易出错的缺陷。
本发明并不仅仅限于说明书和实施方式中所描述,因此对于熟悉领域的人员而言可容易地实现另外的优点和改进,故在不背离权利要求及等同范围所限定的一般概念的精神和范围的情况下,本发明并不限于特定的细节、代表性的方案和这里示出与描述的图示示例。