一种基于转座酶的DNA/RNA共建库方法、试剂盒及应用
文献发布时间:2023-06-19 09:29:07
技术领域
本发明涉及DNA/RNA文库构建,尤其涉及一种基于转座酶的DNA/RNA共建库方法及其试剂盒在微生物鉴定中的应用。
背景技术
高通量测序(NGS)在肿瘤检测方面的应用:
以人基因组为代表的哺乳动物基因组由内含子与外显子区域构成,其中外显子区域只占整个基因组序列的很小一部分。基因转录后产生RNA前体,后者通过可变剪切去除内含子序列,剩下的外显子区域拼接为mRNA序列,最终翻译为蛋白质。
肿瘤基因组的变异主要包括单点突变(SNV)、插入/缺失(InDel)、拷贝数变异(CNV)、基因融合、基因表达差异等。基因的点突变在肿瘤细胞中最为常见,针对性的肿瘤靶向用药也比较成熟。比如EGFR基因的T790M突变导致的非小细胞肺癌病患,使用奥希替尼治疗的平均存活期可到40个月。另一大类的变异是基因融合,主要指的是基因组上两个或两个以上不同区域的编码基因首尾相连,置于一套调控序列控制下。基因融合是一类常见的肿瘤发生机制,是多种肿瘤靶向药的生物标标志物(biomarker),如治疗ALK融合的克唑替尼。
单个或少量的碱基突变可以用ARMS-PCR、一代测序或者数字PCR等技术来检测,但是肿瘤发生发展过程中往往涉及多个基因的多位点变异,上述传统方法难以进行如此广泛的基因检测。而高通量测序技术的出现,很顺理成章的解决了这个难题,该技术直接对整个基因组或者转录组进行测序,能够全面地“扫描”肿瘤相关的基因变异情况,更能在基因组学的维度发现未知的肿瘤靶标。
对肿瘤细胞的基因组进行高通量测序(NGS),可轻松实现单碱基变异的监控,但若想监控基因融合变异就比较捉襟见肘了,原因正是上文中提及的整个基因组上分布有大占比的内含子区域和小占比的外显子区域。基因融合针对的是外显子区域,如果通过基因组测序来监控融合,必须进行跨内含子规模的测序。目前主流的高通量测序是短读长的模式,长度只有300~600bp,无法涵盖完整的内含子区。因此对基因融合的监测,需对肿瘤细胞的转录组进行高通量测序。目前癌症检测还是以实体瘤组织细胞的基因组DNA测序为主,辅以转录组RNA测序来检测融合,但两者的重要程度日趋一致。考虑到肿瘤的临床样本多以福尔马林固定石蜡包埋处理的样本(Formalin-Fixed and Parrffin Embedded,FFPE)为主,这类样本的量比较少,难以分开进行DNA建库和RNA建库实验。近年来有人提出一个方案用于FFPE的DNA/RNA共建库(CN106222164A),该方案仍然使用的是传统的RNA建库流程,流程繁琐、耗时长、产量低,难以解决肿瘤细胞的DNA和RNA共建库的临床难题。
高通量测序在感染检测方面的应用:
宏基因组高通量测序(metagenomics NGS,mNGS)目前在病原微生物感染检测方面的应用如火如荼,特别是对新发的未知感染源检测特别有效,如今年爆发的新冠肺炎疫情,最初就是通过mNGS技术来确诊了感染源是一种新型的冠状病毒。感染相关的病原微生物种类繁多,不过以遗传物质来分类的话,可以分为RNA和DNA病原微生物。常规的mNGS检测一次只能针对一种核酸类型的微生物,比如细菌类的感染病例(肺炎支原体、结核分歧杆菌等)或者RNA病毒类(冠状病毒、艾滋病毒等)的案例。然后在实际的感染病例确诊过程中,特别是对于疑难的新发病例诊断,往往连病原微生物的基本信息都难以获得,换言之无法确认是DNA还是RNA来源的微生物。这时候,如果能同时进行基因组(DNA)和转录组(RNA)建库测序,那必然可以加速感染源的定位。
目前尚未有感染领域mNGS检测的DNA/RNA共建库报道,当然,鉴于感染中病原微生物RNA/DNA共提取的难度大、复杂性高、DNA和RNA丰度差异大等问题,要想实现DNA/RNA共建库,本身就是一个不小的挑战。
综上所述,DNA/RNA共建库是一种比较前沿的技术,相关的文献报道很少,市面上也没有成熟的产品。目前仅有的DNA/RNA共建库报道中RNA建库部分还是使用了传统的RNA建库原理,即先进行RNA逆转录生产cDNA第一链,而后利用RNase H和DNA聚合酶I合成双链cDNA。最后,体系中原有的基因组DNA和后生成的双链cDNA就可以进行常规的DNA建库方案。该方案缺点甚多,主要有如下几个方面:
1)操作非常耗时,进行一次完整建库需要花费1.5天;
2)步骤繁琐,难以适配于自动化建库;手动操作稳定性低、易出错。
3)涉及至少10种酶原料,成本高。
4)底层的建库原理仍采用传统的DNA/RNA建库方法,对于微量DNA/RNA的建库效率低,如从痰液、口拭子、肺泡灌洗液等感染类样本中共提取的DNA/RNA样本。
发明内容
有鉴于现有技术的上述缺陷,本发明提供了一种基于转座酶的DNA/RNA共建库方案,该方案利用转座酶复合物可以识别RNA/DNA杂交链的原理,在片段化杂交链的同时加上接头序列,最后通过PCR进行完整的文库的构建。
本发明一方面提供了一种基于转座酶的DNA/RNA共建库方法,包括以下步骤:
1)从样本中共提取DNA和RNA;
2)以RNA为模板,通过逆转录生成cDNA第一链,最终形成RNA/cDNA杂交双链和原始DNA的混合液;
3)使用包含转座子序列的转座酶复合体对步骤2)中的RNA/cDNA杂交双链和原始DNA进行片段化,并同时加上接头序列;
4)使用逆转录酶和具有链置换活性的DNA聚合酶对步骤3)中的产物进行缺口补齐;
5)用文库扩增引物对步骤4)中的产物进行扩增;
6)步骤5)所得产物经纯化回收后,得到混合RNA/DNA文库;
7)对步骤6)所得的混合RNA/DNA文库进行质检。
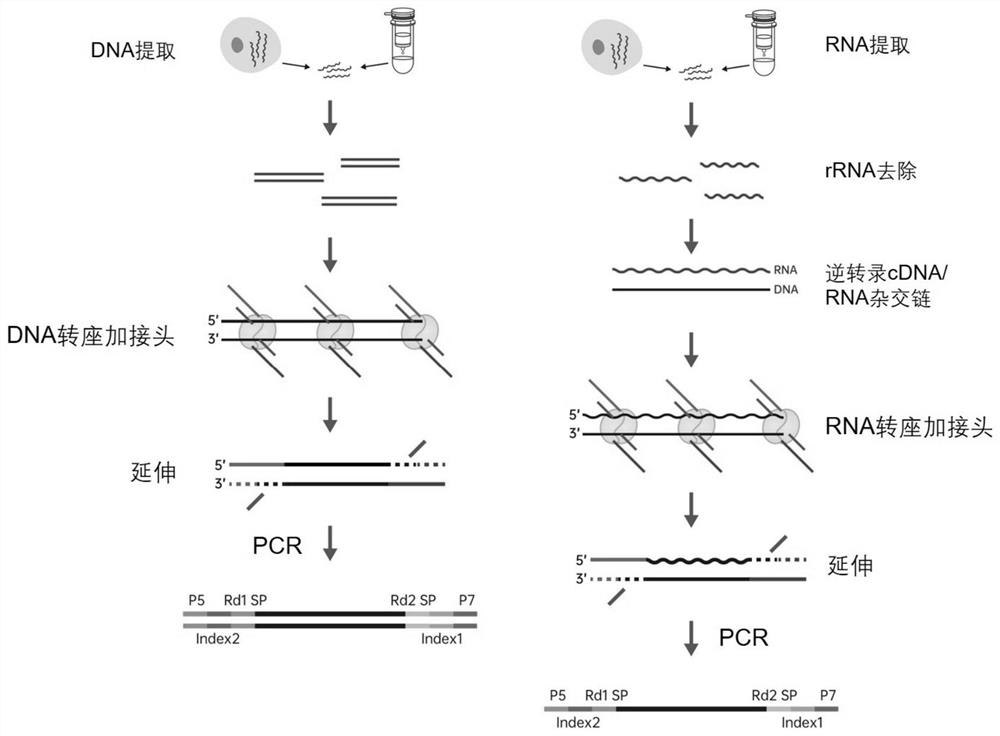
上述方法的共建库流程如图1所示。
其中,步骤3)中所述的转座酶复合体中含有接头序列。
可选地,所述转座酶选自用于NGS文库构建的转座酶及其突变体,例如,商业化的Tn5突变体:Illumina公司的
可选地,步骤4)中所述的逆转录酶和具有链置换活性的DNA聚合酶也可以是具有逆转录能力的Bst 3.0DNA聚合酶(NEB公司,货号M0374M)。
进一步,步骤7)中所述质检包括但不限于文库定量、分布情况检测、鉴定DNA和RNA核酸物质是否都被建成文库;如通过Qubit 3.0核酸定量仪定量,通过毛细管电泳查看文库分布,通过qPCR技术鉴定DNA和RNA核酸物质是否都被建成文库。
优选地,所述鉴定DNA和RNA核酸物质是否都被建成文库的方法包括通过qPCR技术来检测文库中管家基因的含量,其中qPCR引物设计区域位于管家基因的跨内含子与外显子区域,采用不同的荧光探针来加以区别,结果判断依据是只有DNA文库才有内含子区域的荧光信号,而DNA文库和RNA文库都会显示外显子区域的另一种荧光信号。
在本发明的一种较佳实施方式中,所述管家基因为GAPDH。qPCR引物设计区域位于人基因组GAPDH基因的跨内含子与外显子区域,荧光探针分别为FAM和TAMRA,结果判断依据是只有DNA文库才有内含子区域的TAMRA荧光信号,而DNA文库和RNA文库都会贡献外显子区域的FAM信号值。
优选地,qPCR扩增引物和探针的序列如下:
扩增引物F:5’-CCCTTCATTGACCTCAACTAC-3’(SEQ No.1)
扩增引物R:5’-CTCAGCCTTGACGGTGCCATGG-3’(SEQ No.2)
探针TAMRA:5’-TAMRA-AAAGCTGGTGTGGGAGGAGCCA-MGB-3’(SEQ No.3)
探针FAM:5’-FAM-TTTACATGTTCCAATATGATTC-MGB-3’(SEQ No.4)。
进一步优选地,结果合格的指标是:Ct_TAMRA<30,同时Ct_FAM-Ct_TAMRA<-0.5。
优选地,步骤6)中的所述纯化为磁珠纯化,可分多轮进行,如进行两轮纯化。
本发明另一方面提供了一种基于转座酶的DNA/RNA共建库试剂盒,该试剂盒包括:逆转录试剂、包含转座子序列的转座酶复合体片段化试剂、缺口补齐试剂、文库扩增试剂、文库质检试剂。
进一步,所述逆转录试剂用于逆转录RNA模板生成cDNA第一链,优选包括逆转录酶、逆转录缓冲液和逆转录引物。
进一步,所述包含转座子序列的转座酶复合体片段化试剂用于将RNA/cDNA杂交双链和原始DNA片段化并加上接头序列,优选包括包含转座子序列的转座酶复合体、转座缓冲液;且所述转座酶复合体含有接头序列。
进一步,所述缺口补齐试剂包括逆转录酶和具有链置换活性的DNA聚合酶,或者所述缺口补齐试剂包括:具有逆转录能力的DNA聚合酶,如具有逆转录能力的Bst3.0DNA聚合酶(NEB公司,货号M0374M)。
进一步,所述文库扩增试剂包括文库扩增引物、N5标签引物、N7标签引物。
进一步,所述文库质检试剂包括核酸定量、毛细管电泳和qPCR试剂中的一种或几种。优选地,所述文库质检试剂包括鉴定DNA和RNA核酸物质是否都被建成文库的试剂。
进一步优选地,所述鉴定DNA和RNA核酸物质是否都被建成文库的试剂包括通过qPCR技术来检测文库中管家基因的含量的试剂,其中qPCR引物设计区域位于管家基因的跨内含子与外显子区域,采用不同的荧光探针来加以区别,结果判断依据是只有DNA文库才有内含子区域的荧光信号,而DNA文库和RNA文库都会显示外显子区域的另一种荧光信号。
在本发明的一种较佳实施方式中,所述qPCR试剂为通过荧光定量PCR来检测文库中管家基因GAPDH的含量的相关试剂,qPCR引物设计区域位于人基因组GAPDH基因的跨内含子与外显子区域,荧光探针分别为FAM和TAMRA,结果判断依据是只有DNA文库才有内含子区域的TAMRA荧光信号,而DNA文库和RNA文库都会贡献外显子区域的FAM信号值。
优选地,qPCR扩增引物和探针的序列如下:
扩增引物F:5’-CCCTTCATTGACCTCAACTAC-3’(SEQ No.1)
扩增引物R:5’-CTCAGCCTTGACGGTGCCATGG-3’(SEQ No.2)
探针TAMRA:5’-TAMRA-AAAGCTGGTGTGGGAGGAGCCA-MGB-3’(SEQ No.3)
探针FAM:5’-FAM-TTTACATGTTCCAATATGATTC-MGB-3’(SEQ No.4);
进一步优选地,结果合格的指标是:Ct_TAMRA<30,同时Ct_FAM-Ct_TAMRA<-0.5。
进一步,所述基于转座酶的DNA/RNA共建库试剂盒还包括纯化试剂,如磁珠,优选为Beckman公司的AMPure XP磁珠。
本发明第三方面提供了一种鉴定样本中所含微生物种类的方法,该方法包括步骤:
1)从样本中共提取DNA和RNA;
2)以RNA为模板,通过逆转录生成cDNA第一链,最终形成RNA/cDNA杂交双链和原始DNA的混合液;
3)使用包含转座子序列的转座酶复合体对步骤2)中的RNA/cDNA杂交双链和原始DNA进行片段化,并同时加上接头序列;
4)使用逆转录酶和具有链置换活性的DNA聚合酶对步骤3)中的产物进行缺口补齐;
5)用文库扩增引物对步骤4)中的产物进行扩增;
6)步骤5)所得产物经纯化回收后,得到混合RNA/DNA文库;
7)对步骤6)所得的混合RNA/DNA文库进行质检;
8)对质检后的混合RNA/DNA文库进行高通量测序。
其中,步骤1)所述的样本包括至少一种微生物,如包括细菌、RNA病毒中的一种或几种。
在本发明的一些实施例中,所述样本包括细菌、RNA病毒和人基因组DNA,例如人的组织样本、血液样本或痰液、口拭子、肺泡灌洗液等感染类样本。
优选地,步骤7)所述的质检包括但不限于:通过Qubit 3.0核酸定量仪定量,通过毛细管电泳查看文库分布,通过qPCR技术来鉴定DNA和RNA核酸物质是否都被建成文库。
优选地,所述鉴定DNA和RNA核酸物质是否都被建成文库的方法包括通过qPCR技术来检测文库中管家基因的含量,其中qPCR引物设计区域位于管家基因的跨内含子与外显子区域,采用不同的荧光探针来加以区别,结果判断依据是只有DNA文库才有内含子区域的荧光信号,而DNA文库和RNA文库都会显示外显子区域的另一种荧光信号。
在本发明的一种较佳实施方式中,所述管家基因为GAPDH。qPCR引物设计区域位于人基因组GAPDH基因的跨内含子与外显子区域,荧光探针分别为FAM和TAMRA,结果判断依据是只有DNA文库才有内含子区域的TAMRA荧光信号,而DNA文库和RNA文库都会贡献外显子区域的FAM信号值。
优选地,qPCR扩增引物和探针的序列如SEQ No.1-4所示,结果合格的指标是:Ct_TAMRA<30,同时Ct_FAM-Ct_TAMRA<-0.5。
本发明第四方面提供了一种鉴定样本中所含微生物种类的试剂盒,该试剂盒包括:逆转录试剂、包含转座子序列的转座酶复合体片段化试剂、缺口补齐试剂、文库扩增试剂、文库质检试剂。该试剂盒用于鉴定包括至少一种微生物的样本中的微生物种类,如包括细菌、RNA病毒中的一种或几种。
在本发明的一些实施例中,所述样本包括细菌、RNA病毒和人基因组DNA,例如人的组织样本、血液样本或痰液、口拭子、肺泡灌洗液等感染类样本。
进一步,所述逆转录试剂用于逆转录RNA模板生成cDNA第一链,优选包括逆转录酶、逆转录缓冲液和逆转录引物。
进一步,所述包含转座子序列的转座酶复合体片段化试剂用于将RNA/cDNA杂交双链和原始DNA片段化并加上接头序列,优选包括包含转座子序列的转座酶复合体、转座缓冲液;且所述转座酶复合体含有接头序列。
进一步,所述缺口补齐试剂包括逆转录酶和具有链置换活性的DNA聚合酶,或者所述缺口补齐试剂包括:具有逆转录能力的DNA聚合酶,如具有逆转录能力的Bst3.0DNA聚合酶(NEB公司,货号M0374M)。
进一步,所述文库扩增试剂包括文库扩增引物、N5标签引物、N7标签引物。
进一步,所述鉴定样本中所含微生物种类的试剂盒还包括纯化试剂,如磁珠,优选为Beckman公司的AMPure XP磁珠。
进一步,所述文库质检试剂包括核酸定量、毛细管电泳和qPCR试剂中的一种或几种。
优选地,所述文库质检试剂包括鉴定DNA和RNA核酸物质是否都被建成文库的试剂。
进一步优选地,所述鉴定DNA和RNA核酸物质是否都被建成文库的试剂包括通过qPCR技术来检测文库中管家基因的含量的试剂,其中qPCR引物设计区域位于管家基因的跨内含子与外显子区域,采用不同的荧光探针来加以区别,结果判断依据是只有DNA文库才有内含子区域的荧光信号,而DNA文库和RNA文库都会显示外显子区域的另一种荧光信号。
在本发明的一种较佳实施方式中,所述qPCR试剂为通过荧光定量PCR来检测文库中管家基因GAPDH的含量的相关试剂,qPCR引物设计区域位于人基因组GAPDH基因的跨内含子与外显子区域,荧光探针分别为FAM和TAMRA,结果判断依据是只有DNA文库才有内含子区域的TAMRA荧光信号,而DNA文库和RNA文库都会贡献外显子区域的FAM信号值。
优选地,qPCR扩增引物和探针的序列如SEQ No.1-4所示,结果合格的指标是:Ct_TAMRA<30,同时Ct_FAM-Ct_TAMRA<-0.5。
更进一步,所述鉴定样本中所含微生物种类的试剂盒还包括DNA/RNA共提取试剂、高通量测序试剂中的一种或几种。
本发明第五方面提供了一种检测肿瘤基因组变异的方法,该方法包括步骤:
1)从样本中共提取DNA和RNA;
2)以RNA为模板,通过逆转录生成cDNA第一链,最终形成RNA/cDNA杂交双链和原始DNA的混合液;
3)使用包含转座子序列的转座酶复合体对步骤2)中的RNA/cDNA杂交双链和原始DNA进行片段化,并同时加上接头序列;
4)使用逆转录酶和具有链置换活性的DNA聚合酶对步骤3)中的产物进行缺口补齐;
5)用文库扩增引物对步骤4)中的产物进行扩增;
6)步骤5)所得产物经纯化回收后,得到混合RNA/DNA文库;
7)对步骤6)所得的混合RNA/DNA文库进行质检;
8)对质检后的混合RNA/DNA文库进行高通量测序。
可选地,步骤1)所述的样本可以为人的组织样本、血液样本;优选地,所述样本为FFPE样本。
优选地,步骤7)所述的质检包括但不限于:通过Qubit 3.0核酸定量仪定量,通过毛细管电泳查看文库分布,通过qPCR技术来鉴定DNA和RNA核酸物质是否都被建成文库。
优选地,所述鉴定DNA和RNA核酸物质是否都被建成文库的方法包括通过qPCR技术来检测文库中管家基因的含量,其中qPCR技术的引物设计区域位于管家基因的跨内含子与外显子区域,采用不同的荧光探针来加以区别,结果判断依据是只有DNA文库才有内含子区域的荧光信号,而DNA文库和RNA文库都会显示外显子区域的另一种荧光信号。
在本发明的一种较佳实施方式中,所述管家基因为GAPDH。qPCR引物设计区域位于人基因组GAPDH基因的跨内含子与外显子区域,荧光探针分别为FAM和TAMRA,结果判断依据是只有DNA文库才有内含子区域的TAMRA荧光信号,而DNA文库和RNA文库都会贡献外显子区域的FAM信号值。
优选地,qPCR扩增引物和探针的序列如SEQ No.1-4所示,结果合格的指标是:Ct_TAMRA<30,同时Ct_FAM-Ct_TAMRA<-0.5。
本发明最后一方面提供了一种检测肿瘤基因组变异的试剂盒,该试剂盒包括:逆转录试剂、包含转座子序列的转座酶复合体片段化试剂、缺口补齐试剂、文库扩增试剂、文库质检试剂。该试剂盒可用于检测单点突变(SNV)、插入/缺失(InDel)、拷贝数变异(CNV)、基因融合、基因表达差异中的一种或几种。
可选地,所述试剂盒用于检测人的组织样本、血液样本。优选地,所述试剂盒用于检测FFPE样本。
进一步,所述逆转录试剂用于逆转录RNA模板生成cDNA第一链,优选包括逆转录酶、逆转录缓冲液和逆转录引物。
进一步,所述包含转座子序列的转座酶复合体片段化试剂用于将RNA/cDNA杂交双链和原始DNA片段化并加上接头序列,优选包括包含转座子序列的转座酶复合体、转座缓冲液;且所述转座酶复合体含有接头序列。
进一步,所述缺口补齐试剂包括逆转录酶和具有链置换活性的DNA聚合酶,或者所述缺口补齐试剂包括:具有逆转录能力的DNA聚合酶,如具有逆转录能力的Bst3.0DNA聚合酶(NEB公司,货号M0374M)。
进一步,所述文库扩增试剂包括文库扩增引物、N5标签引物、N7标签引物。
进一步,所述检测肿瘤基因组变异的试剂盒还包括纯化试剂,如磁珠,优选为Beckman公司的AMPure XP磁珠。
进一步,所述文库质检试剂包括核酸定量、毛细管电泳和qPCR试剂中的一种或几种。
优选地,所述文库质检试剂包括鉴定DNA和RNA核酸物质是否都被建成文库的试剂。
进一步优选地,所述鉴定DNA和RNA核酸物质是否都被建成文库的试剂包括通过qPCR技术来检测文库中管家基因的含量的试剂,其中qPCR引物设计区域位于管家基因的跨内含子与外显子区域,采用不同的荧光探针来加以区别,结果判断依据是只有DNA文库才有内含子区域的荧光信号,而DNA文库和RNA文库都会显示外显子区域的另一种荧光信号。
在本发明的一种较佳实施方式中,所述qPCR试剂为通过荧光定量PCR来检测文库中管家基因GAPDH的含量的相关试剂,qPCR引物设计区域位于人基因组GAPDH基因的跨内含子与外显子区域,荧光探针分别为FAM和TAMRA,结果判断依据是只有DNA文库才有内含子区域的TAMRA荧光信号,而DNA文库和RNA文库都会贡献外显子区域的FAM信号值。
优选地,qPCR扩增引物和探针的序列如SEQ No.1-4所示,结果合格的指标是:Ct_TAMRA<30,同时Ct_FAM-Ct_TAMRA<-0.5。
更进一步,所述检测肿瘤基因组变异的试剂盒还包括DNA/RNA共提取试剂、高通量测序试剂中的一种或几种。
与现有技术相比,本发明具有如下的几个优点和进步:
1)建库时间短:本发明使用转座酶对RNA和DNA同时进行,整个过程只需要4小时;而现有方案需要1.5天。
2)建库操作简单:转座酶复合物片段化DNA链或RNA/DNA杂交链的同时,即可将接头添加到断裂的DNA片段两边,也称为标签化(tagmentation);整个标签化的过程只需要10分钟,无需纯化即可进行延伸和文库扩增。
3)成本低:转座酶法DNA/RNA共建库全程只需要用到4个分子酶,这些酶都可以用基因工程方法进行大规模表达,成本较低。
4)文库完整性高:通过质检步骤,确定了DNA和RNA都被建成文库,为后续高通量测序结果的完整性打下良好基础。
此外,本发明可实现从同一个生物样本中共提取的DNA和RNA的同时建库,解决了样本量少,难以分开进行DNA建库和RNA建库的问题,并可进一步进行测序,根据测序结果可鉴定所含的微生物种类,从而提高未知样本的微生物鉴定效率,根据测序结果还可获知肿瘤基因组变异情况,从而提高肿瘤的诊断、治疗效率。
以下将结合附图对本发明的构思、具体结构及产生的技术效果作进一步说明,以充分地了解本发明的目的、特征和效果。
附图说明
图1是本发明基于转座酶的DNA和RNA共建库流程图;
图2是HEK293细胞基因组DNA和mRNA共建库文库的分布图;
图3是qPCR法验证DNA/RNA共建文库的原理图;
图4是混合微生物样本经高通量测序后的物种归属分析图。
具体实施方式
实施例1
根据本发明的方法,对同一份组织或者细胞样本所提取的核酸进行DNA/RNA共建库,并在上机测序前进行文库质量的检测。
1.样本处理与准备
取10
2.文库构建
2.1逆转录
2.1.1在200μL PCR管中按下表制备反应体系:
涡旋混匀后4000rpm瞬时离心,在PCR仪中65℃反应5min,4℃保温。
2.1.2在200μL PCR管中按照下表配制反应体系:
涡旋混匀后4000rpm瞬时离心,在PCR仪中按如下程序进行反应:
2.2片段化加接头
按照下表进行反应体系的配制:
涡旋混匀后4000rpm瞬时离心,在PCR仪中55℃反应30min,4℃保温。
2.3延伸
按照下表进行反应体系的配制:
涡旋混匀后4000rpm瞬时离心,在PCR仪中65℃反应20min,4℃保温。
2.4扩增
按照下表进行反应体系的配制:
涡旋混匀后4000rpm瞬时离心,在PCR仪中按如下程序进行反应:
2.5磁珠回收
使用Beckman公司的AMPure XP磁珠进行文库片段分选,选用的分选比例为第一轮0.7×,第二轮为0.2×,所得文库大小为400bp左右。
3.文库质检
3.1DNA/RNA共建库文库通过Qubit 3.0核酸定量仪定量,并进行毛细管电泳查看文库分布,分布结果如图2所示。
3.2为鉴定DNA和RNA核酸物质是否都被建成文库,我们通过qPCR技术来检测文库中管家基因GAPDH的含量。qPCR引物设计区域位于人基因组GAPDH基因的跨内含子与外显子区域,荧光探针分别为FAM和TAMRA(图3),引物和探针的序列如下:
扩增引物F:5’-CCCTTCATTGACCTCAACTAC-3’
扩增引物R:5-CTCAGCCTTGACGGTGCCATGG-3’
探针TAMRA:5’-TAMRA-AAAGCTGGTGTGGGAGGAGCCA-MGB-3’
探针FAM:5’-FAM-TTTACATGTTCCAATATGATTC-MGB-3’
将上一步定量后的DNA/RNA文库进行十倍浓度稀释,按照下表配制体系:
涡旋混匀后4000rpm瞬时离心,在荧光PCR仪中按如下程序进行反应:
结果合格的指标是:Ct_TAMRA<30,同时Ct_FAM-Ct_TAMRA<-0.5,判断依据是只有DNA文库才有内含子区域的TAMRA荧光信号,而DNA文库和RNA文库都会贡献外显子区域的FAM信号值。
实施例2
在本实施例中,人工混合多种细菌和假病毒制备模拟样本,通过DNA/RNA共提取、共建库,高通量测序后进行微生物物种分析。
1.样本处理与准备
1.1将表1中所列的细菌和病毒(假病毒)按照等cfu或等拷贝数进行混合,制备模拟样本,分别设三个平行样。
表1.本实施例中所选细菌和病毒种类
1.2使用AllPrep DNA/RNA Mini Kit(Qiagen)对上述混合样本进行DNA和RNA的共提取。所得的核酸分别用50μL TE buffer溶解,并使用Qubit 3.0荧光计进行浓度测定。最后取10ng DNA和10ng RNA进行混合,按照下述的操作步骤进行共建库。
2.文库构建
按照实施例1中的建库流程进行DNA/RNA共建库。
3.文库质检
DNA/RNA共建库文库通过Qubit 3.0核酸定量仪定量,并进行毛细管电泳查看文库分布。
通过qPCR技术来检测文库中管家基因GAPDH的含量,鉴定DNA和RNA核酸物质是否都被建成文库,具体方法参见实施例1。
4.高通量测序
将质检好的文库送至天津诺禾致源进行高通量测序,下机数据与表1中物种基因组进行比对,分析所测得的物种信息。最终结果如图4所示,其中表1中所列的物种都能正常检出。
实施例3
在本实施例中,等比例混合两种FFPE基因变异标准品,分别是Horizon discovery公司的Quantitative Multiplex Reference Standard标准品(#HD200)和菁良生物的肿瘤融合FFPE标准品(#GW-OPSM001),这两个标准品所涉及的基因融合突变的类型和频率如表2所示。通过对这两个FFPE样本进行DNA/RNA共提取、共建库,共捕获,并在高通量测序后分析点突变和基因融合表达量水平。
表2.两种基因融合突变FFPE标准品的突变类型和频率
1.样本处理与准备
各取一份Quantitative Multiplex Reference Standard标准品(#HD200)和肿瘤融合FFPE标准品(#GW-OPSM001),使用Allprep FFPE DNA/RNA Kit(Qiagen)进行FFPE样本的DNA和RNA的共提取。所得的核酸分别用50μL TE buffer溶解,并使用Qubit 3.0荧光计进行浓度测定。
100ng DNA和100ng RNA进行混合,按照下述的操作步骤进行共建库。
2.文库构建
2.1逆转录
2.1.1在200μL PCR管中按下表制备反应体系:
涡旋混匀后4000rpm瞬时离心,在PCR仪中65℃反应5min,4℃保温。
2.1.2在200μL PCR管中按照下表配制反应体系:
涡旋混匀后4000rpm瞬时离心,在PCR仪中按如下程序进行反应:
2.2片段化加接头
按照下表进行反应体系的配制:
涡旋混匀后4000rpm瞬时离心,在PCR仪中55℃反应30min,4℃保温。
2.3延伸
按照下表进行反应体系的配制:
涡旋混匀后4000rpm瞬时离心,在PCR仪中65℃反应20min,4℃保温。
2.4扩增
按照下表进行反应体系的配制:
涡旋混匀后4000rpm瞬时离心,在PCR仪中按如下程序进行反应:
2.5磁珠回收
使用Beckman公司的AMPure XP磁珠进行文库片段分选,选用的分选比例为第一轮0.7×,第二轮为0.2×,所得文库大小为400bp左右。
3.文库杂交捕获
委托艾吉泰康生物科技(北京)有限公司进行杂交捕获探针的设计,设计的原则包括以下两方面:
A.根据表2,在基因组层面设计针对BRAF、cKIT、EGFR、EGFR、EGFR、EGFR、KRAS、KRAS、NRAS、PI3KCA、PI3KCA基因突变位点的捕获探针。
B.根据表2,在转录组数据库层面设计检测EML4-ALK、CCDC6-RET、SLC34A2-ROS1、TPM3-NTRK1、MET、ETV6-NTRK3、CD74-ROS1基因融合的探针。
3.1文库准备
3.1.1预先从-20℃保存的捕获试剂盒(艾吉泰康生产的
3.1.2按照如下要求配制反应体系,将文库与杂交block混匀,标记为B管。
3.1.3将Hyb Buffer置于室温融化,融解之后有沉淀出现,混匀后置于65℃恒温孵育内预热,完全溶解后取20μL Hyb Buffer置于200μL PCR管中,置于65℃恒温混匀仪孵育待用,标记为A管。
3.1.4取5μL RNase Block与2μL Probe置于200μL PCR管内,轻轻吸打混匀,短暂离心后置于冰上待用,标记为C管。
3.2探针杂交
3.2.1将B管放入真空浓缩仪,打开PCR管盖,启动离心机,打开真空泵,开始浓缩。
3.2.2将B管浓缩至体积小于10μL,之后用无菌水补足至10μL,轻轻混匀,短暂离心后置于冰上待用。
3.2.3设置PCR仪参数如下:Heat lid 105℃,95℃for 5min,65℃hold。
3.2.4将PCR管B管置于PCR仪上,运行以上程序;
3.2.5 PCR仪温度降至65℃,将A管置于PCR仪上孵育,盖上PCR仪热盖;
3.2.6 5min后,将C管置于PCR仪孵育,盖上PCR仪热盖。
3.2.7 2min后,把移液器调至13μL,从A管中吸取13μL Hyb buffer移至C管中,吸取全部B管中样品移至C管中,密封盖子,盖上热盖65℃过夜。
3.3捕获磁珠的平衡
3.3.1捕获磁珠(Dynabeads MyOne Streptavidin T1 magnetic beads)从4℃取出,涡旋震荡重悬。
3.3.2取50μL磁珠置于新的PCR管中,置于磁力加上1min使溶液澄清,移除上清。
3.3.3从磁力架上取下PCR管,加入200μL Binding buffer轻轻吸打数次混匀,重悬磁珠。
3.3.4置磁力架上1min,移除上清。
3.3.5重复步骤3~4两次,共清洗磁珠3次。
3.3.6从磁力架上取下PCR管,加入200μL Binding buffer轻轻吸打6次重悬磁珠待用。
3.4捕获目标区域文库
3.4.1保持杂交产物在PCR仪上,将第6步重悬后的200μL Cap beads加入到杂交产物中,用移液器吸打6次混匀,置于旋转混合仪上室温结合30min。
3.4.2将PCR管置于磁力架上2min使溶液澄清,移除上清。
3.4.3向杂交产物中加入200μL的Wash buffer 1,轻轻吸打6次混匀,置于旋转混合仪上清洗15min,然后短暂离心,将PCR管放于磁力架上2min,使溶液澄清,移除上清。
3.4.4加入200μL的65℃预热的Wash buffer 2,轻轻吸打6次混匀,置于恒温混匀仪上65℃孵育10min,800转/min进行清洗。
3.4.5短暂离心,将PCR管放于磁力架上2min,移除上清。使用Wash buffer清洗两次,共计三次。最后一次彻底移除Wash buffer 2。
3.4.6保持样品在磁力架上,向PCR管内加入200μL 80%乙醇,静置30s后彻底移除乙醇溶液,室温晾干。
3.4.7向PCR管中加入30μL Nuclease-free water,从磁力架上取下PCR管,轻轻吸打6次重悬磁珠待用。
3.5 Post-PCR
3.5.1按照下表配制扩增反应体系
3.5.2使用移液器轻轻吸打混匀6次,然后置于PCR仪上按下表进行反应
3.5.3 PCR结束后向样品中加入55μL Beckman公司的AMPure XP磁珠,用移液器轻轻吸打6次混匀。
3.5.4室温孵育5min,把PCR管置于磁力架上3min使溶液澄清。
3.5.5移除上清,PCR管继续置于磁力架上,加入200μL 80%无水乙醇,静置30s。
3.5.6移除上清,再向PCR管内加入200μL 80%无水乙醇,静置30s后彻底移除上清。
3.5.7室温放置5min,使得残留乙醇彻底挥发。
3.5.8加入25μL无菌水,将PCR管从磁力架拿下,轻轻吹打混匀重悬磁珠,室温放置2min。
3.5.9将PCR管置于磁力架2min。
3.5.10用移液器吸23μL上清液转移到1.5mL离心管,标记样品信息。
3.5.11使用Qubit进行文库产量定量,并用Agilent 2100核酸分析仪检测文库大小,满足大小大约在350bp左右。
3.5.12通过qPCR技术来检测文库中管家基因GAPDH的含量,鉴定DNA和RNA核酸物质是否都被建成文库,具体方法参见实施例1。
4.高通量测序
将质检好的文库送至天津诺禾致源进行高通量测序,下机数据比对参考基因组hg19,分别使用gatk软件和tophat2、cufflinks软件分析基因组单碱基突变和转录组基因表达量的分析。具体数据如表3、表4、表5所示:
分析比对结果,突变碱基的检出率和融合基因表达的检出大致与标准品预计的频率相当,除了个别位点相差较大,可能与建库和探针捕获都有一定关系,后续可以先对相关检测位点的探针数量再进行优化。总体而言,本发明公开的DNA/RNA共建库、共测序的方法有望用于肿瘤样本的检测。
表3.下机数据比对情况
表4.检测的碱基突变频率
表5.检测到的融合基因的表达量
以上详细描述了本发明的较佳具体实施例。应当理解,本领域的普通技术人员无需创造性劳动就可以根据本发明的构思作出诸多修改和变化。因此,凡本技术领域中技术人员依本发明的构思在现有技术的基础上通过逻辑分析、推理或者有限的实验可以得到的技术方案,皆应在由权利要求书所确定的保护范围内。
序列表
<110> 翌圣生物科技(上海)有限公司
<120> 一种基于转座酶的DNA/RNA共建库方法、试剂盒及应用
<160> 4
<170> SIPOSequenceListing 1.0
<210> 1
<211> 21
<212> DNA
<213> 人工序列(Artificial Sequence)
<400> 1
cccttcattg acctcaacta c 21
<210> 2
<211> 22
<212> DNA
<213> 人工序列(Artificial Sequence)
<400> 2
ctcagccttg acggtgccat gg 22
<210> 3
<211> 22
<212> DNA
<213> 人工序列(Artificial Sequence)
<220>
<221> misc_binding
<222> (1)..(1)
<223> TAMRA
<220>
<221> misc_binding
<222> (22)..(22)
<223> MGB
<400> 3
aaagctggtg tgggaggagc ca 22
<210> 4
<211> 22
<212> DNA
<213> 人工序列(Artificial Sequence)
<220>
<221> misc_binding
<222> (1)..(1)
<223> FAM
<220>
<221> misc_binding
<222> (22)..(22)
<223> MGB
<400> 4
tttacatgtt ccaatatgat tc 22