使用氘化右美沙芬和奎尼丁治疗精神分裂症的阴性症状的方法
文献发布时间:2023-06-19 13:46:35
本申请要求2019年3月18日提交的美国临时专利申请第62/820,142号的优先权,所述临时专利申请以引用方式整体并入本文。
本公开涉及精神分裂症的阴性症状的治疗。本公开提供通过施用有效量的氢溴酸氘化[d6]-右美沙芬和硫酸奎尼丁来治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法。
精神分裂症是严重的精神病症,在世界人口中的发病率为约1%并且是功能障碍的主要起因(Switaj等人BMC Psychiatry.2012;12(1):193;World HealthOrganization.Mental Health:Schizophrenia.2012)。疾病通常在青春期或成年早期期间发作并且倾向于在男性中更早地开始。精神分裂症在儿童中很少见,但认识到在儿童期发作的精神分裂症正在增加(World Health Organization.Mental Health:Schizophrenia.2012)。
精神分裂症的症状通常描述为“阳性”或“阴性”。阳性症状包括妄想、思维过程中的干扰、幻觉、无条理性、敌对性和冲动行为。阴性症状包括动机和情绪表达方面的缺陷、情感迟钝、无意志、缺乏动机、情感淡漠和缺乏形成社会关系的欲望。阴性症状还可表达为语言和非语言交流方面的严重表达缺陷。这种交流技能方面的缺损可引起严重的功能性缺陷,会导致适应性亲社会行为减少、社交孤立和退缩(Del-Monte等人Psychia Res.2013;210:29-35;Adamczyk等人Schizophr Res.2016;176(2-3):331-339)。此外,罹患表达缺陷的患者可能远离社交互动并且变得越来越退缩或孤立,进一步增加其疾病的严重程度。交流是个体融入社会以形成并保持关系、上学、寻找和维持职业的能力的主要特征(Del-Monte等人Psychia Res.2013;210:29-35;Adamczyk等人Schizophr Res,2016;176(2-3):331-339)。相信交流缺陷是患者的阴性症状的核心特征以及长期不良结果的主要促成因素。
估计阴性症状影响20%至40%的患有精神分裂症的个体(Pai,Nitte Univ JHealth Sci.2015;5(2):104-115)。约60%的稳定的精神分裂症门诊患者具有至少一种阴性症状,并且41%的患者具有两种或更多种阴性症状(Bobes等人J ClinPsychiatry.2010;71(3):280-6)。阴性症状可影响精神分裂症的病程并且造成大部分患者的长期病态(Fervaha等人Eur Psychiatry.2014;29(7):449-55;Rabinowitz等人Schizophr Res.2012;137(1-3):147-150;Sicras-Mainar等人BMC Psychiatry.2014;14:225)。具有更明显的精神分裂症阴性症状的患者持续展示更坏的功能结果(Alphs等人Schizophr Bull.2006;32(2):225-230;Barnes等人Health Technol Assess.2016;20(29);Blanchard等人Schizophr Res.2005;77(2-3):151-165;Milev等人Am JPsychiatry.2005;162(3):495-506;Ho等人Am J Psychiatry.1998;155(9):1196-1201)。与阳性症状相比,阴性症状还与更大的生活质量下降和更大的家庭/照护者负担相关联(Gonfrier等人J Nutr Health Aging.2012;16(2):134-137;Kirkpatrick和Fischer,Schizophr Bull.2006;32(2):246-249;Rofail等人Qual Life Res.2016;25(1):201-211;Velligan等人Schizophr Res.1997;25(1):21-31;Mantovani等人Trends PsychiatryPsychother.2016;38(2):96-99)。因此,精神分裂症的阴性症状代表疾病管理的重要组成部分和药理学治疗的临床重要目标(Laughren和Levin,Schizophr Bull.2006;32(2):220-222;Marder等人Schizophren Bull.2011;37(2):250-254;Foussias,SchizophrBull.2010;36(2):359-369;Strauss等人J Psychiatr Res.2013;47(6):783-790)。降低这些表达和交流缺陷的严重程度的治疗具有根本上改善患者的功能能力和生活质量的潜力。
国际中枢神经系统临床试验和方法协会(International Society for CentralNervous System Clinical Trials and Methodology;ISCTM)研讨小组考虑使用阳性和阴性综合征量表(Positive and Negative Syndrome Scale;PANSS,例如阴性因子评分)和阴性症状评估-16(Symptom Assessment-16;NSA-16)量表的分量表作为阴性症状的可靠和有效量度,其中这些阴性症状的严重程度反映患者的功能缺损(Marder等人SchizophrenBull.2011;37(2):250-254;Daniel等人Clin Schizophr Relat Psychoses.2011;5(2):87-94;Velligan等人Psychiatry Res.2009;169(2):97-100)。此外,2009 ISCTM共识声明表达与原始PANSS阴性分量表相比,对PANSS阴性因子(例如PANSS Marder阴性因子)的偏好,因为原始分量表包括认为不属于公认的精神分裂症阴性症状领域的N5抽象思维困难和N7刻板思维(Laughren和Levin,Schizophr Bull.2006;32(2):220-222)。
用于治疗精神分裂症的阴性症状的多种介入已成为研究目标,包括使用右美沙芬的药理学策略(Remington等人Curr Treat Options Psychiatry.2016;3:133-150;Veerman等人Drugs.2017;77(13):1423-1459;Lee等人J Psychiatr Res.2015;69:50-56)。“当前所研究的策略的阵列和多样性突出缺乏基于证据的治疗和我们对于与病因学和病理生理学有关的阴性症状的理解的局限性”(Remington等人Curr Treat OptionsPsychiatry.2016;3:133-150,134)。“当前不存在足够的支持用于阴性症状的特定治疗的证据”,同上,144。“尽管如此,其中阴性症状是所鉴别的主要结果的主题以及研究仍显著增加”,同上。当前不存在美国食品和药物管理局(U.S.Food and Drug Administration;FDA)批准的用于精神分裂症的阴性症状的治疗。
因此,仍存在未满足的对于用于治疗精神分裂症的阴性症状的安全且有效的药理学介入的需求。“毫无疑问,至少部分原因是有证据表明阴性症状在功能衰退中发挥了关键作用,而对阳性症状的适当控制并不一定能解决功能衰退这一问题”(Remington等人CurrTreat Options Psychiatry.2016;3:133-150,145)。对具有精神分裂症的阴性症状的患者的有效治疗可显著改善患者的精神健康、生活质量、照护者负担并且降低医疗成本。
在一些实施方案中,本公开提供使用氘化[d6]-右美沙芬或其盐与奎尼丁或其盐的组合来治疗患有精神分裂症的患者中的阴性症状的方法。在一些实施方案中,本公开提供使用氢溴酸氘化[d6]-右美沙芬(d6-DM)与硫酸奎尼丁(Q)的组合来治疗患有精神分裂症的患者中的阴性症状的方法。如本文所用,术语“d6-DM”意指氢溴酸氘化[d6]-右美沙芬。如本文所用,术语“Q”意指硫酸奎尼丁。
更具体而言,在一些实施方案中,本公开提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q。在一些实施方案中,d6-DM以27mg至54mg剂量每天施用两次并且Q以4mg至7.5mg剂量每天施用两次。在一些实施方案中,d6-DM以30mg至45mg剂量每天施用两次并且Q以4mg至6mg剂量每天施用两次。在一些实施方案中,d6-DM以34mg至42.63mg剂量每天施用两次并且Q以4.9mg剂量每天施用两次。在一些实施方案中,d6-DM以34mg剂量每天施用两次并且Q以4.9mg剂量每天施用两次。在一些实施方案中,d6-DM以42.63mg剂量每天施用两次并且Q以4.9mg剂量每天施用两次。
在一些实施方案中,本公开提供治疗患有精神分裂症并且具有临床上稳定的阳性症状的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q。
在一些实施方案中,本公开提供治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者在治疗之前4个月内未进行精神病住院治疗。
在一些实施方案中,本公开提供治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者在治疗之前6个月内未发生精神病院入院或急性加重。
在一些实施方案中,本公开提供治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者已被评估为在阳性和阴性综合征量表(PANSS)的妄想、幻觉和敌对性项目上的评分小于或等于4。在一些实施方案中,患者已被评估为在PANSS的情感迟钝(N1)、情绪退缩(N2)、被动/淡漠社交退缩(N4)和交谈缺乏自发性/流畅性(N6)项目中的任两项上的评分大于或等于4或者任一项上的评分大于或等于5。在一些实施方案中,患者已被评估为具有大于或等于18的PANSS阴性分量表总分(N1至N7)。
在一些实施方案中,本公开提供治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者已被评估为在阳性和阴性综合征量表(PANSS)的妄想、幻觉、猜疑/被害和敌对性项目上的评分小于或等于4。在一些实施方案中,患者已被评估为在PANSS的情感迟钝(N1)、情绪退缩(N2)、被动/淡漠社交退缩(N4)和交谈缺乏自发性/流畅性(N6)项目中的任两项上的评分大于或等于4或者任一项上的评分大于或等于5。在一些实施方案中,患者已被评估为具有大于或等于20的PANSS Marder阴性因子总分(N1:情感迟钝;N2:情绪退缩;N3:交流障碍;N4:被动/淡漠社交退缩;N6:交谈缺乏自发性/流畅性;G7:行动迟缓;和G16:主动回避社交)。
在一些实施方案中,本公开提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者正用非典型抗精神病药治疗。在一些实施方案中,患者已用非典型抗精神病药治疗至少3个月,非典型抗精神病药的剂量已保持稳定至少1个月。在一些实施方案中,患者在用d6-DM和Q治疗之前的4个月内未进行精神病住院治疗。
在一些实施方案中,本公开提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者正用抗抑郁剂治疗。在一些实施方案中,患者已用抗抑郁剂治疗至少3个月并且在用d6-DM和Q治疗之前,抗抑郁剂的剂量已保持稳定至少1个月。
在一些实施方案中,本公开提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者正用安眠药治疗。在一些实施方案中,在用d6-DM和Q治疗之前,安眠药的剂量已保持稳定至少1个月。
在一些实施方案中,本公开提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者正用多达每天2mg的总剂量的劳拉西泮(lorazepam)治疗失眠、焦虑症、坐立不安或躁动。在一些实施方案中,在用d6-DM和Q治疗之前,劳拉西泮的剂量已保持稳定至少1个月。
在一些实施方案中,本公开提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者未正用一种或多种单胺氧化酶抑制剂(MAOI)治疗。
在一些实施方案中,本公开提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者未正用氯氮平(clozapine)治疗。
在一些实施方案中,本公开提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者未正用除劳拉西泮以外的苯二氮
在一些实施方案中,本公开提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者未正用左旋多巴(levodopa)治疗。
在一些实施方案中,本公开提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者未正用典型抗精神病药治疗。
在一些实施方案中,本公开提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者未正用以下剂治疗:
(a)提高奎尼丁的血浆水平的剂;
(b)通过CYP2D6代谢的剂;
(c)与奎尼丁有关的剂;
(d)当与右美沙芬共同施用时产生血清素综合征的剂;
(e)降低右美沙芬和奎尼丁的血浆水平的剂;
(f)为氯氮平的剂;或
(g)为典型抗精神病药的剂。
在一些实施方案中,本公开提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者未正用抗胆碱能药物治疗。
在一些实施方案中,本公开提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者在治疗之前一年内未接受电痉挛治疗、重复经颅磁刺激或深部脑刺激。
在一些实施方案中,本公开提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者未患有重症肌无力。
在一些实施方案中,本公开提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者未患有分裂情感性障碍。
在一些实施方案中,本公开提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者未患有双相障碍。
在一些实施方案中,本公开提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者未患有抑郁障碍并且/或者不具有大于或等于6的卡尔加里精神分裂症抑郁量表(CalgaryDepression Scale for Schizophrenia;CDSS)评分。
在一些实施方案中,本公开提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者在辛普森-安格斯量表(Simpson-Angus Scale;SAS)的以下八个项目的总和上的评分不大于3:步态、落臂、摇肩、肘强直、腕强直、腿摆动、头部旋转和眉间轻敲。
在一些实施方案中,本公开提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者未患有并发性的临床上显著或不稳定的全身性疾病、神经病症、认知障碍、神经退化性病症、肝脏病症、肾脏病症、代谢病症、血液病症、免疫病症、心血管病症、肺部病或胃肠道病症,如由处方医生确定。
在一些实施方案中,本公开提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者没有自杀风险。在一些实施方案中,通过以下中的一者或多者确定自杀风险:
(a)处方医生的判断;
(b)患者在哥伦比亚自杀严重程度评级量表(Columbia Suicide SeverityRating Scale)(C-SSRS)自杀意念项目4(有某些行动意图但无具体计划的主动自杀意念)上回答是并且患者的最近一次符合此C-SSRS项目4的发作在六个月内发生;
(c)患者在C-SSRS自杀行为项目5(有具体计划和意图的主动自杀意念)上回答是并且患者的最近一次符合此C-SSRS项目5的发作在六个月内发生;以及
(d)患者在5个C-SSRS自杀行为项目(主动尝试、被中断的尝试、放弃的尝试、预备的行动或行为)中的任一项上回答是并且患者的最近一次符合这些C-SSRS项目中的任一项的发作在治疗之前的两年内发生。举例来说,在一些此类实施方案中,通过所有(a)、(b)、(c)和(d)确定自杀风险。举例来说,在一些此类实施方案中,通过(a)、(b)和(c)确定自杀风险。举例来说,在一些此类实施方案中,通过(a)和(b)确定自杀风险。举例来说,在一些此类实施方案中,通过(a)、(b)、(c)或(d)中的任一者确定自杀风险。
在一些实施方案中,本公开提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者没有以下任一者或多者的心血管病史:
(a)完全性心脏传导阻滞、心室性心动过速、如由中央读取器评估的临床上显著的心室性早期收缩(PVC)的存在、QTc延长或尖端扭转型心室心动过速的病史或证据;
(b)基于中央审查,使用弗氏公式(Fridericia′s formula)的QTc(QTcF)对于男性而言大于450msec并且对于女性而言大于470msec,除非归因于心室起搏;
(c)先天性QT间期延长综合征的家族病史;以及
(d)临床上显著的晕厥、直立性低血压或体位性心动过速的病史或存在。举例来说,在一些此类实施方案中,患者没有(a)、(b)、(c)和(d)中的全部的病史。举例来说,在一些此类实施方案中,患者没有(a)、(b)和(c)的病史。举例来说,在一些此类实施方案中,患者没有(a)和(b)的病史。举例来说,在一些此类实施方案中,没有(a)、(b)、(c)或(d)中的任一者的病史。
在一些实施方案中,本公开提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者未患有继发于抗精神病药治疗的假性帕金森症(pseudoparkinsonism)。
在一些实施方案中,本公开提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者没有物质和/或酒精滥用史,但可使用烟草和/或烟碱产品。
在一些实施方案中,本公开提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者不使用休闲或药用大麻,如由大麻尿液药检呈阴性所证明。
在一些实施方案中,本公开提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者的乙型肝炎表面抗原、丙型肝炎抗体或HIV抗体的测试不呈阳性。
在一些实施方案中,本公开提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中在治疗的第一周期间,d6-DM以24mg剂量每天施用一次并且Q以4.9mg剂量每天施用一次;在治疗的第二周期间,d6-DM以24mg剂量每天施用两次并且Q以4.9mg剂量每天施用两次;并且在治疗的其余时间期间,d6-DM以34mg剂量每天施用两次并且Q以4.9mg剂量每天施用两次。
在一些实施方案中,本公开提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中在治疗的前三天期间,d6-DM以28mg剂量每天施用一次并且Q以4.9mg剂量每天施用一次;在治疗的接下来四天期间,d6-DM以28mg剂量每天施用两次并且Q以4.9mg剂量每天施用两次;并且在治疗的其余时间期间,d6-DM以42.63mg剂量每天施用两次并且Q以4.9mg剂量每天施用两次。
在本文所公开的方法的一些实施方案中,还向患者施用除氯氮平以外的非典型抗精神病药。
在本文所公开的方法的一些实施方案中,患者是年龄为18至60岁的男性或女性患者。
在本文所公开的方法的一些实施方案中,患者是具有生育潜力的女性。在一些实施方案中,患者:(a)尿液妊娠测试呈阴性;(b)在直至最后一次给药之后30天的治疗持续时间内不在哺乳或计划怀孕;并且(c)在治疗之前禁欲或愿意使用避孕方法并且继续同样的方法直至最后一次给药之后28天。
在本文所公开的方法的一些实施方案中,患者对右美沙芬、奎尼丁、鸦片药物、d6-DM、Q或它们的任何成分没有过敏反应。
在本文所公开的方法的一些实施方案中,患者对一种或多种药物没有过敏反应或超敏反应。
在本文所公开的方法的一些实施方案中,患者不具有一种或多种临床上显著的实验室异常、一种或多种临床关注的安全性值或大于正常值上限的两倍的天冬氨酸转氨酶(AST)或丙氨酸转氨酶(ALT)水平,如由处方医生所确定。
在本文所公开的方法的一些实施方案中,基于精神分裂症的精神病症诊断与统计手册(Diagnostic and Statistical Manual of MentalDisorders;DSM)标准,患者已被诊断为患有精神分裂症。在一些实施方案中,基于DSM标准的诊断已通过简明国际神经精神访谈(Mini International Neuropsychiatric Interview;M.I.N.I.)证实。
在本文所公开的方法的一些实施方案中,患者未患有分裂情感性障碍。在一些实施方案中,患者未患有双相障碍。
在本文所公开的方法的一些实施方案中,治疗导致PANSS Marder阴性因子评分与治疗之前的基线相比降低至少20%。在一些实施方案中,治疗导致PANSS Marder阴性因子评分与治疗之前的基线相比降低至少2分。
本文所公开的方法还可任选地包括施用d6-DM和Q以及其他治疗剂,诸如一种或多种可用于治疗精神分裂症的治疗剂。在一些实施方案中,d6-DM和Q可与除氯氮平以外的非典型抗精神病药(例如第二代非典型抗精神病药物(SGA))组合施用。
本文还提供d6-DM和Q的治疗用途。在一些实施方案中,本公开提供用于特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的d6-DM和Q。在一些实施方案中,本公开提供d6-DM和Q在特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状中的用途。在一些实施方案中,本公开提供d6-DM和Q在制造用于特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的药剂的方法中的用途。还提供可用于治疗精神分裂症的阴性症状的组合物。
在一些实施方案中,本公开提供用于治疗患有精神分裂症的患者中的精神分裂症的阴性症状的包含治疗有效量的d6-DM的药剂,所述药剂与治疗有效量的Q同时、单独或依序组合使用。
在一些实施方案中,本公开提供用于治疗患有精神分裂症的患者中的精神分裂症的阴性症状的治疗有效量的d6-DM,其特征在于d6-DM与治疗有效量的Q组合施用,其中两种药剂同时、单独或依序施用。
在一些实施方案中,本公开提供治疗有效量的d6-DM与治疗有效量的Q的组合,所述组合用于治疗患有精神分裂症的患者中的精神分裂症的阴性症状,其中两种药剂同时、单独或依序施用。
在一些实施方案中,本公开提供包含治疗有效量的d6-DM的药物组合物,所述药物组合物与治疗有效量的Q同时、单独或依序组合使用,以用于治疗患有精神分裂症的患者中的精神分裂症的阴性症状。
附图说明
图1示出氢溴酸氘化[d6]-右美沙芬(d6-DM)的化学结构。
图2示出来自实施例1的研究的研究设计和评估时间表。研究药物(活性剂或安慰剂)以上午1个胶囊和晚上1个胶囊的形式施用,间隔约12小时。M:上午剂量(mg d6-DM/mgQ);E:晚上剂量(mg d6-DM/mg Q);*:第4次访视(第43天)分级是基于治疗应答标准,随后进行再随机化(1∶1)。
图3示出来自实施例1的研究中调整意向治疗(mITT)群体中的患者的部署。
图4示出来自实施例1的研究中各次访视时的NSA-16总平均评分(顺序并行比较设计(SPCD),mITT群体)。
图5示出来自实施例1的研究中各次访视时的PANSS总平均评分(SPCD,mITT群体)。
图6示出来自实施例1的研究中各次访视时的PANSS阴性分量表平均评分(SPCD,mITT群体)。
图7示出来自实施例1的研究中各次访视时的PANSS Marder阴性因子平均评分(SPCD,mITT群体)。
图8示出来自实施例1的研究中各次访视时的PANSS亲社会因子平均评分(SPCD,mITT群体)。
图9示出来自实施例1的研究中各次访视时的NSA-16总体阴性症状评级(SPCD,mITT群体)。
图10示出“显著”或“极显著”改善的PGI-C评级:来自实施例1的研究中的第1阶段(基线至第6周)和第2阶段(第6周至第12周)。
具体实施方式
以下详细说明和实施例说明本公开的某些实施方案。本领域技术人员将认识到,存在涵盖于本公开的范围内的本公开的大量变化和修改。因此,不应认为某些实施方案的描述限制本公开的范围。
为了使本公开可更易于理解,在整个详细说明中定义某些术语。除非本文另外定义,否则结合本公开使用的所有科学和技术术语应具有与本领域普通技术人员通常所理解相同的含义。
本文所引用的所有参考文献,包括但不限于已公布和未公布的申请、专利和参考文献,都以引用方式整体并入本文并且在此成为本说明书的一部分。在所引用的参考文献与本文的公开内容相冲突的情况下,以本说明书为准。
如本文所用,除非上下文另外明确规定,否则词语的单数形式还包括复数形式;作为实例,术语“一个”、“一种”和“所述”应当理解为单数或复数。举例来说,“元件”意指一个或多个元件。除非特定上下文另外指示,否则术语“或”应意指“和/或”。
在一些实施方案中,本公开提供通过向患者施用氢溴酸氘化[d6]-右美沙芬(d6-DM)和硫酸奎尼丁(Q)来治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法。还提供d6-DM和Q例如在治疗精神分裂症的阴性症状中的用途。还公开包含d6-DM和Q的组合物(例如药物组合物)并且所述组合物可用于本文所述的治疗方法和用途。
如本文所用,术语“d6-DM”意指氢溴酸氘化[d6]-右美沙芬。如本文所用,术语“Q”意指硫酸奎尼丁。
如本文所用,术语“患有精神分裂症的患者”意指已被诊断为患有精神分裂症的患者。
“精神分裂症的阴性症状”包括以下中的一者或多者:情感迟钝、情绪退缩、交流障碍、被动/淡漠社交退缩、交谈缺乏自发性/流畅性、行动迟缓、主动回避社交、抽象思维困难、刻板思维、失语症、无社会性、快感缺乏和无意志。在一些实施方案中,精神分裂症的阴性症状是情感迟钝。在一些实施方案中,精神分裂症的阴性症状是情绪退缩。在一些实施方案中,精神分裂症的阴性症状是交流障碍。在一些实施方案中,精神分裂症的阴性症状是被动/淡漠社交退缩。在一些实施方案中,精神分裂症的阴性症状是交谈缺乏自发性/流畅性。在一些实施方案中,精神分裂症的阴性症状是行动迟缓。在一些实施方案中,精神分裂症的阴性症状是主动回避社交。在一些实施方案中,精神分裂症的阴性症状是抽象思维困难。在一些实施方案中,精神分裂症的阴性症状是刻板思维。在一些实施方案中,精神分裂症的阴性症状是失语症。在一些实施方案中,精神分裂症的阴性症状是无社会性。在一些实施方案中,精神分裂症的阴性症状是快感缺乏。在一些实施方案中,精神分裂症的阴性症状是无意志。
除阴性症状以外,患有精神分裂症的患者还可表现出一种或多种阳性症状。精神分裂症的示例性阳性症状包括妄想、概念紊乱、幻觉、兴奋、夸大、猜疑/被害和敌对性。
如本文所用,术语“治疗精神分裂症的阴性症状”意指改善精神分裂症的一种或多种阴性症状。
如本文所用,术语“治疗亲社会因子”意指改善一种或多种亲社会因子。
如本文所用,术语“特异性地治疗精神分裂症的阴性症状”意指改善精神分裂症的一种或多种阴性症状而与对以下一者或多者的作用无关:精神分裂症的任何阳性症状;抑郁症;和任何锥体外系症状。
术语“治疗有效量的氢溴酸氘化[d6]-右美沙芬(d6-DM)和硫酸奎尼丁(Q)”是指在组合施用时足以改善精神分裂症的一种或多种阴性症状的d6-DM的量和Q的量。如本文所用,应用于d6-DM和Q的术语“组合”意指包含d6-DM和Q的单一药物组合物(配制物),或两种独立的药物组合物(配制物),其各自包含待组合施用的d6-DM或Q。
如本文所用,“组合”施用或“共同施用”是指在一种组合物中同时施用d6-DM和Q,或在不同组合物中同时或依序施用。对于视为“组合”施用或“共同施用”的依序施用,d6-DM和Q以能够产生用于治疗患者中的精神分裂症的一种或多种阴性症状的有利作用的时间间隔分开施用。
术语“患者”和“受试者”意指人。在一些实施方案中,患者是患有精神分裂症的人。
在某些替代性实施方案中,除氢溴酸盐以外的氘化[d6]-右美沙芬的盐形式和除硫酸盐以外的奎尼丁的盐形式可用于本文所述的实施方案中。
除非另外说明,否则本文所述的剂量分别是指氘化[d6]-右美沙芬的氢溴酸盐形式和奎尼丁的硫酸盐形式(即,d6-DM和Q)。基于此类信息,本领域技术人员可计算活性成分的个别游离碱形式的相应剂量。本领域技术人员可计算右美沙芬的盐的分子量和右美沙芬的游离碱的分子量并且使用比率计算游离碱以及盐的合适的剂量。
治疗方法
在一些实施方案中,本公开提供治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的氢溴酸氘化[d6]-右美沙芬(d6-DM)和硫酸奎尼丁(Q)。
在一些实施方案中,本说明书提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中d6-DM以27mg至54mg剂量每天施用两次并且Q以4mg至7.5mg剂量每天施用两次。
在一些实施方案中,本说明书提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中d6-DM以30mg至45mg剂量每天施用两次并且Q以4mg至6mg剂量每天施用两次。
在一些实施方案中,本公开提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中d6-DM以34mg至42.63mg剂量每天施用两次并且Q以4.9mg剂量每天施用两次。在一些实施方案中,d6-DM以34mg剂量每天施用两次并且Q以4.9mg剂量每天施用两次。在一些实施方案中,d6-DM以42.63mg剂量每天施用两次并且Q以4.9mg剂量每天施用两次。
在一些实施方案中,本公开提供治疗患有精神分裂症并且具有临床上稳定的阳性症状的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q。
在一些实施方案中,本公开提供治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者在治疗之前4个月内未进行精神病住院治疗。
在一些实施方案中,本公开提供治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者在治疗之前6个月内未发生精神病院入院或急性加重。
在一些实施方案中,本公开提供治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者已被评估为在阳性和阴性综合征量表(PANSS)的妄想、幻觉和敌对性项目上的评分小于或等于4。在一些实施方案中,患者已被评估为在PANSS的情感迟钝(N1)、情绪退缩(N2)、被动/淡漠社交退缩(N4)和交谈缺乏自发性/流畅性(N6)项目中的任两项上的评分大于或等于4或者任一项上的评分大于或等于5。在一些实施方案中,患者已被评估为具有大于或等于18的PANSS阴性分量表总分(N1至N7)。
在一些实施方案中,本公开提供治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者已被评估为在阳性和阴性综合征量表(PANSS)的妄想、幻觉、猜疑/被害和敌对性项目上的评分小于或等于4。在一些实施方案中,患者已被评估为在PANSS的情感迟钝(N1)、情绪退缩(N2)、被动/淡漠社交退缩(N4)和交谈缺乏自发性/流畅性(N6)项目中的任两项上的评分大于或等于4或者任一项上的评分大于或等于5。在一些实施方案中,患者已被评估为具有大于或等于20的PANSS Marder阴性因子总分(N1:情感迟钝;N2:情绪退缩;N3:交流障碍;N4:被动/淡漠社交退缩;N6:交谈缺乏自发性/流畅性;G7:运动迟缓;和G16:主动回避社交)。
在一些实施方案中,本公开提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者正用非典型抗精神病药治疗,其中患者已使用非典型抗精神病药治疗至少3个月,非典型抗精神病药的剂量已保持稳定至少1个月。在一些实施方案中,患者在用d6-DM和Q治疗之前的4个月内未进行精神病住院治疗。
在一些实施方案中,本公开提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者正用抗抑郁剂治疗,其中患者已使用抗抑郁剂治疗至少3个月,并且在用d6-DM和Q治疗之前,抗抑郁剂的剂量已保持稳定至少1个月。
在一些实施方案中,本公开提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者正用安眠药治疗,其中在用d6-DM和Q治疗之前,安眠药的剂量已保持稳定至少1个月。
在一些实施方案中,本公开提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者正用多达每天2mg的总剂量的劳拉西泮治疗失眠、焦虑症、坐立不安或躁动,其中在用d6-DM和Q治疗之前,劳拉西泮的剂量已保持稳定至少1个月。
在本文所公开的方法的一些实施方案中,未在用d6-DM和Q治疗的同时用某些其他治疗剂治疗患者。在一些实施方案中,在开始用d6-DM和Q治疗之前,患者在2周或其他治疗剂的5个半衰期内(以较长者为准)未使用某些其他治疗剂。
在一些实施方案中,本公开提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者未正用一种或多种单胺氧化酶抑制剂(MAOI)治疗。示例性MAOI包括卡马西平(carbamazepine)、环丙孕酮(cyproterone)、贯叶金丝桃素(hyperforin)、奥卡西平(oxcarbazepine)、苯巴比妥(phenobarbital)、苯妥英(phenytoin)、利福平(rifampicin)和圣约翰草(St.John′sWort)。
在一些实施方案中,本公开提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者未正用氯氮平治疗。
在一些实施方案中,本公开提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者未正用除劳拉西泮以外的苯二氮
在一些实施方案中,本公开提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者未正用左旋多巴治疗。
在一些实施方案中,本公开提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者未正用典型抗精神病药治疗。示例性典型抗精神病药包括但不限于氟哌啶醇(haloperidol)、洛沙平(loxapine)、硫利达嗪(thioridazine)、吗茚酮(molindone)、胺砜噻吨(thiothixene)、氟非那嗪(fluphenazine)、美索哒嗪(mesoridazine)、三氟吡啦嗪(trifluoperazine)、奋乃静(perphenazine)和氯丙嗪(chlorpromazine)。
在一些实施方案中,本公开提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者未正用以下剂治疗:
(a)提高奎尼丁的血浆水平的剂;
(b)通过CYP2D6代谢的剂;
(c)与奎尼丁有关的剂;
(d)当与右美沙芬共同施用时产生血清素综合征的剂;
(e)降低右美沙芬和奎尼丁的血浆水平的剂;
(f)为氯氮平的剂;或
(g)为典型抗精神病药的剂。
在一些实施方案中,患者未正用可提高奎尼丁的血浆水平的剂治疗。可提高奎尼丁的血浆水平的示例性剂包括但不限于胺碘酮(amiodarone)、碳酸酐酶抑制剂、西咪替丁(cimetidine)、地尔硫卓(diltiazem)、伊曲康唑(itraconazole)、酮康唑(ketoconazole)、大环内酯类抗生素、蛋白酶抑制剂和伏立康唑(voriconazole)。大环内酯类抗生素的非限制性实例包括红霉素(erythromycin)、阿奇霉素(azithromycin)、克拉霉素(clarithromycin)、地红霉素(dirithromycin)和罗红霉素(roxithromycin)。蛋白酶抑制剂的非限制性实例包括沙奎那韦(saquinavir)、利托那韦(ritonavir)、阿扎那韦(atazanavir)和茚地那韦(indinavir)。
在一些实施方案中,患者未正用通过CYP2D6代谢并且在与奎尼丁共同施用时可具有提高的血浆水平的剂治疗。通过CYP2D6代谢并且在与奎尼丁共同施用时可具有提高的血浆水平的示例性剂包括但不限于右美沙芬(非处方或处方)、三环抗抑郁剂(TCA)和阿托西汀(atomoxetine)。TCA的非限制性实例包括丙咪嗪(imipramine)、地昔帕明(desipramine)、阿米替林(amitriptyline)和去甲替林(nortriptyline)。
在一些实施方案中,患者未正用与奎尼丁有关的剂治疗。与奎尼丁有关的示例性剂包括但不限于奎宁(quinine)和甲氟喹(mefloquine)。
在一些实施方案中,患者未正用在与右美沙芬共同施用时可产生血清素综合征的剂治疗。在与右美沙芬共同施用时可产生血清素综合征的示例性剂包括MAOI。MAOI的非限制性实例包括卡马西平、环丙孕酮、贯叶金丝桃素、奥卡西平、苯巴比妥、苯妥英、利福平和圣约翰草。
在一些实施方案中,本公开提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者未正用抗胆碱能药物治疗。
在一些实施方案中,本公开提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者在治疗之前一年内未接受电痉挛治疗、重复经颅磁刺激或深部脑刺激。
在一些实施方案中,本公开提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者未患有重症肌无力。
在一些实施方案中,本公开提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者未患有抑郁障碍并且/或者不具有大于或等于6的卡尔加里精神分裂症抑郁量表(CDSS)评分。
在一些实施方案中,本公开提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者在辛普森-安格斯量表(SAS)的八个项目的总和上的评分不大于3:步态、落臂、摇肩、肘强直、腕强直、腿摆动、头部旋转和眉间轻敲。
在一些实施方案中,本公开提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者未患有并发性的临床上显著或不稳定的全身性疾病、神经病症、认知障碍、神经退化性病症、肝脏病症、肾脏病症、代谢病症、血液病症、免疫病症、心血管病症、肺部病或胃肠道病症,如由处方医生确定。
在一些实施方案中,本公开提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者未患有分裂情感性障碍。
在一些实施方案中,本公开提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者未患有双相障碍。
在一些实施方案中,本公开提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者没有自杀风险。在一些实施方案中,通过以下中的一者或多者确定自杀风险:
(a)处方医生的判断;
(b)患者在哥伦比亚自杀严重程度评定量表(C-SSRS)自杀意念项目4(有某些行动意图但无具体计划的主动自杀意念)上回答是,并且患者的最近一次符合此C-SSRS项目4的发作在六个月内发生;
(c)患者在C-SSRS自杀行为项目5(有具体计划和意图的主动自杀意念)上回答是,并且患者的最近一次符合此C-SSRS项目5的发作在六个月内发生;以及
(d)患者在5个C-SSRS自杀行为项目(主动尝试、被中断的尝试、放弃的尝试、预备的行动或行为)中的任一项上回答是,并且患者的最近一次符合这些C-SSRS项目中的任一项的发作在治疗之前的两年内。举例来说,在一些此类实施方案中,通过所有(a)、(b)、(c)和(d)确定自杀风险。举例来说,在一些此类实施方案中,通过(a)、(b)和(c)确定自杀风险。举例来说,在一些此类实施方案中,通过(a)和(b)确定自杀风险。举例来说,在一些此类实施方案中,通过(a)、(b)、(c)或(d)中的任一者确定自杀风险。
在一些实施方案中,本公开提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者没有以下任一者或多者的心血管病史:
(a)完全性心脏传导阻滞、心室性心动过速、如由中央读取器评估的临床上显著的心室性早期收缩(PVC)的存在、QTc延长或尖端扭转型心室心动过速的病史或证据;
(b)基于中央审查,使用弗氏公式的QTc(QTcF)对于男性而言大于450msec并且对于女性而言大于470msec,除非归因于心室起搏;
(c)先天性QT间期延长综合征的家族病史;以及
(d)临床上显著的晕厥、直立性低血压或体位性心动过速的病史或存在。举例来说,在一些此类实施方案中,患者没有(a)、(b)、(c)和(d)中的全部的病史。举例来说,在一些此类实施方案中,患者没有(a)、(b)和(c)的病史。举例来说,在一些此类实施方案中,患者没有(a)和(b)的病史。举例来说,在一些此类实施方案中,没有(a)、(b)、(c)或(d)中的任一者的病史。
在一些实施方案中,本公开提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者未患有继发于抗精神病药治疗的假性帕金森症。
在一些实施方案中,本公开提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者没有物质和/或酒精滥用史,但可使用烟草和/或烟碱产品。
在一些实施方案中,本公开提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者不使用休闲或药用大麻,如由大麻尿液药检呈阴性所证明。
在一些实施方案中,本公开提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中患者的乙型肝炎表面抗原、丙型肝炎抗体或HIV抗体的测试不呈阳性。
在一些实施方案中,本公开提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中在治疗的第一周期间,d6-DM以24mg剂量每天施用一次并且Q以4.9mg剂量每天施用一次;在治疗的第二周期间,d6-DM以24mg剂量每天施用两次并且Q以4.9mg剂量每天施用两次;并且在治疗的其余时间期间,d6-DM以34mg剂量每天施用两次并且Q以4.9mg剂量每天施用两次。
在一些实施方案中,本公开提供特异性地治疗患有精神分裂症的患者中的精神分裂症的阴性症状的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q,其中在治疗的前三天期间,d6-DM以28mg剂量每天施用一次并且Q以4.9mg剂量每天施用一次;在治疗的接下来四天期间,d6-DM以28mg剂量每天施用两次并且Q以4.9mg剂量每天施用两次;并且在治疗的其余时间期间,d6-DM以42.63mg剂量每天施用两次并且Q以4.9mg剂量每天施用两次。
在一些实施方案中,本公开提供治疗患有精神分裂症的患者中的亲社会因子的方法,所述方法包括向患者施用治疗有效量的d6-DM和Q。亲社会因子包括以下PANSS因子:G16(主动回避社交);N2(情绪退缩);N4(被动/淡漠社交退缩);N7(刻板思维);P3(幻觉行为);和P6(猜疑/被害)。
在本文所公开的方法的一些实施方案中,基于精神分裂症的精神病症诊断与统计手册(DSM)标准,患者已被诊断为患有精神分裂症。在一些实施方案中,DSM标准是美国精神病协会(American Psychiatric Association)(2000)Diagnostic and StatisticalManual of Mental Disorders,第4版,修订版(DSM-IV-TR)中所阐述的标准,此类标准的公开内容以引用方式并入本文。在一些实施方案中,DSM标准是美国精神病协会(2013)Diagnostic and Statistical Manual of Mental Disorders,第5版(DSM-V)中所阐述的标准,此类标准的公开内容以引用方式并入本文。
在一些实施方案中,患者的基于DSM标准的精神分裂症的诊断已通过简明国际神经精神访谈(M.I.N.I.)证实。M.I.N.I.是用于精神障碍的简要的结构式诊断性访谈,包括DSM-IV和DSM-5中的访谈。在一些实施方案中,用于证实精神分裂症的诊断的M.I.N.I.是6.0版M.I.N.I.,基于DSM-IV-TR标准。在一些实施方案中,用于证实精神分裂症的诊断的M.I.N.I.是7.0.2版M.I.N.I.,基于DSM-V标准。
在本文所公开的方法的一些实施方案中,患者满足一条、超过一条或所有来自实施例1的研究的章节1.3.1中所述的示例性包含标准。在一些实施方案中,患者满足一条、超过一条或所有来自实施例2的研究的章节2.1中所述的示例性包含标准。
在本文所公开的方法的一些实施方案中,患者不具有来自实施例1的研究的章节1.3.2中所述的示例性排除标准中的一者或多者。在一些实施方案中,患者不具有来自实施例2的研究的章节2.2中所述的示例性排除标准中的一者或多者。
在本文所公开的方法的一些实施方案中,患者是年龄为18至60岁的男性或女性患者。
在本文所公开的方法的一些实施方案中,患者是具有生育潜力的女性。在一些实施方案中,患者:(a)尿液妊娠测试呈阴性;(b)在直至最后一次给药之后30天的治疗持续时间内不在哺乳或计划怀孕;并且(c)在治疗之前禁欲或愿意使用避孕方法并且继续同样的方法直至最后一次给药之后28天。
在本文所公开的方法的一些实施方案中,患者对右美沙芬、奎尼丁、鸦片药物、d6-DM、Q或它们的任何成分没有过敏反应。
在本文所公开的方法的一些实施方案中,患者对一种或多种药物没有过敏反应或超敏反应。
在本文所公开的方法的一些实施方案中,患者不具有一种或多种临床上显著的实验室异常、一种或多种临床关注的安全性值或大于正常值上限的两倍的天冬氨酸转氨酶(AST)或丙氨酸转氨酶(ALT)水平,如由处方医生所确定。
在本文所公开的方法的一些实施方案中,向患者施用d6-DM和Q,与患者的ALDH2基因型无关。
Lee等人(Psychiatr Res.2015;69:50-56)评估右美沙芬是用抗精神病药利培酮(risperidone)进行的治疗的附加物。在该研究中,Lee等人使用阳性和阴性综合征量表(PANSS)和阴性症状评估量表(SANS),同上,52评估患者。未报告在用利培酮和右美沙芬治疗的患者与用利培酮和安慰剂治疗的患者之间存在PANSS和SANS评分的显著差异,同上。
Lee等人(Psychiatr Res.2015;69:50)还评估具有ALDH2基因的ALDH2*2等位基因的13名患者的子群(7名施用利培酮和右美沙芬;6名施用利培酮和安慰剂),同上,52-53,包括表2。约50%的亚洲人群具有ALDH2等位基因,但在高加索人中较罕见,同上,54。未报告两个组(右美沙芬对比安慰剂)中存在PANSS评分(包括PANSS阴性症状分量表)的显著差异,但报告了SANS总分的显著差异,同上,52-53。根据Lee等人,这些结果表明ALDH2基因可能影响SANS总分的变化,同上,54。Lee等人还提及研究的若干局限性并且质疑所述结果是否适用于西方人群,同上。尚未知晓是否存在任何进一步评估具有ALDH2*2等位基因的此患者子群的后续研究。
在本文所描述和例示的研究中,未测定患者的ALDH2基因型。施用包含d6-DM和Q的组合物(d6-DM/Q)以治疗精神分裂症的阴性症状,与患者的ALDH2基因型无关(参见例如来自实施例1的研究;还参见来自实施例2的研究)。
在本文所公开的方法的一些实施方案中,向患者施用d6-DM和Q以及其他治疗剂,诸如一种或多种已知或经鉴别用于治疗精神分裂症的治疗剂。
在本文所公开的方法的一些实施方案中,还向患者施用除氯氮平以外的非典型抗精神病药。在一些实施方案中,根据非典型抗精神病药的用于治疗精神分裂症的美国药品说明书中的剂量指南来施用非典型抗精神病药。在一些实施方案中,非典型抗精神病药是口服和长效肌肉内可注射剂。在一些实施方案中,非典型抗精神病药是第二代非典型抗精神病药物(SGA)。示例性SGA包括但不限于奥氮平(olanzapine)、利培酮、帕潘立酮(paliperidone)、喹硫平(quetiapine)、阿立哌唑(aripiprazole)和鲁拉西酮(lurasidone)。在一些实施方案中,患者未正用超过一种SGA治疗。在一些实施方案中,除用于失眠的低剂量喹硫平(例如在夜晚至多50mg)以外,患者未正用超过一种SGA治疗。
在一些实施方案中,d6-DM以24mg、28mg、34mg或42.63mg剂量施用,例如每天一次或两次。在一些实施方案中,d6-DM以24mg剂量施用,例如每天一次或两次。在一些实施方案中,d6-DM以28mg剂量施用,例如每天一次或两次。在一些实施方案中,d6-DM以34mg剂量施用,例如每天一次或两次。在一些实施方案中,d6-DM以42.63mg剂量施用,例如每天一次或两次。在一些实施方案中,d6-DM以34mg剂量每天施用两次。在一些实施方案中,d6-DM以42.63mg剂量每天施用两次。
在一些实施方案中,Q以4.9mg剂量施用,例如每天一次或两次。
在一些实施方案中,d6-DM和Q以单位剂型施用或使用。在一些实施方案中,单位剂型包括24mg、28mg、34mg或42.63mg d6-DM和4.9mg Q。在一些实施方案中,单位剂型包括24mg d6-DM和4.9mg Q。在一些实施方案中,单位剂型包括28mg d6-DM和4.9mg Q。在一些实施方案中,单位剂型包括34mg d6-DM和4.9mg Q。在一些实施方案中,单位剂型包括42.63mgd6-DM和4.9mg Q。在一些实施方案中,d6-DM和Q的单位剂型呈片剂或胶囊形式。在一些实施方案中,d6-DM和Q的单位剂型呈胶囊形式。
在一些实施方案中,d6-DM和Q以组合剂量或单独剂量施用或使用。在一些实施方案中,单独剂量基本上同时施用。在一些实施方案中,每天一次、每天两次、每天三次、每天四次或更频繁地施用d6-DM和Q的组合剂量(或同时施用的单独剂量)。举例来说:在单个剂量中提供每天24mg d6-DM和4.9mg Q;在两个剂量中提供每天48mg d6-DM和9.8mg Q,每个剂量含有24mg d6-DM和4.9mg Q;在两个剂量中提供每天68mg d6-DM和9.8mg Q,每个剂量含有34mg d6-DM和4.9mg Q;在单个剂量中提供每天28mg d6-DM和4.9mg Q;在两个剂量中提供每天56mg d6-DM和9.8mg Q,每个剂量含有28mg d6-DM和4.9mg Q;或在两个剂量中提供每天85.26mg d6-DM和9.8mg Q,每个剂量含有42.63mg d6-DM和4.9mg Q。在一些实施方案中,取决于每天施用的剂量数目,可调整组合剂量中的d6-DM和Q的总量,以向患者提供合适的每日总剂量。
在一些实施方案中,在两个剂量中提供每天68mg d6-DM和9.8mg Q,每个剂量含有34mg d6-DM和4.9mg Q。在一些实施方案中,两个剂量以6、8、10、12、14或16小时间隔施用,在一些实施方案中,两个剂量以12小时间隔施用(例如上午和晚上)。
在一些实施方案中,在两个剂量中提供每天85.26mg d6-DM和9.8mg Q,每个剂量含有42.63mg d6-DM和4.9mg Q。在一些实施方案中,两个剂量以约6、约8、约10、约12、约14或约16小时间隔施用。在一些实施方案中,两个剂量以约12小时间隔施用(例如上午和晚上)。
在一些实施方案中,治疗以较低日剂量开始,例如每天24mg或28mg d6-DM与4.9mgQ的组合,并且增加至每天85.26mg d6-DM与9.8mg Q的组合。
举例来说,在一些实施方案中,在治疗的第一周期间,d6-DM以24mg剂量每天施用一次并且Q以4.9mg剂量每天施用一次;在治疗的第二周期间,d6-DM以24mg剂量每天施用两次并且Q以4.9mg剂量每天施用两次;并且在治疗的其余时间期间,d6-DM以34mg剂量每天施用两次并且Q以4.9mg剂量每天施用两次。
在一些实施方案中,在治疗的前三天期间,d6-DM以28mg剂量每天施用一次并且Q以4.9mg剂量每天施用一次;在治疗的接下来四天期间,d6-DM以28mg剂量每天施用两次并且Q以4.9mg剂量每天施用两次;并且在治疗的其余时间期间,d6-DM以42.63mg剂量每天施用两次并且Q以4.9mg剂量每天施用两次。
如本领域技术人员将显而易见,在一些情况下,可施用不属于这些所公开的剂量和范围内的剂量。此外应注意,普通技术临床医师或治疗医师将知晓如何和何时考虑到个体反应而中断、调整或终止疗法。
可采用口服施用向患者提供有效剂量的d6-DM与Q的组合,以用于治疗患有精神分裂症的患者中的精神分裂症的阴性症状。在一些实施方案中,配制物可含有d6-DM和Q与本领域技术人员已知的药学上可接受的载体或稀释剂的组合。在一些实施方案中,d6-DM和Q经口服施用。在一些实施方案中,d6-DM和Q以单位剂型口服施用。在一些实施方案中,d6-DM和Q的单位剂型呈胶囊形式。
d6-DM和Q可配制为一种或多种药物组合物中的活性成分。此类药物组合物还可含有药学上可接受的载体和任选的其他治疗成分。
药物组合物可以诸如散剂、胶囊、片剂、悬浮液、药囊、扁囊剂、溶液和酏剂形式制备。诸如淀粉、糖、微晶纤维素、稀释剂、造粒剂、润滑剂、结合剂、崩解剂等的载体可用于口服固体制剂中。在一些实施方案中,组合物以口服固体制剂(诸如散剂、胶囊和片剂)形式制备。在一些实施方案中,组合物以口服液体制剂形式制备。在一些实施方案中,口服固体制剂是胶囊或片剂。如果需要,胶囊或片剂可通过标准水性或非水性技术来包覆。
适于口服施用的药物组合物可以离散单元形式提供,诸如胶囊、扁囊剂、药囊、贴剂、片剂和气溶胶喷雾剂,各自含有预定量的活性成分,以粉末或颗粒形式,或以于水性液体、非水性液体、水包油乳液或油包水液体乳液中的溶液或悬浮液形式。此类组合物可通过任何常规药学方法制备,但大部分方法通常包括使活性成分与组成一种或多种成分的载体相关联的步骤。一般来讲,通过将活性成分与液体载体、细粉状固体载体或两者均匀并且紧密地掺合在一起,并且接着任选地使产物成形为所需呈现形式来制备组合物。
举例来说,片剂可通过压缩或模制,任选地与一种或多种其他成分一起制备。压缩片剂可通过在合适的机器中压缩呈自由流动形式的任选地与结合剂、润滑剂、惰性稀释剂、表面活性剂或分散剂混合的活性成分(诸如粉末或颗粒)来制备。模制片剂可通过使经惰性液体稀释剂湿润的粉末化合物的混合物在合适的机器中模制来制造。
在一些实施方案中,d6-DM和Q以胶囊形式共同施用。在一些实施方案中,包含d6-DM和Q的胶囊是直接释放型胶囊。在一些实施方案中,胶囊是硬明胶胶囊。在一些实施方案中,胶囊是3号尺寸。
在一些实施方案中,各胶囊含有24mg、28mg、34mg或42.63mg d6-DM和4.9mg Q。在一些实施方案中,各胶囊含有24mg d6-DM和4.9mg Q。在一些实施方案中,各胶囊含有28mgd6-DM和4.9mg Q。在一些实施方案中,各胶囊含有34mg d6-DM和4.9mg Q。在一些实施方案中,各胶囊含有42.63mgd6-DM和4.9mgQ。在一些实施方案中,各胶囊每天施用一次。在一些实施方案中,各胶囊每天施用两次。
在一些实施方案中,各胶囊含有34mg d6-DM和4.9mg Q并且每天施用两次。
在一些实施方案中,各胶囊含有42.63mg d6-DM和4.9mg Q并且每天施用两次。
在一些实施方案中,各胶囊(或其他包含d6-DM和Q作为活性成分的组合物)还含有非活性成分。在一些实施方案中,非活性成分可包括交联羧甲纤维素钠、微晶纤维素、胶态二氧化硅和/或硬脂酸镁。在一些实施方案中,非活性成分由交联羧甲纤维素钠、微晶纤维素、胶态二氧化硅和硬脂酸镁组成或包含交联羧甲纤维素钠、微晶纤维素、胶态二氧化硅和硬脂酸镁。
氢溴酸氘化右美沙芬
右美沙芬(DM)是(+)-3-甲氧基-N-甲基吗啡烷的通用名,其是一类作为吗啡样类鸦片的右旋类似物的分子中的一员。图1示出氢溴酸氘化[d6]-右美沙芬(d6-DM)的结构,它是其中氘置换图1中所示的位置处的6个氢原子的DM的氘化同位素。
在一些实施方案中,d6-DM是经分离或纯化的,例如d6-DM分别以d6-DM的同位素体的总量的按重量计至少50%(例如至少55%、60%、65%、70%、75%、80%、85%、90%、95%、97%、98%、98.5%、99%、99.5%或99.9%)的纯度存在。因此,在一些实施方案中,包含d6-DM的组合物可包括化合物的同位素体的分布,前提条件是至少50重量%的同位素体是d6-DM。
在一些实施方案中,d6-DM中任何指示为具有D的位置在d6-DM中的指定位置处具有至少80%、至少85%、至少87%、至少90%、至少95%、至少97%、至少99%或至少99.5%的最小氘并入。因此,在一些实施方案中,包含d6-DM的组合物可包括化合物的同位素体的分布,前提条件是至少80%的同位素体在一个或多个指定位置处包括D。
在一些实施方案中,d6-DM基本上不含化合物的其他同位素体,例如存在小于20%、小于10%、小于5%、小于2%、小于1%或小于0.5%的其他同位素体。
普通技术的合成化学家可容易地实现d6-DM的合成。相关程序和中间体公开于例如Kim等人(Bioorg Med Chem Lett 2001,11:1651)和Newman等人(J Med Chem 1992,35:4135)中。
根据一些实施方案的用于合成d6-DM的便利方法取代用于制备右美沙芬的合成方法中的合适的氘化中间体和试剂。这些方法描述于例如美国专利第7,973,049号中,其以引用方式整体并入本文。
其他用于合成d6-DM的方法属于本领域普通技术化学家的手段内。可用于合成d6-DM的合成化学转变和保护基方法(保护和去保护)是本领域已知的并且包括例如以下所述方法:Larock,Comprehensive Organic Transformations,VCH Publishers(1989);Greene等人Protective Groups in Organic Synthesis,第3版,John Wiley and Sons(1999);Fieser等人Fieser and Fieser′s Reagents for Organic Synthesis,John Wiley andSons(1994);和Paquette编,Encyclopedia of Reagents for Organic Synthesis,JohnWiley and Sons(1995);以及它们的后续版本。
硫酸奎尼丁
本公开设想使用硫酸奎尼丁(Q)与d6-DM的组合。在大部分人中(预计在美国包括约90%的一般人群),右美沙芬经历由CYP2D6催化的广泛肝O-脱甲基化成为右羟吗喃并且被身体快速清除(Ramachander等人J.Pharm.Sci.1977;66(7):1047-1048;Vetticaden等人Pharm.Res.1989;6(1):13-19)。然而,奎尼丁是强效CYP2D6抑制剂并且已特别研究此项用途(参见例如美国专利第5,206,248号)。奎尼丁的硫酸盐形式Q((C
奎尼丁施用可将具有广泛右美沙芬代谢表型的受试者转化为弱代谢表型(Inaba等人Br.J.Clin.Pharmacol.1986;22:199-200)。
示例性量表
在本文所公开的方法的一些实施方案中,可使用本文所述的一种或多种量表或本领域已知的其他量表。示例性量表包括阳性和阴性综合征量表(PANSS)、16项目型阴性症状评估(NSA-16)、临床总体印象(CGI)量表(例如临床总体印象-严重程度(CGI-S)、临床总体印象-变化(CGI-C))、患者总体印象-变化(PGI-C)、用于改善精神分裂症中的认知的测量和治疗研究(MATRICS)认知功能成套测验(MCCB)、卡尔加里精神分裂症抑郁量表(CDSS)和奖励与努力付出任务(EEfRT)。在一些实施方案中,使用所述量表中的一者或多者评估患者在治疗之前的基线状态。在一些实施方案中,使用所述量表中的一者或多者评估患者在治疗期间的多个点处的进程。在一些实施方案中,将患者在治疗期间的一个或多个点处的进程与患者在治疗之前的基线状态进行比较。
PANSS是经验证的临床量表,其已广泛用作精神分裂症的阴性和阳性症状的可靠和有效量度(Daniel,Schizophr Res.2013;150(2-3):343-345)。所述量表包含30个不同的项目,这些项目共同评估精神分裂症的阳性和阴性综合征,包括其彼此之间的关系以及与总体精神病理学的关系。各项目以“1”(无)至“7”(极重度)评分。
PANSS的现有心理测量特性使得能够评估阳性、阴性和一般精神病理学作为精神分裂症的类别或维度视角的一部分(Kay,Schizophr Bull.1987;13(2):261-276;Kumari等人J Addict Res Ther.2017;8(3))。因此,通常以因子结构形式分析项目的不同组合以对精神分裂症的阴性综合征的特定方面进行评分。
PANSS的五因子解决方案的概念已成功地用于临床试验中,并且鉴别精神分裂症的五个因子或维度:1)阴性症状;2)阳性症状;3)思维混乱;4)不受控的敌对性/兴奋;和5)焦虑症/抑郁症(Lindenmayer等人Psychopathology.1995;28(1):22-31;Marder等人JClin Psychiary.1997;58(12):538-546)。Marder在两项受控试验中研究五因子解决方案并且发现与氟哌啶醇相比,利培酮对所有五个维度产生显著更大的改善,其中与氟哌啶醇相比,利培酮对因子1(阴性症状)具有尤其强效的作用。因子1,即,PANSS Marder阴性因子,相对于阴性分量表具有若干个经改善的内容效度的方面,更一致地符合在2006 NIMH-MATRICS共识声明中鉴别的领域(Kirkpatrick等人Schizophr Bull.2006;32(2):214-219)。
PANNS Marder阴性因子评分
PANSS Marder阴性因子评分是精神分裂症的阴性症状的可靠且经验证的量度,并且包含30项目型PANSS中的以下7个项目:
Marder阴性因子:
·N1:情感迟钝
·N2:情绪退缩
·N3:交流障碍
·N4:被动/淡漠社交退缩
·N6:交谈缺乏自发性/流畅性
·G7:行动迟缓
·G16:主动回避社交
PANSS Marder阴性因子中的每一者与阴性症状的五个主要领域中的一者相关(Kirkpatrick等人Schizophr Bull.2006;32(2):214-219)。PANSS项目N1:情感迟钝,与情感迟钝相关;N6:交谈缺乏自发性/流畅性,与失语症相关;而N4:被动/淡漠社交退缩;G16:主动回避社交;和N3:交流障碍,是与无社会性相关的因子。PANSS项目N2:情绪退缩,与快感缺乏相关;并且G7:行动迟缓,与快感缺乏和无意志相关(Daniel,Schizophr Res.2013;150(2-3):343-345)。
PANSS Marder阴性因子评分中的两个项目(N4和G16)仅基于从信息提供者获得的信息。在一些实施方案中,患者鉴别可靠的信息提供者(例如病例管理人员、社会工作者、家庭成员),其与患者一起度过足够长的时间以能够向PANSS评级者提供信息。
在一些实施方案中,基于PANSS评分评估精神分裂症的一种或多种阴性症状。在一些实施方案中,仅测定PANSS评分。在一些实施方案中,测定PANSS评分和一种或多种其他量表(例如本文所述的示例性量表中的任一者或多者)。
认为NSA-16是与精神分裂症相关的阴性症状的存在、严重程度和范围的有效且可靠的量度;其跨越语言和文化具有高评级者间和测试-再测试可靠性(Daniel,SchizophrRes.2013;150(2-3):343-345;Axelrod等人J Psychiatr Res.1993;27(3):253-258)。NSA-16使用5因子模型描述阴性症状:(1)交流,(2)情绪/反应,(3)社会参与,(4)动机和(5)迟缓。由结构式访谈评估的这些因子是全面的并且经明确定义以帮助标准化评估。作为25项目型NSA的截短版本,NSA-16仍捕获阴性症状的多维性,但可在约15至20分钟内完成(Axelrod等人JPsychiatr Res.1993;27(3):253-258)。NSA-4(Alphs等人Int J MethodsPsychiatr Res.2011;20(2):e31-37)包含如下4个NSA-16项目:1)言语量受限,2)情绪:范围缩窄,3)社交动力下降,和4)兴趣减少,以及整体的总体阴性症状评级。
当定义为通常存在于健康年轻人中的行为消失或减少时,NSA总体阴性症状评分将阴性症状的整体严重程度评级。在一些实施方案中,评级不取决于来自NSA或任何其他类似工具的任何特定项目。反而是,在一些实施方案中,评级衡量评级者的访谈的完形并且在NSA-16访谈完成之后评估(Alphs等人Int J Methods Psychiatr Res.2011;20(2):e31-37)。
在一些实施方案中,使用NSA-16评估精神分裂症的一种或多种阴性症状。在一些实施方案中,NSA-16是单独使用的。在一些实施方案中,NSA-16与一种或多种其他量表(例如本文所述的示例性量表中的任一者或多者)组合使用。
研发CGI以在开始治疗之前和之后,提供临床医师对患者的总体功能的观察的简要、独立评估(Busner和Targum,Psychiatry(Edgmont).2007;4(7):28-37)。CGI提供全面的临床医师测定的概述性量度,其考虑所有可用的信息,包括患者病史、心理社会环境、症状、行为和症状对患者的功能能力的影响的知识。CGI包含2个并用1项目型量度,CGI-S(严重程度)和CGI-C(变化)。
CGI-S是7点量表,其需要临床医师在评估时对患者的疾病的严重程度进行评级,与临床医师对具有相同诊断的患者的过往经验有关(Guy,ECDEU Assessment Manual forPsychopharmacology.1976:76-338)。考虑全部临床经验,在评级时患者的精神疾病的严重程度被评估为1,正常,完全无疾病;2,处于疾病边缘;3,轻度疾病;4,中度疾病;5,明显疾病;6,重度疾病;或7,疾病程度最严重的患者。
CGI-C是7点量表,其需要临床医师在评估时对患者的疾患的变化进行评级,与临床医师对患者在入院时的疾患的过往经验有关。考虑全部临床经验,患者的精神疾病的变化被评估为1,极显著改善;2,显著改善;3,极小改善;4,无变化;5,极小恶化;6,显著恶化;或7,极显著恶化。
在一些实施方案中,使用CGI(例如CGI-S和/或CGI-C)评估精神分裂症的一种或多种阴性症状。在一些实施方案中,CGI(例如CGI-S和/或CGI-C)是单独使用的。在一些实施方案中,CGI(例如CGI-S和/或CGI-C)与一种或多种其他量表(例如本文所述的示例性量表中的任一者或多者)组合使用。
PGI-S是7点(1-7)、患者评级量表,其用于如下评估患者的精神分裂症的严重程度:1)正常,完全无疾病;2)处于疾病边缘;3)轻度疾病;4)中度疾病;5)明显疾病;6)重度疾病;7)极重度疾病。
在一些实施方案中,使用PGI-S评估精神分裂症的一种或多种阴性症状。在一些实施方案中,PGI-S是单独使用的。在一些实施方案中,PGI-S与一种或多种其他量表(例如本文所述的示例性量表中的任一者或多者)组合使用。
PGI-C是7点(1-7)、患者评级量表,其用于如下评估与患者的精神分裂症有关的治疗应答:极显著改善、显著改善、极小改善、无变化、极小恶化、显著恶化或极显著恶化。
在一些实施方案中,使用PGI-C评估精神分裂症的一种或多种阴性症状。在一些实施方案中,PGI-C是单独使用的。在一些实施方案中,PGI-C与一种或多种其他量表(例如本文所述的示例性量表中的任一者或多者)组合使用。
MCCB是用于评估精神分裂症中认知增强剂的试验中的认知变化的标准工具。MCCB(Nuechterlein等人Am J Psychiatry.2008;165(2):203-213)意欲提供与精神分裂症和相关病症相关的重要认知领域的相对简要评估。MCCB包括10项测试,其测量7个认知领域:处理速度、注意力/警惕性、工作记忆、语言学习、视觉学习、推理和问题解决以及社会认知。
在一些实施方案中,使用MCCB评估认知领域。在一些实施方案中,MCCB是单独使用的。在一些实施方案中,MCCB与一种或多种其他量表(例如本文所述的示例性量表中的任一者或多者)组合使用。
CDSS是来源于汉密尔顿抑郁量表(Hamilton Depression Scale;Ham-D)的9项目型量表,其被设计用于特异性地评估患有精神分裂症的患者中的抑郁症(Addington等人Schizophr Res.1996;19(2-3):205-12)。与Ham-D不同,CDSS不含与精神分裂症的阴性症状重叠的抑郁症状,诸如快感缺乏和社交退缩。CDSS展示极佳的心理测量特性。量表中的每个项目评分为0,无;1,轻度;2,中度;或3,重度。通过将每个项目评分相加来获得CDSS评分。对于预测存在重性抑郁发作,高于6的评分具有82%特异性和85%敏感性。
在一些实施方案中,使用CDSS评估抑郁症。在一些实施方案中,CDSS是单独使用的。在一些实施方案中,CDSS与一种或多种其他量表(例如本文所述的示例性量表中的任一者或多者)组合使用。
奖励与努力付出任务(EEfRT)(Treadway等人PLoS One.2009;4(8):e6598)是多试验计算机化任务,其中向患者提供在具有不同困难水平并且与不同水平的货币奖励相关的2项任务之间进行选择的机会。此任务检查响应于不同奖励方案以及为获得奖励而付出的努力(按压按钮)的机率学习。在EEfRT中操作机率,因为与努力动员类似,机率贴现似乎可很好地预测阴性症状。此外,包含机率操作可改善任务的整体生态效度,因为需要动机的大部分现实世界中的选择通常与结果的某种程度的不确定性相关联。EEfRT可靠地测量药物对付出与奖励量或奖励机率相关的努力的意愿的作用。举例来说,安非他命增加响应于低和中等机率奖励的努力(Wardle等人J Neurosci.2011;31(46):16597-16602)。尽管认为奖励显著性和行为反应与纹状体中的多巴胺释放有关,但还需要经由NMDA受体进行的对中脑多巴胺神经元的谷氨酸能输入以用于奖励调整(Stuber等人Science.2008;321(5896):1690-1692)。在一些实施方案中,使用困难任务选择与中等机率奖励的比率作为阴性症状的结果量度。
在一些实施方案中,使用EEfRT评估精神分裂症的一种或多种阴性症状。在一些实施方案中,EEfRT是单独使用的。在一些实施方案中,EEfRT与一种或多种其他量表(例如本文所述的示例性量表中的任一者或多者)组合使用。
在本文所公开的方法的一些实施方案中,使用NSA-16总分评估精神分裂症的一种或多种阴性症状。在一些实施方案中,使用以下任一者或多者评估精神分裂症的一种或多种阴性症状:PANSs总分;PANSS分量表(例如阳性、阴性、一般精神病理学、Marder阴性因子、兴奋成分和/或亲社会因子);NSA-16因子领域;NSA-16总体症状/功能评分;NSA-16个别项目评分;NSA-4评分;CGI-S评分;CGI-C评分;PGI-C评分;和EEfRT评分。在一些此类实施方案中,仅使用一个量表。在一些此类实施方案中,使用所有量表。在一些此类实施方案中,使用两种或更多种量表的组合。在一些实施方案中,使用MCCB综合评分评估认知。在一些实施方案中,使用CDSS评估抑郁症。在一些实施方案中,使用来自实施例1的研究中所描述的功效终点和/或量表中的任一者或多者评估精神分裂症的一种或多种阴性症状。
在本文所公开的方法的一些实施方案中,使用PANSS Marder阴性因子评分评估精神分裂症的一种或多种阴性症状。在一些实施方案中,使用NSA-16总体阴性症状评分评估精神分裂症的一种或多种阴性症状。在一些实施方案中,使用PGI-S评分评估精神分裂症的一种或多种阴性症状。在一些实施方案中,使用PGI-C评分评估精神分裂症的一种或多种阴性症状。在一些实施方案中,使用PANSS阳性分量表评估精神分裂症的一种或多种阴性症状并且使用CDSS评估抑郁症。在一些实施方案中,使用来自实施例2的研究中所描述的功效终点和/或量表中的任一者或多者评估精神分裂症的一种或多种阴性症状。
在本文所公开的方法的一些实施方案中,患者具有临床上稳定的阳性症状。在一些实施方案中,基于诸如精神病住院治疗、精神病院入院或急性加重和/或阳性和阴性综合征量表(PANSS)的某些方面中的特定评分,患者已被诊断为患有临床上稳定的阳性症状。
在一些实施方案中,PANSS阳性分量表(P1-P7)指示精神病症状的变化并且包括P1:妄想;P2:概念紊乱;P3:幻觉行为;P4:兴奋;P5:夸大;P6:猜疑/被害;和P7:敌对性。在一些实施方案中,基于PANSS阳性分量表,患者具有临床上稳定的阳性症状。
临床研究设计
在以下实施例1中的临床研究中,评估通过施用d6-DM和Q来治疗精神分裂症的阴性症状的益处。该研究包括安慰剂组和被施用d6-DM和Q的组。在此研究中具有安慰剂组是尤其具有信息性的,因为在精神障碍的研究中通常观测到高安慰剂应答(参见例如Fava等人“The Problem of the Placebo Response in Clinical Trials for PsychiatricDisorders:Culprits,Possible Remedies,and a Novel Study Design Approach,”Psychother Psychosom.2003;72(3):115-127,115-116)。“安慰剂应答表示随机指定安慰剂治疗的患者的临床疾患的明显改善......”,同上,116。因此,为避免患者中的改善是由药物的治疗益处提供还是归因于安慰剂应答的不确定性,涉及施用用于治疗精神障碍的药物的研究应包括安慰剂组。
另外,以下实施例1中的临床研究考虑某些可加重精神分裂症的阴性症状的其他因素,诸如阳性症状、抑郁症(通过卡尔加里精神分裂症抑郁量表评估)和锥体外系症状。在考虑这些其他因素之后,研究得出以下结论:药物治疗特异性地治疗精神分裂症的阴性症状,而非改善此类其他加重症状。参见例如Kirkpatrick等人,“The NIMH-MATRICSConsensus Statement on Negative Symptoms”,Schizophrenia Bulletin,32(2):214-219(2006)(Kirkpatrick);Arango等人,“Pharmacological approaches to treatingnegative symptoms:A review of clinical trials”,Schizophrenia Research,150(2-3):346-352(2013)。将在不清楚药物是否对阴性症状具有直接作用或还是通过改善加重症状而间接影响阴性症状时产生的不确定性称为“假特异性问题”。Kirkpatrick,同上,216,“临床上稳定的患者(其精神病症状已经治疗达到常见临床标准并且不显著变化)将实现明确说明”。
在以下实施例1中的临床研究中,患者具有低基线PANSS阳性评分并且在研究期间几乎不显示变化。另外,在以下实施例1中的研究中,患者具有低抑郁症基线症状(通过卡尔加里精神分裂症抑郁量表评估)和锥体外系症状,并且在整个研究期间,这些症状未发生显著变化。此类结果支持施用d6-DM和Q可特异性地治疗精神分裂症的阴性症状的结论。
以下实施例提供本公开的说明性实施方案。本领域普通技术人员将认识到,可在不改变本公开的精神或范围的情况下进行大量修改和变化。此类修改和变化涵盖于本公开的范围内。所提供的实施例不以任何方式限制本公开。
实施例
多中心、随机化、双盲、安慰剂对照、用于评估d6-DM/Q(氢溴酸氘化[d6]-右美沙芬[d6-DM]/硫酸奎尼丁[Q])作为用于患有精神分裂症的患者的辅助治疗的功效、安全性和耐受性的研究
为评估d6-DM/Q在用作具有精神分裂症的阴性症状的患者中的辅助疗法时的功效、安全性和耐受性,进行随机化、安慰剂对照、顺序平行比较设计(SPCD)研究。
所研究的患者群体具有多种精神分裂症的阴性症状并且研究的主要功效终点是16项目型阴性症状评估(NSA-16)总分,这是一种经验证的且广泛使用的精神分裂症的阴性症状的量度(Daniel,Schizophr Res.2013;150(2-3):343-5;Axelrod等人J PsychiatrRes.1993;27(3):253-8)。所研究的患者群体具有临床上稳定的用背景第二代非典型抗精神病药物治疗的阳性症状。以34mg d6-DM/4.9mg Q(d6-DM/Q-34/4.9)每天两次(BID)的剂量测试d6-DM/Q。
1研究性计划
1.1整体研究设计
此为2期、多中心、随机化、双盲、安慰剂对照、SPCD研究,其由至多4周的筛选期、具有2个连续阶段的12周双盲治疗期(第1阶段和第2阶段)以及5天电话随访组成。每位患者参与研究的持续时间为约12周,其中最大值为17周,包括筛选阶段和研究结束后的5天电话访问。
在诊断患有精神分裂症的美国患者中,约120名患者计划在15个中心参与研究,临床上稳定、处于疾病的残留(非急性)阶段并且符合所有包含标准并且不符合所有排除标准的患者符合入选条件。在第1阶段中,患者以1∶2(活性剂∶安慰剂)比率随机分配成接受d6-DM/Q或匹配安慰剂胶囊。随机分配至d6-DM/Q组的患者在第1天开始每天一次(QD)接受d6-DM 24mg/Q 4.9mg(d6-DM/Q-24/4.9)保持第一个7天。在第8天(达到d6-DM/Q-24/4.9BID)和第14天(达到d6-DM/Q-34/4.9BID)进行预定剂量递增,而随机分配至安慰剂组的患者在第1阶段中每天两次接受安慰剂。
完成第1阶段的患者符合参与第2阶段的条件。在第1阶段中接受d6-DM/Q的患者在第2阶段中继续接受d6-DM/Q 34/4.9BID,无进一步剂量递增。在第1阶段中接受安慰剂的患者在第2阶段基线(第4次访视[第43天])处分配至2个治疗应答子群(应答者与无应答者)中的1个中并且在每个子群内以1∶1(活性剂∶安慰剂)比率再随机分配。如果患者的阳性和阴性综合征量表(PANSS)总分的变化百分比相对于基线降低≥20%,则将其视为应答者,并且将不符合此标准的患者视为无应答者。从第1阶段中的安慰剂组再随机分配至第2阶段中的d6-DM/Q组的患者使用与第1阶段中所使用相同的剂量递增时间表在第2阶段中开始接受d6-DM/Q,而再随机分配至安慰剂组的患者在整个第2阶段持续时间内每天两次接受安慰剂。研究的示意图示于图2中。
在基线访视之前,患者参加至多4周(28天)的筛选访视以确定研究的适用性。在研究期间,患者在基线/第1次访视(第1天)、第2周/第2次访视(第15天)、第3周/第3次访视(第22天)、第6周/第4次访视(第43天)、第8周/第5次访视(第57天)、第9周/第6次访视(第64)天和第12周/第7次访视/提前终止访视(第85天)时参加临床访视,如表1中所概述。患者还在第8和50天接受电话随访访问以询问相关不良事件(AE)和研究药物依从性。此外,在第86至90天每天进行研究结束后的随访电话访问,以评估健康(即,AE)和药物(即,并用药物)的任何变化。
1.2研究设计的论述,包括对照组的选择
采用随机化、安慰剂对照、双盲研究设计以减少精神分裂症研究所固有的偏差源。此外,使用SPCD降低混淆安慰剂应答的影响,所述SPCD在第2阶段中根据经安慰剂治疗的患者在第1阶段结束时的应答状态将其分级。通常在行为和精神障碍的研究中观测到高安慰剂应答并且造成对这些适应症中的药物研发的显著挑战(Fava等人PsychotherPsychosom.2003;72(3):115-127;Chen等人Contemp Clin Trials.2011;32(4):592-604)。所选择的用于此研究的SPCD(Chen等人Contemp Clin Trials.2011;32(4):592-604)通过根据在第1阶段结束时的治疗应答将经安慰剂治疗的患者分级,以降低安慰剂应答对信号检测的不利影响来尝试克服这些挑战。因此,此设计基本上包含依序进行的两项随机化试验,预期信号检测将由在主要分析中仅包括安慰剂无应答者而得到增强。此设计保留比较在整个试验持续时间内使用药物或安慰剂的患者(内置式平行研究比较)的机会。
选择第1阶段和第2阶段具有6周持续时间,以确保在第1阶段中随机分配至d6-DM/Q组的患者暴露于所靶向的d6-DM/Q的最佳剂量达至少4周并且至多10周。此治疗持续时间还使得具有足够的时间以观测治疗应答(预期其在研究治疗之前几周内出现)以及评估应答的持续时间。包含来自国家心理卫生研究所(National Institute of Mental Health)、FDA、学院和行业的代表的共识组鉴别6至12周治疗持续时间适用于设计精神分裂症的阴性症状的研究(Laughren和Levin,Schizophr Bull.2006;32(2):220-222)。
此研究中使用的安全性评估是临床研究中的标准。用于评估功效的评级量表是公认的工具,其经临床验证并且已广泛用于精神分裂症以及其他精神与行为失常的临床研究中。主要功效分析基于由Chen等人评述并且在先前进行的使用SPCD的研究中使用的方法(Chen等人Contemp Clin Trials.2011;32(4):592-604)。整体作用的计算基于在第1阶段中观测到的和在第2阶段中观测到的仅安慰剂无应答者群体的作用的组合(Fava等人Psychother Psychosom.2003;72(3):115-127)。
1.3研究群体的选择
1.3.1包含标准
1.在签署知情同意书时年龄为18至60岁(包括端点)的男性和女性。
2.使用6.0版简明国际神经精神访谈(M.I.N.I.)(附录11A)符合精神分裂症的DSM-IV-TR诊断标准并且符合残余型精神分裂症的DSM-IV-TR诊断标准的患者。
3.患者在妄想、幻觉和敌对性的PANSS项目上的评分必须≤4。患者必须在PANSS的以下项目中的任2项中具有≥4(中度)或在任1项中具有≥5的PANSS评分:情感迟钝、情绪退缩、被动/淡漠社交退缩或交谈缺乏自发性/流畅性,并且在筛选和基线时的PANSS阴性分量表总分(N1至N7)≥18。
4.如果是具有生育潜力的女性,则患者必须
a.尿液妊娠测试呈阴性(所有女性都需提交妊娠测试,与生育潜力无关),并且
b.在直至最后一次给药访视之后的第30天的研究持续时间内未进行哺乳或计划怀孕,并且
c.从筛选访视开始禁欲或愿意使用可靠的避孕方法,并且继续同样的方法直至最后一次给药访视之后的第28天
符合研究要求的可靠的避孕方法是:
·子宫内装置
·切除输精管搭配物
·手术绝育(去除子宫和/或两个卵巢,和/或进行双侧输卵管结扎)
·激素避孕药(含有雌激素的避孕丸、阴道环、贴剂、注射剂或植入物)
·使用2种屏障避孕法(即,共同使用以下中的2者):男性保险套和阴道内杀精子剂、子宫帽和杀精子剂;子宫颈帽和杀精子剂
注意:对于此研究,小型丸剂(不含雌激素的微量给药孕酮制剂)并非可接受的避孕形式。
进行禁欲的具有生育潜力的女性可参与研究。
除非绝经后(即,绝经史[即,报告无月经≥12个月]并且无其他生物学/手术起因),否则女性视为具有生育潜力。
从筛选访视至最后一次给药访视、第7次访视/提前终止访视(第85天)之后的第28天,所有男性患者必须与具有生育潜力的配偶一起遵循相同避孕方法。
5.当前根据美国药品说明书的剂量指导接受非典型抗精神病药(口服和长效肌肉内可注射剂)(例如第二代抗精神病药[SGA],诸如奥氮平、利培酮、帕潘立酮、喹硫平、阿立哌唑和鲁拉西酮)以用于治疗精神分裂症病症的患者是符合条件的,前提条件是其已用所述药物治疗至少3个月(90天)、在筛选访视之前剂量保持稳定至少1个月(30天)(从筛选至基线/第1次访视[第1天]无变化)并且在筛选之前的过去4个月内未进行精神病住院治疗。
6.允许并用抗抑郁剂,诸如选择性血清素再吸收抑制剂(SSRI;例如氟西汀(fluoxetine)、舍曲林(sertraline)、西酞普兰(citalopram))、血清素-去甲肾上腺素再吸收抑制剂(SNRI;例如文拉法辛(venlafaxine)、去甲文拉法辛(desvenlafaxine)、度洛西汀(duloxetine)、沃托西汀(vortoxetine)、维拉唑酮(vilazodone)),只要患者使用优化剂量达3个月(90天),前提条件是在基线之前,剂量保持稳定至少1个月(30天)并且所使用的剂量在该药物的美国说明书的指导范围内。允许使用帕罗西汀(Paroxetine),一种CYP2D6底物,前提条件是剂量不超过10毫克/天。
7.允许在就寝时并用安眠药(例如右佐匹克隆(eszopiclone)、唑吡坦(zolpidem)、扎来普隆(zaleplon)、曲唑酮(trazodone)[至多100毫克/天])以用于失眠的夜间治疗,前提条件是在基线之前,剂量保持稳定至少1个月(30天)并且在整个研究期间保持稳定。在参与研究之前使用劳拉西泮治疗焦虑症、坐立不安或躁动的患者应在研究期间保持相同治疗方案。除用于失眠和行为障碍的短期或必要治疗的劳拉西泮以外,不允许使用任何其他苯二氮
8.根据研究者,在完全说明研究参与的性质和风险之后,有能力并且签署和接受患者知情同意书(ICF)的复本的患者。
9.患者必须具有研究者认为合适的可靠的信息提供者。
1.3.2排除标准
如果患者符合以下标准中的任一者,则其不应参与研究:
1.患有重症肌无力的患者。
2.在筛选访视时患有当前重性抑郁障碍和/或卡尔加里精神分裂症抑郁量表(CDSS)评分≥6的患者。
3.在筛选或基线时具有心血管问题的患者,诸如:
a.完全性心脏传导阻滞、心室性心动过速、如由中央读取器评估的临床上显著的心室性早期收缩的存在、QTc延长或尖端扭转型心室心动过速的病史或证据;
b.基于筛选访视时的中央审查,在筛选时使用弗氏公式的QTc(QTcF)对于男性而言>450msec并且对于女性而言>470msec,除非归因于心室起搏。
c.先天性QT间期延长综合征的任何家族病史。
d.临床上显著的晕厥、直立性低血压或体位性心动过速的病史或存在。
4.对DM、Q、鸦片药物(可待因(codeine)等)或研究药物的任何其他成分具有已知的超敏反应的患者。
5.具有对若干种药物的过敏反应或超敏反应的病史的患者。
6.在基线之前的3个月(90天)内接受与Q共同施用的DM的患者。
7.基于PI判断,患有继发于其正在使用的抗精神病药物的假性帕金森症的患者。
8.在基线之前的3个月(90天)内用任何典型抗精神病药物治疗的患者。在研究期间不允许使用典型抗精神病药。
9.在基线之前的3个月(90天)内具有氯氮平使用史的患者。在研究期间不允许使用氯氮平。
10.当前正在使用或在基线之前的1个月(30天)内使用抗胆碱能药物以用于治疗与抗精神病药物相关的AE的患者。
11.当前或在基线之前的2周内用单胺氧化酶抑制剂(MAOI)治疗的患者。
12.在基线之前的2周或5个半衰期内使用研究方案的禁止并用药物的患者。
13.患有可混淆研究的安全性结果的说明的并发性的临床上显著或不稳定的全身性疾病(例如恶性疾病[除皮肤基底细胞癌或未经治疗的前列腺癌以外]、控制不佳的糖尿病、控制不佳的高血压、肺不稳定、肾或肝疾病、不稳定的局部缺血性心脏疾病、扩张型心肌病或不稳定的心脏瓣膜病)、认知和其他神经退化性病症的患者。一些情况可能已由研究者和医学监测员个别地评估。
14.当前自杀风险,如由以下中的任一者证明:
a.研究者判断患者可能具有自杀风险。
b.患者在筛选和基线时,在哥伦比亚自杀严重程度评级量表(C-SSRS)的问题4或问题5中被评级为“是”,并且最近一次发作在筛选和基线之前的6个月内发生。
c.患者在筛选和基线之前的12个月内尝试自杀。
15.在筛选的4个月内进行精神病住院治疗的患者。
16.在筛选访视时,具有临床上显著的实验室异常(血液学、化学和尿分析)或者具有临床潜在关注的安全性值或者天冬氨酸转氨酶(AST)或丙氨酸转氨酶(ALT)>正常值上限的2倍的患者。
17.当前正在参与或在基线之前的30天内参与其他介入性(药物或装置)临床研究的患者。
18.以研究者的观点,不愿意或不能遵守研究指示。
19.在基线之前的6个月内具有物质和/或酒精滥用或在基线之前的1年内具有物质和/或酒精依赖性的历史的患者,不包括香烟(根据研究者判断,可能允许使用大麻)。
20.在筛选之前的一年内接受电痉挛治疗、重复经颅磁刺激或深部脑刺激的患者。
21.在CTS数据库中发现与在基线之前的30天内参与另一项介入性药物或装置研究的患者几乎确定匹配的患者。
1.3.3从疗法或评估去除患者
口头并且在ICF中告知患者,其具有在任何时间退出研究的权力而不会遭受偏见或损失其在其他方面获取的权益,并且无需提供原因。
研究者或赞助商可出于以下原因中的任一者而中止患者参与研究:
·在间发病、AE、其他与患者的健康或康乐有关的原因的情况下
·在缺乏合作、无依从性、违反协议或其他行政原因的情况下。
·在基线之后的任何时间,呈现QTcF>500msec(除非归因于心室起搏)或相比给药前基线ECG的QTcF变化>60msec的患者。记录QTcF值并且针对临床显著性进行评估。
·在研究期间开始禁止的并用药物、心理疗法或体细胞疗法(光疗法、重复经颅磁刺激和其他非侵袭性脑部刺激技术)的患者。结合医学监测员,基于个案分析来作出使患者退出的决定。
要求出于任何原因在研究完成之前退出的患者返回诊所以完成第7次访视(提前终止)评估。如果患者未返回进行预定访视,则尽最大努力联系患者。在任何情况下,在可能的情况下尽最大努力记录患者结果。
如果患者退出研究并且撤回公开其他信息的同意书,则不进行进一步评估并且不再收集数据。
不替换退出研究的患者。
1.4治疗
1.4.1所施用的治疗
以硬、蓝色不透明明胶胶囊(3号尺寸)形式提供临床研究药物。提供三种不同胶囊强度,如下:
·d6-DM/Q-24/4.9(d6-DM 24mg/Q 4.9mg)
·d6-DM/Q-34/4.9(d6-DM 34mg/Q 4.9mg)
·d6-DM/Q安慰剂,具有与研究药物相同的赋形剂
此研究中使用的所有药物根据良好生产规范(Good Manufacturing Practice)指南、ICH指南、GCP指南以及适用的法律和法规制备、封装和标记。
1.4.2研究产品的身份
以固体口服剂型(明胶胶囊)形式提供d6-DM/Q和匹配安慰剂。各研究产品的组成示于表2中。
表2.研究药物的组成
EP=欧洲药典(European Pharmacopoeia);NF=国家处方集(NationalFormulary);USP=美国药典(United States Pharmacopoeia)
研究药物以现成封装、盲式、预先标记、单个预先封装的泡壳包装卡(blistercard)形式提供。每个泡壳包装卡含有足以维持3周的研究药物,即,2种活性研究药物或安慰剂中的1者的48个胶囊。清楚地标记每个3周量的泡壳包装卡以鉴别上午和晚上剂量。
1.4.3将患者分配至治疗组的方法
在第1阶段基线时,将符合条件的患者以1∶2比率的d6-DM/Q或匹配安慰剂随机分配。在第1阶段基线时随机分配至安慰剂组的患者在第2阶段开始时,以1∶1比率再随机分配至d6-DM/Q和安慰剂组。由患者应答状态(应答者和无应答者)将再随机分配分级。应答者定义为PANSS总分自基线的变化≥20%(第1阶段)的患者。在第1阶段中提前退出的在第1阶段中被分配至安慰剂组的患者还以与另一安慰剂患者相同的方式随机分配第2阶段治疗,以用于统计分析目的;其应答状态基于其提前终止访视时的测量值。
双盲研究药物分配遵守随机化流程并且使用交互式应答技术(IRT)管理。由IRT进行分派,其根据需要将随机化区组动态分配至研究中心。在整个研究期间,入选是中央随机化的。
1.4.4选择研究中的剂量
依据d6-DM评估受体药理学的若干体外研究,假设此研究中所选择用于评估的剂量潜在地有效治疗精神分裂症。依据来自已完成的d6-DM/Q的1期研究的数据,还预期d6-DM/Q的剂量具有良好的安全性和耐受性概况。因此,选择此研究中使用的d6-DM/Q的剂量(d6-DM/Q-24/4.9和d6-DM/Q-34/4.9),以在此患者群体中提供最佳效益-风险比。
在可提高耐受性的假设下,使用固定滴定流程,进行递增至d6-DM/Q的较高剂量(d6-DM/Q-34/4.9)。
1.4.5每个患者的剂量选择和给药时序
除在访视当天,在有工作人员存在的情况下施用研究药物的上午剂量以外,患者约每12小时一次(上午和晚上)自行喝水口服施用研究药物。
在第1阶段中随机分配至d6-DM/Q组的患者,在第一周(第1至7天)期间上午使用d6-DM/Q-24/4.9QD并且晚上使用安慰剂,在下一周(第8至13天)使用d6-DM/Q-24/4.9BID并且在研究的其余10周(第14至85天)使用d6-DM/Q-34/4.9BID。
在第1阶段中随机分配至安慰剂组的患者,在第1阶段的6周持续时间(第1至42天)内使用安慰剂BID。那些再随机分配至安慰剂组的患者在第2阶段的6周持续时间(第43至85天)内继续使用安慰剂BID,并且那些再随机分配至d6-DM/Q组的患者使用与第1阶段中相同的剂量递增时间表来使用d6-DM/Q,即,在第2阶段的第一周(第43至50天)期间,上午使用d6-DM/Q-24/4.9QD并且晚上使用安慰剂,在下一周(第51至58天)使用d6-DM/Q-24/4.9BID,并且在第2阶段的其余4周(第59至85天)使用d6-DM/Q-34/4.9BID。
1.4.6盲式
所有研究药物(包括d6-DM/Q胶囊和安慰剂胶囊)具有相同外观以保持盲式的完整性,包括在剂量递增期间。赞助商、患者、研究者和其他研究人员都不知道患者的治疗分配。在变得在医学上必须鉴别患者接受的治疗的情况下,可打破盲式。在该情况下,研究者尽最大努力联系医学监测员或代表以申请患者的揭盲。IRT管理员无需盲化,并且其有权知晓研究药物列表和随机化代码。
1.4.7先前和并用疗法
1.4.7.1允许的并周药物
允许的并用药物由研究者评估并且根据需要由医学监测员论述以确定在研究期间使用是否会存在任何问题。
允许在就寝时并用安眠药(例如右佐匹克隆、唑吡坦、扎来普隆、曲唑酮[至多100毫克/天])以用于失眠的夜间治疗,前提条件是在基线之前,剂量保持稳定至少1个月并且在整个研究期间保持稳定。在参与研究之前使用劳拉西泮治疗焦虑症、坐立不安或躁动的患者在研究期间保持相同治疗方案。
除用于失眠和行为障碍的短期或必要治疗的劳拉西泮以外,不允许使用任何其他苯二氮
1.4.7.2禁止药物
在研究期间或在第1天开始给药之前的2周或5个半衰期(以较长者为准)内,患者不允许使用任何禁止药物。禁止药物的实例列举于表3中。在每次访视时,询问患者是否使用任何并用药物并且如果使用,则研究者记录所使用的药物及其使用原因。在筛选访视之后,根据需要使周镇静剂/安眠药或苯二氮
表3.禁止药物
这些是不允许的药物的实例而非全面列表。
CYP2D6=细胞色素P450同功酶2D6;DM=右美沙芬;MAOI=单胺氧化酶抑制剂;Q=硫酸奎尼丁;TCA=三环抗抑郁剂。
1.5功效和安全性变量
1.5.1所评估的功效和安全性测量值以及流程图
程序和评估的时间表呈现于表1中。
1.5.1.1功效量度
主要功效量度是16项目型NSA-16总分。次要量度包括来自PANSS、临床总体印象-严重程度(CGI-S)、临床总体印象-变化(CGI-C)、患者总体印象-变化(PGI-C)、MCCB、CDSS、EEfRT和戒烟问题的评分。用于测量功效的量表的描述提供于下文中。
1.5.1.1.1 16项目型阴性症状评估(NSA-16)
认为NSA-16(附录3A)是与精神分裂症相关的阴性症状的存在、严重程度和范围的有效且可靠的量度;其跨越语言和文化具有高评级者间和测试-再测试可靠性(Daniel,Schizophr Res.2013;150(2-3):343-345;Axelrod等人J Psychiatr Res.1993;27(3):253-258)。NSA-16使用5因子模型描述阴性症状:(1)交流,(2)情绪/反应,(3)社会参与,(4)动机和(5)迟缓。由结构式访谈评估的这些因子是全面的并且经明确定义以帮助标准化评估。作为25项目型NSA的截短版本,NSA-16仍捕获阴性症状的多维性,但可在约15至20分钟内完成(Axelrod等人J Psychiatr Res.1993;27(3):253-258)。NSA-4(Alphs等人Int JMethods Psychiatr Res.2011;20(2):e31-37)包含如下4个NSA-16项目:1)言语量受限,2)情绪:范围缩窄,3)社交动力下降,和4)兴趣减少,以及整体的总体阴性症状评级。
在筛选(第-28天至第-1天)、基线/第1次访视(第1天)、第3次访视(第22天)、第4次访视(第43天)、第6次访视(第64天)和第7次访视/提前终止访视(第85天)进行NSA-16评估。
1.5.1.1.2阳性和阴性综合征量表(PANSS)
PANSS(附录2)是30项目型临床量表,其已广泛用作阴性症状试验的可靠和有效量度(Daniel,Schizophr Res.2013;150(2-3):343-345)。各项目以“1”(不存在)至“7”(极重度)进行评分。PANSS的分量表包括:
·阳性分量表(P1-P7),
·阴性分量表(N1-N7),
·一般精神病理学分量表(G1-G16),
·亲社会因子(G16.主动回避社交,N2.情绪退缩,N4.被动/淡漠社交退缩,N7.刻板思维,P3.幻觉行为,P6.猜疑/被害),
·Marder阴性因子(N1.情感迟钝,N2.情绪退缩,N3.交流障碍,N4.被动/淡漠社交退缩,N6.交谈缺乏自发性/流畅性,G7.行动迟缓,G16.主动回避社交),
·兴奋成分(P4.兴奋,P7.敌对性,G4.紧张,G8.不合作,G14.冲动控制障碍)。
在筛选(第-28天至第-1天)、基线/第1次访视(第1天)、第3次访视(第22天)、第4次访视(第43天)、第6次访视(第64天)和第7次访视/提前终止访视(第85天)时进行PANSS评估。
1.5.1.1.3临床总体印象(CGI)量表
研发CGI以在开始使用研究药物之前和之后,提供临床医师对患者的总体功能的观察的简要、独立评估(Busner和Targum,Psychiatry(Edgmont).2007;4(7):28-37)。CGI提供全面的临床医师测定的概述性量度,其考虑所有可用的信息,包括患者病史、心理社会环境、症状、行为和症状对患者的功能能力的影响的知识。CGI包含2个并用1项固型量度,CGI-S(严重程度)和CGI-C(变化)。CGI表格可由有经验的评级者在小于1分钟内完成。
CGI-S是7点量表,其需要临床医师在评估时对患者的疾病的严重程度进行评级,与临床医师对具有相同诊断的患者的过往经验有关(Guy W.ECDEU Assessment Manualfor Psychopharmacology.1976:76-338)。考虑全部临床经验,在评级时患者的精神疾病的严重程度被评估为1,正常,完全无疾病;2,处于疾病边缘;3,轻度疾病;4,中度疾病;5,明显疾病;6,重度疾病;或7,疾病程度最严重的患者。
在基线/第1次访视(第1天)、第4次访视(第43天)和第7次访视/提前终止访视(第85天)时进行CGI-S评估。
CGI-C是7点量表,其需要临床医师在评估时对患者的疾患的变化进行评级,与临床医师对患者在入院时的疾患的过往经验有关。考虑全部临床经验,患者的精神疾病的变化被评估为1,极显著改善;2,显著改善;3,极小改善;4,无变化;5,极小恶化;6,显著恶化;或7,极显著恶化。
在第4次访视(第43天)和第7次访视/提前终止访视(第85天)时进行CGI-C评估。在第43天(第4次访视)时,完成CGI-C以评估自基线访视(第1天)的变化。在第85天(第7次访视)时,完成CGI-C以评估自第43天(第4次访视)的变化和自基线访视(第1天)的变化。
1.5.1.1.4患者总体印象-变化(PGI-C)
PGI-C(附录5A)是7点(1-7)、患者评级量表,其用于评估治疗应答为:极显著改善、显著改善、极小改善、无变化、极小恶化、显著恶化或极显著恶化。
在第4次访视(第43天)和第7次访视/提前终止访视(第85天)时进行PGI-C评估。
1.5.1.1.5用于改善精神分裂症中的认知的测量和治疗研究(MATRICS)认知功能成套测验(MCCB)
MCCB是用于评估精神分裂症中认知增强剂的试验中的认知变化的标准工具。MCCB(Nuechterlein等人Am J Psychiatry.2008;165(2):203-213)意欲提供与精神分裂症和相关病症相关的重要认知领域的相对简要评估。MCCB包括10项测试,其测量7个认知领域:处理速度、注意力/警惕性、工作记忆、语言学习、视觉学习、推理和问题解决以及社会认知。
在筛选(第-28天至第-1天)、基线/第1次访视(第1天)、第4次访视(第43天)和第7次访视/提前终止访视(第85天)时进行MCCB评估。在一天中大致相同的时间(+/-2小时)并且优选在上午进行MCCB。在不同访视时使用成套测验的替代性版本以减少学习混淆。
1.5.1.1.6卡尔加里精神分裂症抑郁量表(CDSS)
CDSS(附录6)是来源于汉密尔顿抑郁量表(Ham-D)的9项目型量表,其被设计用于特异性地评估患有精神分裂症的患者中的抑郁症(Addington等人Schizophr Res.1996;19(2-3):205-212)。与Ham-D不同,CDSS不含与精神分裂症的阴性症状重叠的抑郁症状,诸如快感缺乏和社交退缩。CDSS展示极佳的心理特性。量表中的每个项目评分为0,不存在;1,轻度;2,中度;或3,重度。通过将每个项目评分相加来获得CDSS评分。对于预测存在重性抑郁发作,高于6的评分具有82%特异性和85%敏感性。
在筛选(第-28天至第-1天)、基线/第1次访视(第1天)、第3次访视(第22天)、第4次访视(第43天)、第6次访视(第64天)和第7次访视/提前终止访视(第85天)时进行CDSS评估。
1.5.1.1.7奖励与努力付出任务(EEfRT)
奖励与努力付出任务(EEfRT)(Treadway等人PLoS One.2009;4(8):e6598)是多试验计算机化任务,其中在每次试验中向参与者提供在具有不同困难水平并且与不同水平的货币奖励相关的2项任务之间进行选择的机会。此任务检查响应于不同奖励方案以及为获得奖励而付出的努力(按压按钮)的机率学习。在EEfRT中操作机率,因为与努力动员类似,机率折现似乎可很好地预测阴性症状。此外,包含机率操作可改善任务的整体生态效度,因为需要动机的大部分现实世界中的选择通常与结果的某种程度的不确定性相关联。EEfRT可靠地测量药物对付出与奖励量或奖励机率相关的努力的意愿的作用。举例来说,安非他命增加响应于低和中等机率奖励的努力(Wardle等人J Neurosci.2011;31(46):16597-16602)。尽管认为奖励显著性和行为反应与纹状体中的多巴胺释放有关,但还需要经由NMDA受体进行的对中脑多巴胺神经元的谷氨酸能输入以用于奖励调整(Stuber等人Science.2008;321(5896):1690-1692)。使用困难任务选择与中等机率奖励的比率作为阴性症状的结果量度。
在基线/第1次访视(第1天)、第4次访视(第43天)和第7次访视/提前终止访视(第85天)时进行EEfRT评估。
1.5.1.1.8戒烟问题
在基线/第1次访视(第1天)、第4次访视(第43天)和第7次访视/提前终止访视(第85天)时询问戒烟问题。在基线访视时,询问患者的烟草(香烟)用量;例如从未使用、正在使用、曾经使用。接着,询问其吸烟数量和频率。在第4次访视和第7次访视时,询问受试者是否存在任何用量变化。
1.5.1.2功效变量
主要功效变量是使用加权普通最小平方法测试统计,基于SPCD方法分析的NSA-16总分的从基线/第1次访视至第6周/第4次访视(第43天,第1阶段)的变化和从第6周/第4次访视(第43天)至第12周/第7次访视(第85天,第2阶段)的变化,以及来自第1和2阶段的治疗作用。
次要功效变量包括以下功效量度的从基线至第6周/第4次访视(第43天,第1阶段)的变化和从第6周/第4次访视(第43天)至第12周/第7次访视(第85天,第2阶段)的变化:
·PANSS总分
·PANSS分量表(阳性、阴性、一般精神病理学、Marder阴性因子、兴奋成分和亲社会因子)
·NSA-16(因子领域、总体评分、个别项目和NSA-4因子)
·PANSS总分降低≥20%的患者的比例
·MCCB综合评分
·CGI-S评分
·CGI-C评分(测量在基线后访视时的变化)
·PGI-C评分(测量在基线后访视时的变化)
·CDSS
·EEfRT
1.5.1.3安全性量度
通过所报告的AE、严重AE(SAE)、体检(预定仅在筛选访视时进行)、生命体征、体重、妊娠测试、临床实验室评估和静息12导联ECG来评估安全性。此外,使用以下量表评估安全性:
·哥伦比亚自杀严重程度评级量表(C-SSRS)
·异常不自主运动量表(AIMS)
·巴恩斯静坐不能量表(BAS)
·辛普森安格斯锥体外系症状量表(SAS)
1.5.1.3.1不良事件
AE定义为从签署ICF时出现的任何不适当的医学结果或不希望的变化(包括身体、精神或行为),包括在开始治疗之后的临床试验过程期间出现的间发病,无论是否视为与治疗相关。因此,AE是在时间上与药品的使用相关的任何不利的和不希望的体征(包括例如异常实验室结果)、症状或疾病,无论是否视为与药品相关。
治疗引发不良事件(TEAE)定义为在第一次施用研究药物之后并且在永久性停止研究药物之后的30天内(在AE开始日期当天或之前的第一次给药日期,所述AE开始日期是最后一次给药+30天当天或之前)首次出现或恶化的AE。与临床上通常预期的发生率或量值相比没有差异的与正常生理学相关的变化不视为AE(例如,在生理学上合适的时间发生的月经)。故意或无意地以高于方案中指定的剂量且高于已知治疗剂量的剂量施用治疗视为过度剂量。无关结果报告过度剂量(即使未观测到毒性作用)。
跟踪在患者接受研究药物的最后一次给药之后并且直至在接受研究药物的最后一次给药之后的第30天所报告的任何AE直至消退(患者的健康恢复至其基线状态或者所有变量恢复至正常值)或直至所发生的事件稳定(研究者不预期事件的任何进一步改善或恶化)。
在3点量表中对所有AE进行分级并且如以下电子病例报告表(eCRF)中所指示进行详细报告:
由研究者使用以下解释来确定每种AE与研究药物的关系:
·无关:此类别适用于明确地与其他因素(诸如患者的临床状态、治疗性介入或向患者施用的并用药物)相关的AE。
·不太可能相关:此类别适用于最可能由其他因素(诸如患者的临床状态、治疗性介入或向患者施用的并用药物)产生的AE;并且不遵循已知的对研究药物的反应模式。
·可能相关:此类别适用于满足以下条件的AE:遵循从药物施用时间开始的合理的时间序列;和/或遵循已知的对研究药物的反应模式;但可能由其他因素(诸如患者的临床状态、治疗性介入或向患者施用的并用药物)产生。
·相关:此类别适用于满足以下条件的AE:遵循从药物施用时间开始的合理的时间序列;并且遵循已知的对研究药物的反应模式;并且不能由其他因素(诸如患者的临床状态、治疗性介入或向患者施用的并用药物)合理地说明。
1.5.1.3.2严重不良事件
SAE定义为在任何剂量下发生的引起以下结果中的任一者的任何AE:
·死亡;
·危及生命的经历(在初始报告者看来,从AE出现时便使患者具有立即死亡的风险的经历;即,其不包括在以更严重的形式出现时才可能引起死亡的AE);
·持续性或显著功能障碍/失能(功能障碍是受试者进行正常生活功能的能力的实质性破坏);
·住院治疗或住院时间延长;
·先天性异常/天生缺陷。
当基于合适的医学判断,不引起死亡、不会危及生命或无需住院的重要医学事件会危害患者或需要医学或手术介入以防止定义中所列举的结果中的一者时,将其视为SAE。对于任何SAE(包括异常实验室测试值),必须立即(在24小时内)联系医学监测员。
应报告在研究期间出现或在停止治疗之后的30天内引起研究者的注意的死亡,无论是否视为与治疗相关。妊娠不视为AE或SAE,除非出现符合AE或SAE要求的并发症;然而,其在妊娠报告表中报告。术语“癌症”和“过度剂量”不视为SAE,除非满足SAE的其他标准;然而,癌症和过度剂量报告为AE。
1.5.1.3.3身体和神经检查
在筛选(第-28天至第-1天)时并且在后续访视时由研究者决定进行身体和神经检查。检查包括头部、眼睛、耳、鼻、咽喉、淋巴结、皮肤、四肢、呼吸道、胃肠道、肌肉骨胳、心血管和神经系统的评估。在可能时,每次由同一个人进行身体和神经检查。
在筛选时由研究者测定为是临床上显著的身体和神经检查异常记录为医疗史。与筛选检查相比,身体和神经检查结果的任何临床上显著的变化记录为AE。
1.5.1.3.4生命体征和体重
在筛选(第-28天至第-1天)时,进行立位血压(BP)和心率(HR)测量。在患者以仰卧体位休息至少5分钟后测量仰卧BP和HR。每项测量进行和记录两次。在测量仰卧BP和HR之后,患者静站长达3分钟并且在此站立的3分钟内记录站立BP和HR的单次测量值。还记录呼吸速率(呼吸/分钟)、体温、身高和体重。
呈现直立性低血压(在体位由仰卧变成站立时,在3分钟内测量的收缩压[SBP]降低≥20mm Hg或舒张压[DBP]降低≥10mm Hg)和/或体位性心动过速(与仰卧测量值相比,HR增加≥30次跳动/分钟(bpm),或在站立时HR≥120bpm)的患者符合排除标准3。
在所有后续访视时,在休息至少5分钟之后,获取和记录仰卧/半卧收缩和舒张BP和HR(跳动/分钟)两次。还记录呼吸速率(呼吸/分钟)、体温和体重。
在所有访视时获取生命体征和体重,如表1中所指示。
1.5.1.3.5妊娠测试
指示所有具有生育潜力的女性患者使用合适的避孕方法直至研究药物的最后一次给药之后4周。
除筛选访视(其中进行血清β-hCG测试)以外,在所有临床访视时对所有女性进行尿液妊娠测试(β-hCG),与生育潜力无关。
1.5.1.3.6临床实验室评估
在表1中呈现的访视时进行临床实验室评估,包括血液化学、血液学和尿分析。临床实验室评估包括:
·血液化学(钙、镁、磷、葡萄糖、钠、钾、氯离子、二氧化碳、血尿素氮、血清肌酐、尿酸、白蛋白、总胆红素、碱性磷酸酶、乳酸脱氢酶、天冬氨酸转氨酶/血清谷氨酸草酰乙酸转氨酶[AST/SGOT]、丙氨酸转氨酶/血清谷氨酸丙酮酸转氨酶[ALT/SGPT]、肌酸激酶、γ-谷氨酰基转移酶、三酸甘油酯、总蛋白质、总胆固醇和糖基化血红蛋白[HbA1c,在筛选和第7次访视时])。
·血液学(红细胞计数、血红蛋白、血细胞比容、白细胞计数、中性粒细胞、杆状核粒细胞、淋巴细胞、单核细胞、嗜酸性粒细胞、嗜碱性粒细胞、血小板计数和形态)。
·尿分析(pH值、比重、蛋白质、葡萄糖、酮、胆红素、尿胆素原、亚硝酸盐、白细胞和血液)。对血液、蛋白质、白细胞酯酶或硝酸盐呈阳性的那些样品进行微观分析。
·针对是否存在乙醇和滥用物质(苯环己哌啶(pheneyelidine)、PCP、苯二氮
·甲状腺功能测试TSH、T3、T4(仅筛选访视时)。
·在筛选、第6周和第12周时测量血浆抗精神病药水平。
如果在医学上指示,则由医学监测员要求任何具有临床上显著的异常实验室测试结果的患者在1周后或更早的时间进行重复测试。临床上显著的实验室异常可以是禁止参与研究的依据。通过从中央实验室进行数据传送来将非eCRF数据(包括但不限于实验室测试和结果)发送至合约研究组织(CRO)以用于同化至数据库中。
1.5.1.3.7心电图
ECG设备通过中央读取器提供。在研究中心记录ECG数据并且其包括一般结果、HR(跳动/分钟)、综合QRS以及PR和QTc间期(毫秒)。结果由中央读取器在72小时内提供给研究者并且在24小时内报告任何重要结果。在筛选时存在的ECG异常记录为医疗史。研究者认为临床上显著的相比筛选时的ECG状态的任何变化记录为AE。与研究医学监测员一起论述任何临床上显著的异常ECG并且必要时在1周期间内重复进行。通过从中央读取器进行数据传送来将非eCRF数据(包括但不限于ECG测试和结果)发送至CRO以同化至数据库中。在所有访视时进行静息12导联ECG,如表1中所指示。在第1阶段基线(第1天)和第2阶段基线第4次访视(第43天)时,进行2次ECG;1次在施用研究药物之前进行,并且另一次在给药之后2-3小时进行。在筛选时重复进行三次ECG。
1.5.1.3.8哥伦比亚自杀严重程度评级量表(C-SSRS)
C-SSRS(附录10)是一系列自杀意念和行为的低负担量度,其由国家青少年自杀尝试者心理卫生治疗研究所(National Institute of Mental Health Treatment ofAdolescent Suicide Attempters Study)的哥伦比亚大学(Columbia University)研究人员研发以评估严重程度并经由任何治疗追踪自杀事件。它是提供意念和行为的概述的临床访谈,可在任何评估或风险评估期间施用以鉴别所存在的自杀倾向的程度和类型。C-SSRS还可在治疗期间使用以监测临床恶化。在所有临床访视时进行C-SSRS评级。
1.5.1.3.9辛普森安格斯锥体外系症状量表(SAS)
SAS(附录9A)由10个项目构成并且用于评估假性帕金森症。使用5点量表对每个项目的严重程度的等级进行评级。SAS评分可在0至40范围内。所评估的体征包括步态、落臂、摇肩、肘强直、腕强直、腿摆动、垂头、眉间轻敲、震颤和流涎。在基线/第1次访视(第1天)、第4次访视(第43天)和第7次访视/提前终止访视(第85天)时进行SAS评估。
1.5.1.3.10巴恩斯静坐不能量表(BAS)
BAS(附录8)由评估静坐不能的客观存在和发生率、个体主观意识和苦恼程度以及总体严重程度的项目组成。BAS评分如下:客观静坐不能、坐立不安的主观意识和与坐立不安相关的主观苦恼在4点量表中以0-3进行评级并且相加得到在0至9范围内的总分。静坐不能的总体临床评估使用在0-4范围内的5点量表。在基线/第1次访视(第1天)、第4次访视(第43天)和第7次访视/提前终止访视(第85天)时进行BAS评估。
1.5.1.3.11异常不自主运动量表(AIMS)
AIMS(附录7A)由12个项目构成并且用于评估运动障碍。项目与口颌面、四肢和躯干运动的严重程度、与失能有关的总体判断相关,并且使用5点量表对患者意识进行评级(0=无至4=重度)。使用“是”或“否”回答对与牙齿状态相关的两个项目进行评分。在基线/第1次访视(第1天)、第4次访视(第43天)和第7次访视/提前终止访视(第85天)时进行AIMS评估。
1.5.2药物浓度测量
在基线/第1次访视(第1天,给药后)、第4次访视(第43天)和第7次访视/提前终止访视(第85天时)时,在研究药物的上午给药之后的2至3小时收集所有患者的血浆样品以用于分析d6-DM、d3-DX、d3-3-MM和Q血浆浓度。对于使用d6-DM/Q的患者,基于用于d6-DM、其代谢物氘化(d3)-右羟吗喃(d3-DX)和Q的PK模型评估最大浓度(C
通过离心来分离血浆样品并且接着在-20℃下冷冻直至在分析型单元处分祈。
1.5.3细胞色素P4502D6和基因型评估
在基线访视(第1天)时,在研究药物施用之前收集用于CYP2D6基因分型的血液样品。
还在已抽取血液的任何访视时收集全血的样品以用于探索性生物标记物分析。处理样品并且以等分试样形式储存于生物数据库中长达5年(或直至等分试样耗尽)。
1.5.4测量的适当性
用于评估研究药物的功效的评级量表是公认的工具,其经临床验证并且已广泛用于精神分裂症以及其他精神与行为失常的临床研究中。此研究中使用的安全性评估是临床研究中的标准并且公认为是可靠、精确和恰当的。
1.6数据质量保证
1.6.1研究施用和指导
常规地监测研究以确保遵守研究方案和所收集的数据的整体质量。
对于每位参与研究的患者,由研究者完成并且以电子方式签署eCRF以保证各eCRF内的数据是完整且正确的。这也适用于未能完成研究的患者。如果患者由于治疗限制性AE而退出研究,则将尽最大努力记录结果。由研究监测员在研究点审查eCRF的完整性和对方案的依从性。通过后续内部数据审查检测到的误差可能需要说明或校正误差,并且由研究者记录并批准更改。
任何具有数据输入、查询解析或eCRF审批职责的研究点人员在访问eCRF之前完成训练。电子数据采集供应商在确认训练完成之后提供使用者名称并且接着批准账户。经由审计轨迹来追踪在初始保存之后的对数据的更改,并且强制性需要更改原因。审计轨迹还包括关于进行更改的人员和日期/时戳的信息。
进行研究者会议和研究点启动访视(初始研究点研究方案和程序训练)以使研究者做好准备并标准化功效。经由Web-Ex训练和季度简讯使研究者和研究工作人员保持最新信息并且知晓重要研究更新,诸如方案修正案,并且现场监测员在间歇性监测访视期间审查季度简讯和研究点。由临床研究管理员指导的内部临床研究专员(CRA)定期地访问研究点以审查重要研究更新、患者筛选和入选状态并解答任何研究点问题。基于特定基础,内部CRA还可用于任何研究点和监测员支持。
1.7方案中计划使用的统计方法和样品尺寸的测定
使用9.3版或更高版本的统计分析软件
1.7.1终点
1.7.1.1功效终点
主要功效终点是NSA-16总分相比基线的变化。
次要终点是以下列举的评估的评分的自基线的变化(或CGI-C和PGI-C的实际评分):
·PANSS总分、PANSS分量表(阳性、阴性、一般精神病理学、Marder阴性因子、兴奋成分和亲社会因子)
·NSA-16因子领域、总体症状/功能评分、个别项目和NSA-4
·MCCB综合评分
·CGI-S、CGI-C和PGI-C评分
·CDSS总分
·EEfRT评分
·PANSS总分降低达20%或更大的患者的比例
·戒烟问题
1.7.1.2安全性终点
此研究中的安全性终点包括所报告的AE的发生率和性质、生命体征的随时间推移的变化、体重、尿液妊娠测试、临床实验室评估、静息12导联ECG、C-SSRS、AIMS、BAS和SAS。
172分析群体
1.7.2.1调整意向治疗群体
调整意向治疗(mITT)群体是SPCD分析的主要功效分析群体。单独地确定此群体中所包括的患者的第1阶段和第2阶段。mITT群体的第2阶段分析中所包括的患者是第1阶段分析中所包括的患者的子集。mITT群体定义如下:
·第1阶段:在第1阶段中随机分配的在第1阶段中具有至少1次基线后NSA-16总分评估的所有患者。
·第2阶段:再随机分配至第2阶段中(与第1阶段治疗组无关)并且在第2阶段中具有至少1次NSA-16总分评估(在第4次访视[第6周]之后)的所有患者。
当实施方案修正案4中的变化时,进行IVRS的IRT供应商发现约14名第1阶段安慰剂患者在第3周而非第6周再随机分配至第2阶段中。从mITT分析群体排除具有随机化误差的患者(13名患者)。
由第1阶段安慰剂应答者状态确定SPCD分析中使用的第2阶段mITT群体子集:
·第1阶段安慰剂无应答者(这是SPCD分析的主要功效第2阶段mITT子集)
第1阶段安慰剂应答者
·第1阶段安慰剂应答者和无应答者。
1.7.2.2符合方案的群体
符合方案(PP)的群体包括没有可能实质上影响功效评估的严重方案违规的mITT患者。使用以下标准作为指导以从每个方案分析群体排除患者。
·可能实质上影响功效评估的包含和排除标准的违规。
·研究药物依从性<80%。
·在研究期间接受不当的治疗。具体而言,活性剂治疗组患者接受安慰剂或安慰剂治疗组患者接受活性剂再治疗。
·在基线后使用可能实质上影响功效评估的禁止药物。
·在接受活性剂治疗时,在第6周或第12周的d6-DM或Q浓度低于定量极限。
1.7.2.3意向治疗群体
使用意向治疗(ITT)群体进行SPCD方法的敏感性分析。该群体包括在第1阶段中随机分配的所有患者和正确地再随机分配至第2阶段中的所有患者。从该ITT群体排除13名具有随机化误差的患者。
1.7.2.4安全性群体
使用安全性群体进行所有安全性分析。该群体包括所有接受至少一次研究药物给药的患者。
1.7.2.5 12周平行组群体
12周平行组群体包含随机分配至安慰剂/安慰剂组或d6-DM/Q/d6-DM/Q组(在第1阶段中随机分配至d6-DM/Q组)的患者。意欲在12周研究持续时间下评估功效或安全性,如在研究具有平行组设计时进行。应注意,随机分配至安慰剂/d6-DM/Q组的患者(无论其是否在第1阶段中退出)不是此群体的一部分。
mITT 12周平行组群体:此群体包含12周平行组群体中的具有至少一个基线后NSA-16总分的患者。
安全性12周平行组群体:此群体包含12周平行组群体中的接受至少一次研究药物给药的患者。
符合方案的12周平行组群体:此群体包含mITT 12周平行组群体中的还属于章节1.7.2.1中定义的PP群体的患者。
1.7.3功效分析
所有统计测试是2边的并且在0.05水平下进行。使用描述性统计(平均值、标准偏差[SD]、中值、最小值和最大值)概述定量显示。
1.7.3.1主要功效分析(SPCD,混合模型重复测量)
使用SPCD加权测试统计来分析主要功效终点(NSA-16总分的自基线的变化),其中通过对所观测的数据进行基于可能性的混合模型重复测量(MMRM)分析来评估每个阶段中的治疗作用(Chen等人Contemp Clin Trials.2011;32(4):592-604)。此分析包括mITT群体(第1阶段mITT群体和安慰剂无应答者第2阶段mITT子集)中的患者。MMRM分析包括用于治疗、访视、治疗与访视相互相用、基线NSA-16值以及基线与访视相互相用的条款。使用非结构化协方差。从模型结果直接获得治疗作用和标准误差。
进行独立的MMRM以产生第1阶段的第6周时与第2阶段的第12周(第6周至第12周)时的治疗组之间的最小平方(LS)平均差,以产生组合加权测试统计Z
1.7.3.2主要功效终点的敏感性分析
对主要终点进行以下敏感性分析,以证实在使用不同统计分析、设算方法或患者群体进行分析时的数据的稳定性:
·具有普通最小平方(OLS)协方差分析(ANCOVA)的SPCD,使用mITT群体和用于缺失值的末次观测值结转(LOCF)。
·似乎不相关回归(SUR)方法(Tamura和Huang,Clin Trials.2007;4(4):309-17),其考虑对于两个阶段中具有数据的患者,来自研究中的2个阶段的随机误差可能相关的事实。对mITT群体进行此分析并且使用LOCF进行缺失值的设算。
·对PP群体进行的具有MMRM的SPCD。
1.7.3.3次要功效终点分析
使用下文所描述的方法分析章节1.7.1.1中列举的次要终点。此外,对NSA-16总分(主要功效量度)进行一些功效分析。
1.7.3.3.1通过MMRM SPCD OLS ANCOVA SPCD进行的次要功效终点分析
对于mITT群体,使用SPCD OLS ANCOVA方法分析在基线、第6周和第12周时测量的所有定量次要终点(除CGI-C和PGI-C以外)的自基线的变化,并且研究阶段内的LOCF用于第6周或第12周时的缺失值。ANCOVA模型包括治疗作为因子以及基线值作为协变量。此外,使用MMRM SPCD方法分析来源于NSA-16、PANSS和CDSS的次要终点(其在基线、第3周、第6周、第9周和第12周时评估)。
对于CGI-C和PGI-C,使用具有治疗作为因子以及基线NSA-16总分作为协变量的ANCOVA模型。
1.7.3.3.2 PANSS应答分析
按阶段和治疗组概述mITT群体中的在第6周和第12周时具有有利的治疗应答(即,PANSS总分降低≥20%)的患者的数目和百分比。LOCF用于患者的缺失数据。经由假设第2阶段和第1阶段治疗作用比ρ=1的SPCD 1自由度评分测试来测试整体第1阶段和第2阶段治疗差异(Ivanova等人Stat Med.2011;30(23):2793-2803)。此外,在每次访视时通过卡方检验(Chi-square test)或费希尔精确检验(Fisher′s Exact)来测试治疗作用。
对于治疗应答的二元应答变量,对所观测的数据进行广义估算方程式(GEE)模型分析。广义估算方程式模型包括治疗、访视、治疗与访视相互相用、基线值以及基线与访视相互相用的条款。提供每次访视的P值、优势比(OR)及其95%置信区间(CI)。
1.7.3.3.3 12周平行组分析
为评估在暴露于相同治疗保持12周的情况下的治疗作用,对mITT 12周平行组群体和PP 12周平行组群体进行使用来自所有预定访视的所观测的数据进行的NSA-16总分的重复测量分析。此分析使用线性混合作用模型随时间推移比较治疗组。模型包括治疗、访视、治疗与访视相互相用、基线NSA-16总分以及基线与访视相互相用的固定效应。使用非结构化协方差作为第一偏好,与主要终点相同。
对NSA-16总分以及mITT 12周群体的所有其他次要终点进行此分析。用于CGI-C和PGI-C的模型含有治疗、访视和治疗与访视相互相用。
1.7.3.3.4使用PP群体进行的分析
除对PP群体进行的主要功效终点NSA-16分析(敏感性分析)以外,还使用PP群体分析以下次要功效终点:PANSS总分、PANSS阴性分量表、PANSS Marder阴性因子、MCCB综合评分和CGI-C。进行SPCD和12周平行组比较分析。
1.7.3.4补充分析
1.7.3.4.1 12周平行组ANCOVA
对于所有功效变量,对mITT 12周平行组群体进行独立的访视ANCOVA模型。通过LOCF设算缺失值(与阶段无关)。ANCOVA模型包括治疗作为因子以及基线值作为协变量。对于CGI-C和PGI-C,模型不含基线值。对于CGI-C,用于第12周的模型评估相对于基线的差异。
1.7.3.4.2 CGI-C和PGI-C比例优势比
对于CGI-C和PGI-C,在每次访视时经由比例优势回归模型获得优势比并且表示与安慰剂相比,在d6-DM/Q情况下的有利应答的优势。优势比>1指示使用d6-DM/Q的患者中的有利应答的可能性增加。对mITT群体和12周平行组群体进行此分析。
1.7.3.4.3戒烟分析
呈现每种烟草使用类别(从未使用、正在使用和曾经使用)中的患者的数目和百分比的描述性概述。在曾经使用烟草的患者中,计算使用速率(定义为每天的单位数目)的描述性统计。在基线时正在使用或在基线后开始吸烟的患者中,计算在每次访视时的速率和速率的自基线的变化并且以描述性方式概述。概述各治疗组的所有分析并且对mITT(第1阶段)和mITT 12周平行组群体进行所有分析。
1.7.3.4.4通过子群进行的SPCD分析
对于mITT群体,使用OLS ANCOVA方法,使用由以下各类别定义的子群分析主要终点。
·年龄组(<45,≥45)
·性别(男性,女性)
·基线MCCB综合评分(<30,≥30)
·基线并用苯二氮
·精神分裂症(<20年,≥20年)和残余型精神分裂症(<4年,≥4年)的发作
1.7.3.4.5基线并用药物的治疗作用
对使用视为CYP2D6主要底物的并用精神病药物(抗抑郁剂、抗精神病药)(阿立哌唑、利培酮、度洛西汀、氟西汀、氟伏沙明、米氮平、帕罗西汀和文拉法辛)的患者的子群和使用不视为CYP2D6主要底物的并用精神病药物的子群进行主要终点(NSA-16总分)的分析。使用mITT(第1阶段)和mITT 12周平行组群体进行ANCOVA分析(LOCF)。
此外,针对TEAE(诸如心血管相关AE、跌倒等)分析作为CYP2D6底物的β阻断剂的并用。对使用视为CYP2D6的主要底物的并用β阻断剂的患者的子群和使用不视为CYP2D6的主要底物的β阻断剂的患者进行分析。
1.7.3.4.6 NSA-16带通滤波器分析
带通滤波是一种统计方法,其从产生过高或过低程度的安慰剂应答的试验点滤出数据,由此产生更精确的效应值和活性药物(当有效时)与安慰剂的更好的区分(Targum等人Eur Neuropsychopharmacol.2014;24(8):1188-1195)。使用带通滤波器(>0或<-7)分析NSA-16总分的自基线的变化(对所观测的数据进行)。计算每个点的NSA-16的相比基线评分的变化的平均值并且将具有超过带通滤波器阈值的边界的评分的点视为非信息性并且从分析排除。在应用带通滤波器之后,使用SPCD方法(mITT)并且对12周平行组群体进行NSA-16总分的自基线的变化的分析。
1.7.4药物动力学和药效学分析
以整体描述方式并且按CYP2D6代谢者组概述从在基线(第1天)、第4次访视(第6周)和第7次访视(第12周/ET)时收集的血液样品获得的d6-DM、d3-DX、d3-3-MM和Q的血浆浓度。针对d6-DM、d3-DX和Q进行PK参数(C
提供所估算的d6-DM、d3-DX和Q的PK参数之间的相关性和NSA-16的自基线的变化。
在筛选、第6周和第12周时进行用于评估SGA浓度的血液抽取,并且以描述方式概述血浆浓度的结果。
1.7.5安全性分析
除非另外说明,否则使用以下治疗组显示包括数目和百分比的概述(例如AE)的安全性分析:
·安慰剂/安慰剂:在研究期间接受安慰剂/安慰剂的患者。应注意,此群体中不包括随机分配至安慰剂/d6-DM/Q组,但在第1阶段中退出的患者。而是在‘全部安慰剂’治疗组的情况下概述这些患者。
·d6-DM/Q/d6-DM/Q:在整个研究期间接受d6-DM/Q的患者。
·安慰剂/d6 DM/Q:从安慰剂变换成d6-DM/Q的患者。此组进一步分成在使用安慰剂时产生的数据和在使用d6-DM/Q时产生的数据。
·全部安慰剂:此组包括来自在患者接受安慰剂时的阶段的数据。
·全部d6-DM/Q:此组包括来自在患者接受d6-DM/Q时的阶段的数据。
对于定量概述(例如ECG、实验室),不包括全部安慰剂和全部d6-DM/Q组。
1.7.5.1不良事件
使用18.1版监管活动医学词典(Medical Dictionary for RegulatoryActivities;MedDRA)对AE进行编码。通过系统器官分类(SOC)和优选术语(PT)概述TEAE、引起中止的AE、治疗相关AE和SAE的表格概述。仅使用d6-DM/Q或仅使用安慰剂并且在相同SOC或PT内具有多个AE的患者在MedDRA的水平内仅计数一次。如果患者从安慰剂变成d6-DM/Q并且在两个研究阶段中具有相同AE起点,则其在安慰剂和d6-DM/Q下都计数。
1.7.5.2临床实验室评估
使用在各次访视时和在各阶段时自基线的变化和自基线的变化百分比,以描述方式概述血液学、化学和尿分析评估。经由偏移表评估超出范围的值。基于由中央实验室提供的正常范围,将每个值评估为低、正常或高。提供各治疗组的各偏移组合的发生率。
创建各阶段以及安全性12周平行组群体(安慰剂/安慰剂和d6-DM/Q/d6-DM/Q)的偏移表。在两个阶段中,比较使用d6-DM/Q的患者与使用安慰剂的患者。第1阶段偏移表包括安全性群体中的所有患者,而第2阶段偏移表仅包括再随机分配的患者。所有偏移表中的基线是在各阶段中第一次给药之前的最后一次评估。
概述各治疗组的在基线后的任何时间符合PCS标准的患者的数目和百分比。
在研究过程中,可能进行方案中未提及的一些实验室测试。这些测试未加以概述,但包括于列表中并且标记为非方案测试。
1.7.5.3心电图
通过中央读取器报告HR、PR间期、QRS持续时间、QT间期(未校正)和QTcF的定量参数。计算各参数的自基线的变化和自基线的变化百分比并且提供各治疗组的概述。此外,因为在基线、第6周和第12周时,在给药前和给药后记录ECG,所以在这些访视时概述从给药前至给药后的变化。
概述各治疗组的符合PCS标准的患者的数目和百分比。提供基线后的任何时间时和各次访视时的概述。对于QTcF,分别评估男性和女性。患者可参与其有资格参与的所有类别。
通过正常或异常的数值和百分比来概述ECG整体解释。使用心脏病专家解释(即,中央ECG)进行这些概述。列举各患者的所有解释和相应细节。
1.7.5.4生命体征
在患者处于仰卧/半卧体位时概述的参数包括SBP、DBP和HR。记录这些测量值两次,由此获得2个测量值的平均值并且用于下文所提及的所有概述。还对体重进行概述。以与ECG参数类似的方式,经由自基线的变化和自基线的变化百分比来概述这些参数。
还经由PCS标准评估生命体征。如果患者在基线后的任何时间符合给定标准,则对其进行计数。在各患者列表中包括所有生命体征。
1.7.5.5身体和神经检查
仅在筛选时计划进行身体和神经检查以用于评估并且呈现于各患者列表中。
1.7.5.6哥伦比亚自杀严重程度评级量表(C-SSRS)
使用以下指标分析C-SSRS的评分:
·意念严重程度:与意念类型有关的5个问题中的每一者(是/否)
·最严重意念的强度:5个强度项目的总和
·自杀行为类型:4种自杀行为类型中的每一者(是/否)
·自杀行为:自杀行为问题
·实际致死性:关于致死性最高的尝试的实际致死性问题(0-5级)
·潜在致死性:关于致死性最高的尝试的潜在致死性问题(0-2级)
概述各治疗组和各次访视时的上文列举的具有是/否回答的个别问题。以描述方式概述各治疗组和各次访视时的大部分或严重意念的强度。经由描述性统计、数值和百分比来概述含有顺序回答的项目。在各患者列表中包括所有C-SSRS数据,包括个别文本说明(作为一些问题的一部分而包括)。
1.7.5.7辛普森安格斯锥体外系症状量表(SAS)
概述SAS的个别项目以及总分。分别提供各治疗组和各次访视时的男性和女性以及整体上两种性别的描述性统计。对于每个项目,在整体上并且分别对于男性和女性概述从基线至第6周以及从基线至第12周的每个评分(0至4)的患者数目和百分比的偏移表。
1.7.5.8巴恩斯静坐不能量表(BAS)
概述BAS的客观评估、主观意识、主观苦恼和总体临床评估以及总分。分别提供各治疗组和各次访视时的男性和女性以及整体上两种性别的描述性统计。分别对于男性和女性以及对于整体上两种性别,呈现基线至第6周以及基线至第12周的总分和总体临床评估的患者数目和百分比的偏移表。
1.7.5.9异常不自主运动量表(AIMS)
分别对于男性和女性以及对于整体上两种性别,使用描述性统计概述各治疗组和各次访视时的AIMS的每个分量表的评分。分别对于男性和女性以及对于整体上两种性别,呈现基线至第6周以及基线至第12周的总分和总体临床评估的患者数目和百分比的偏移表。
1.7.6样品尺寸的测定
基于公开的研究,诸如Kane等人(Arch Gen Psychiatry.1988;45(9):789-796)和Buchanan等人(Schizophr Bull.2015;41(4):900-908),假设用于主要终点的双变量正态分布,进行样品尺寸计算。假设第1阶段和第2阶段治疗差都为-2.5,并且假设药物和安慰剂组的标准偏差等于5(效应值为-0.5)。进一步假设82%的患者将完成第1阶段,70%的这些分配至安慰剂组的患者将是安慰剂无应答者,并且91%的这些患者将完成第2阶段。对于此SPCD研究,在第1阶段中以1∶2比率(活性剂∶安慰剂)随机分配的120名患者的样品尺寸将具有约80%功效,其中2边I型误差α=0.05。如果假设两个阶段中的效应值都为-0.425,则功效将为约70%。
1.7.7统计/分析问题
1.7.7.1协变量的调整
此研究中的计划和实际协变量以及分析中使用的方法无不同之处。
1.7.7.2退出或缺失数据的处理
取决于参数和分析以不同方式处理缺失数据。应注意,对‘所观测的情况’进行的分析(诸如MMRM分析)不遵循以下任何设算规则。关于考虑因素,参见下文:
·在任何情形中都不设算缺失基线值。
·对于SPCD以及各阶段和访视功效分析(不包含所观测的情况分析),在研究阶段内通过LOCF设算缺失数据。对于在第2阶段中没有数据的患者,不进行设算。
·对于mITT 12周平行组群体,还使用LOCF方法进行功效分析,意指将最后一次非缺失基线后观测转入每次访视。
1.7.7.3期中分析和数据监测
对于此研究,未指定数据监测安全性委员会并且未计划或进行期中分析。
1.7.7.4多中心研究
整体上分析此研究中收集的数据。
1.7.7.5多重比较/多重性
对于测试多个次要结果量度,未进行调整。因为有可能一些显著结果仅偶然出现,不过度地考虑孤立的显著差异;反而应基于显著差异的模式及其与主要终点分析的一致性来进行解释。
1.7.7.6使用患者的“功效子集”
进行事后分析(排除来自安慰剂和d6-DM/Q治疗组的所有具有超过一位评级者的患者)以评估在整个研究期间,使用单个评级者与多个评级者评估相同终点的影响。此分析的结果呈现于章节3.4.1.4.1中。
1.7.7.7子群的检查
用于功效的子群分析的因子描述于章节1.7.3.4.4中。类别包括年龄、性别、基线MCCB综合评分、基线并用苯二氮
1.8研究或计划分析的指导的变化
在章节1.7.3.4.1中指定所有功效终点的12周平行组ANCOVA,但仅对NSA-16相关功效终点进行。这是因为如章节1.7.3.3.3中所指定来进行类似的12周平行组MMRM并且MMRM是优选方法。
在数据库锁定之后,一名研究者(研究点117,主要研究者)告知在所述研究点发现一些在数据库锁定之前未校正的数据输入错误。审查这些偏差并且得出以下结论:所述变化对数据的功效结果或整体完整性无影响并且不存在安全性问题,因此未解锁和再锁定数据库。根据SOP,在试验总档案中提交偏差报告。
2研究患者
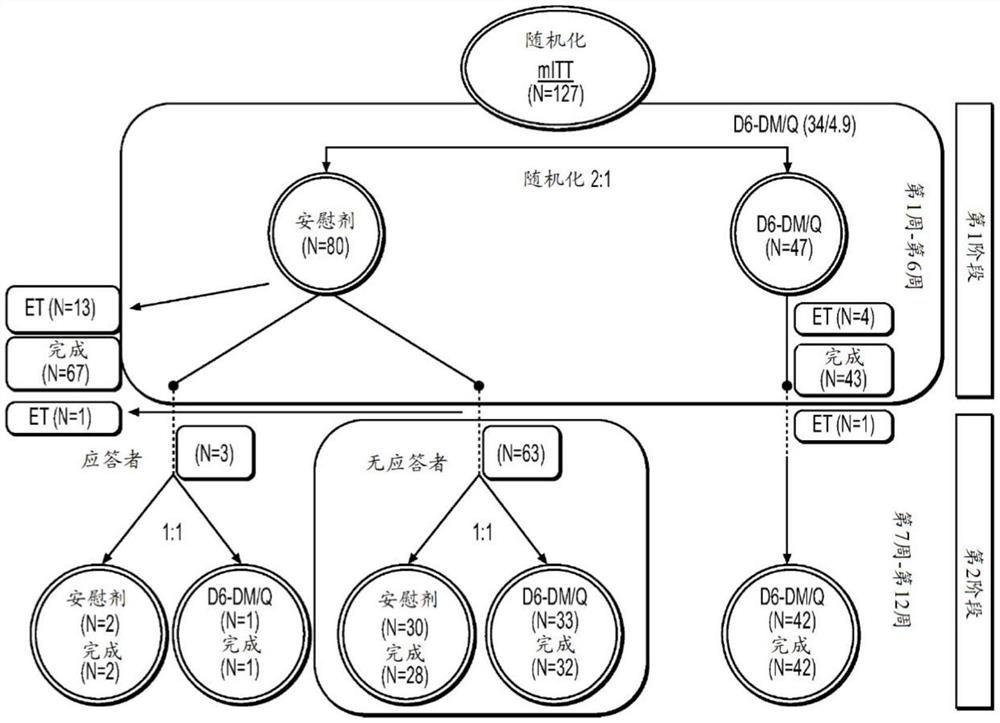
2.1患者的部署
整体患者部署概述于表4中并且mITT群体的患者部署描绘于图3中。在所筛选的286名患者中,145名患者参与并且在第1阶段中随机分配。在随机分配的145名患者中,在第1阶段中,48名患者随机分配至d6-DM/Q组并且97名患者随机分配至安慰剂组;一名患者随机分配至安慰剂组但未接受研究药物。对于所有随机分配的患者,总共123名患者完成第1阶段(d6-DM/Q:43名患者;安慰剂:80名患者)并且其余21名患者在第1阶段期间中止。对于mITT群体,在第1阶段中接受d6-DM/Q的47名患者中,42名患者进入第2阶段并且继续接受d6-DM/Q,而在第1阶段中接受安慰剂的80名患者中,66名进入第2阶段并且再随机分配(d6-DM/Q:32名患者;安慰剂:34名患者)。在第1阶段结束时,在安慰剂组中,64名患者是无应答者(PANSS总分降低<20%)并且3名患者是应答者(PANSS总分降低≥20%),并且在d6-DM/Q组中,42名是无应答者并且5名是应答者。
总体而言,对于所有随机分配的患者,118名患者完成研究(在第1阶段和第2阶段中使用d6-DM/Q:42名患者;在第1阶段和第2阶段中使用安慰剂:37名患者;在第1阶段中使用安慰剂并且在第2阶段中使用d6-DM/Q:39名患者)。在中止研究的27名患者(18.6%)中,10名是归因于AE,8名是患者退出,4名是未能随访,3名由于研究药物的非依从性而退出,1名由于医师决策而退出,并且1名由于未列举的其他原因而退出。各治疗组中的中止原因是类似的。
表4.整体患者部署-所有患者
筛选失败的共同点和筛选失败的原因为所筛选的患者的数目。所有其他类别的共同点是随机分配的患者的数目(N=145)。--=不适用。
3功效评估
3.1所分析的数据集
在表5中概述各治疗组的分析群体。在研究开始时随机分配的145名患者中,132名包括于ITT群体中,144名包括于安全性群体中,并且110名包括于PP群体中。在随机分配的患者中,127名患者符合mITT群体的标准并且包括于第1阶段mITT群体中。总共108名患者包括于第2阶段ITT群体和第2阶段mITT群体中。mITT群体中所包括的患者的部署提供于图3中。
对于此研究中分析的所有数据集,各群体的患者标准描述于章节1.7.2中。
表5.分析群体和第2阶段子集的概述-所有随机分配的患者
对12周平行组和安全性群体的分析不考虑研究阶段,因此无需定义第2阶段N。
ITT=意向治疗;mITT=调整意向治疗;pP=符合方案;--=不适用。
3.2人口统计和其他基线特征
3.2.1人口统计
第1阶段mITT群体的基线和人口统计特征概述于表6中。入选时平均(SD)年龄为45.5(11.19)岁。mITT群体中有88名男性患者(69.3%)和39名女性患者(30.7%)。mITT群体中所包括的大部分患者是黑人(69名患者[54.3%])或白人(48名患者[37.8%])。各治疗组中的基线和人口统计特征是类似的。在各治疗组中,mITT 12周平行组和安全性群体的人口统计和基线特征与在表6中发现的mITT群体类似。
表6.人口统计和基线特征-mITT群体
对于各类别参数,百分比的分母是进行参数评估的患者的数目。
BMI=身体质量指数;mITT=调整意向治疗;SD=标准偏差。
3.2.2医疗史
由于此研究中研究的群体,医疗史的最常报告的SOC和PT分别为精神障碍(144名患者[100.0%])和残余型精神分裂症(144名患者[100.0%])。患者医疗史中第二频繁报告的PT是高血压(35名患者[36.5%],在安慰剂组中和12名患者[25.0%],在d6-DM/Q组中)。
3.2.3先前和并用药物
通过表67中的解剖学治疗子群和安全性群体的优选基本名称来呈现先前药物。通过表68中的解剖学治疗子群和安全性群体的优选基本名称来呈现并用药物。表17至表94包括于附录1中。
表17中概述安全性群体的SGA的基线并用。安慰剂和d6-DM/Q治疗组中的患者在基线时最常使用的SGA是阿立哌唑(16.7%安慰剂,31.3%d6-DM/Q)、奥氮平(27.1%安慰剂,25.0%d6-DM/Q)和利培酮(26.0%安慰剂,20.8%d6-DM/Q)。
3.2.4其他基线特征
用于mITT群体中的功效测量的第1阶段的基线值概述并呈现于表7中。2个治疗组的基线值相当。
mITT研究群体中的安慰剂无应答者和安慰剂应答者的第2阶段基线功效评估分别呈现于表19和表20中。
表7.第1阶段基线功效评估-mITT群体
CDSS=卡尔加里精神分裂症抑郁量表;CGI-S=临床总体印象-疾病严重程度;mITT=调整意向治疗;NSA-16=16项目型阴性症状评估;PANSS=阳性和阴性综合征量表;SD=标准偏差
来源:表18
3.3治疗依从性的测量
安全性群体的研究药物依从性概述并呈现于表8中。各治疗组中的大部分患者使用其预定剂量的80%至120%并且考虑其依从性。
表8.治疗依从性-安全性群体
SD=标准偏差。
3.4个体患者数据的功效结果和列表
3.4.1功效分析
3.4.1.1主要功效终点(16项目型阴性症状评估[NSA-16]总分)
3.4.1.1.1主要分析
主要功效分析是d6-DM/Q对比安慰剂,NSA-16总分的自基线的变化的SPCD分析。通过使用mITT群体的SPCD分析,与安慰剂相比,在d6-DM/Q情况下观测到NSA-16总分的自基线的变化的数值上更高的改善(SPCD加权Z统计=-1.79,p=0.073,表9)。在第1阶段(其模仿平行组设计)中,NSA-16总分的自基线的平均(SD)变化为-5.0(5.64)(d6-DM/Q)和-3.4(5.54)(安慰剂),导致LS平均治疗差为-1.79(95%CI-3.86,0.29;p=0.091)。在仅包括随机分配至d6-DM/Q或安慰剂组的安慰剂无应答者的第2阶段中,NSA-16总分的自基线的平均变化(SD)为-3.7(6.41)(d6-DM/Q)和-2.4(5.88)(安慰剂),导致LS平均治疗差为-1.28(95%CI-4.39,1.83;p=0.413;图4)。
表9.NSA-16总分:自基线的变化SPCD MMRM(所观测的数据)-mITT群体
可能的NSA-16总分范围为16至96;较高评分指示较差状况。
CI=置信区间;NSA-16=阴性症状评估量表;mITT=调整意向治疗;MMRM=混合模型重复测量;SD=标准偏差;SPCD=顺序平行比较设计。
3.4.1.1.2敏感性分析
使用不同统计分析方法(SUR方法[LOCF]和SPCD与OLS ANCOVA[LOCF])和不同分析群体(PP群体)对主要终点进行的敏感性分析证实主要分析的结果;此外,在SUR方法(p=0.048,表23)和SPCD OLS ANCOVA(p=0.042,表24)(但不使用PP群体)的情况下都观测到d6-DM/Q与安慰剂之间的统计显著治疗差(有利于d6-DM/Q)(表25)。结果的概述提供于表10中。
3.4.1.1.3 12周分析
随机分配并且在两个阶段中保持(或提前退出)相同治疗分配的患者群组的NSA-16总分的变化的分析(模仿传统12周平行比较设计而非2阶段SPCD)表明d6-DM/Q与安慰剂治疗之间不存在差异。对于mITT 12周平行组群体,NSA-16总分的自基线的平均(SD)变化为-6.6(7.81)(d6-DM/Q/d6-DM/Q组)和-7.0(7.71)(安慰剂/安慰剂组)。
在使用mITT 12周平行组群体通过ANCOVA(LOCF)或使用PP 12周平行组群体通过MMRM(所观测的数据)进行分析时观测到类似结果。
3.4.1.1.4子群分析
对于mITT群体,使用OLS ANCOVA SPCD方法,使用由以下各类别定义的子群分析主要终点。
·年龄组(<45;≥45)
·性别(男性;女性)
·基线MCCB综合评分(<30;≥30)
·基线并用苯二氮
·精神分裂症(<20年;≥20年)和残余型精神分裂症(<4年;≥4年)的发作
对于按年龄、性别、基线MCCB综合评分、基线并用药物使用或精神分裂症和残余型精神分裂症的发作进行的子群分析,未观测到d6-DM/Q与安慰剂之间存在有意义的差异治疗作用。
3.4.1.1.5带通滤波器分析
使用一种带通滤波器分析主要终点,所述带通滤波器从分析排除具有超过边界(>0或<-7)的安慰剂的相比基线评分的平均NSA-16变化的研究点。在SPCD ANCOVA分析情况下,此分析的结果展示有利于d6-DM/Q的统计显著治疗差(SPCD加权OLS Z统计=-2.25,p=0.025,表26)。
对mITT 12周平行组群体进行的类似分析的结果呈现于表27中。
3.4.1.2次要终点
3.4.1.2.1阳性和阴性综合征量表(PANSS)
PANSS的分析包括所有30个项目的总分和各种来源于这些30个项目的分量表,其包括:阴性分量表(N1-N7)、阳性分量表(P1-P7)、一般精神病理学分量表(G1-G16)、亲社会因子(G16、N2、N4、N7、P3和P6)、Marder阴性因子(N1、N2、N3、N4、N6、G7和G16)和兴奋成分(P4、P7、G4、G8和G14)。表11中包括PANSS总分和分量表的结果的概述。
3.4.1.2.1.1 PANSS总分
PANSS总分在30至210范围内,其中评分越高指示症状的严重程度越高。通过主要SPCD MMRM分析,观测到有利于d6-DM/Q的PANSS总分的d6-DM/Q与安慰剂之间的统计显著差异(SPCD加权Z统计=-2.25,p=0.025,表28)。在第1阶段中,PANSS总分的自基线的平均(SD)变化为-4.7(6.98)(d6-DM/Q)和-2.5(6.50)(安慰剂),导致LS平均治疗差为-2.36(95%CI-4.77,0.06;p=0.055)。在仅包括随机分配至d6-DM/Q或安慰剂组的安慰剂无应答者的第2阶段中,PANSS总分的自基线的平均(SD)变化为-4.0(7.71)(d6-DM/Q)和-1.4(7.64)(安慰剂),导致LS平均治疗差为-2.53(95%CI-6.51,1.45;p=0.209;图5)。使用mITT群体通过OLS ANCOVA SPCD(p=0.024,表36)和使用PP群体通过SPCD MMRM分析(p=0.022,表38)观测到类似结果。
PANSS总分的自基线的变化的分析概述于表76(mITT 12周平行组群体)和表39(PP12周平行组群体)中。
3.4.1.2.1.2 PANSS阴性分量表
阴性分量表包含PANSS的7个项目并且评分在7至49范围内,其中评分越高指示阴性症状的严重程度越高。在mITT群体(图6)和PP群体(p=0.019,表40)中,通过主要SPCDMMRM分析(SPCD加权Z统计=-2.20,p=0.027,表30),在PANSS阴性分量表中观测到有利于d6-DM/Q的d6-DM/Q与安慰剂之间的统计显著差异。在mITT群体中,通过ANCOVA SPCD分析观测到类似结果(p=0.019,表41)。
PANSS阴性分量表评分的自基线的变化的分析概述于表42(mITT 12周平行组群体)和表43(PP12周平行组群体)中。
3.4.1.2.1.3 PANSS Marder阴性因子
PANSS Marder阴性因子包含PANSS的7个项目并且评分在7至49范围内,其中评分越高指示精神分裂症的阴性症状的严重程度越高。在mITT群体(图7)和PP群体(p=0.019,表44)中,通过主要SPCD MMRM分析(SPCD加权Z统计=-2.26,p=0.024,表32)观测到PANSSMarder阴性因子中有利于d6-DM/Q的d6-DM/Q与安慰剂之间的统计显著差异。在mITT群体中,通过SPCD ANCOVA分析观测到类似结果(p=0.019,表45)。
PANSS Marder阴性分量表评分的自基线的变化的分析概述于表46(mITT 12周平行组群体)和表47(PP 12周平行组群体)中。
3.4.1.2.1.4 PANSS亲社会因子
PANSS亲社会因子包含PANSS的6个项目并且评分在6至42范围内,其中评分越高指示特定阴性症状的严重程度越高。在mITT群体中,通过主要SPCD MMRM分析(SPCD加权Z统计=-2.60,p=0.009,表34)观测到PANSS亲社会因子中有利于d6-DM/Q的d6-DM/Q与安慰剂之间的统计显著差异(图8)。在mITT群体中,通过SPCDANCOVA分析观测到类似结果(p=0.007,表48)。
对于mITT 12周平行组群体,PANSS亲社会因子评分的自基线的变化的分析概述于表49中。
3.4.1.2.1.5 PANSS阳性分量表
阳性分量表包含PANSS的7个项目并且评分在7至49范围内,其中评分越高指示精神分裂症的阳性症状的严重程度越高。通过SPCD MMRM分析(表29)、SPCD ANCOVA分析(表50)或使用mITT 12周平行组群体的12周分析(表51),未观测到经d6-DM/Q治疗的患者与经安慰剂治疗的患者之间存在PANSS阳性分量表评分的显著差异。
3.4.1.2.1.6 PANSS一般精神病理学分量表
一般精神病理学分量表包含PANSS的16个项目并且评分在16至112范围内,其中评分越高指示精神分裂症的症状的严重程度越高。通过使用mITT群体的SPCD分析(SPCD加权Z统计=-1.93,p=0.054),与安慰剂相比,在d6-DM/Q情况下观测到PANSS一般精神病理学分量表评分的自基线的变化的数值上更高的改善。在第1阶段中,相比基线评分的平均(SD)变化为-1.7(4.04)(d6-DM/Q)和-0.7(3.21)(安慰剂),导致LS平均治疗差为-1.14(95%CI-2.40,0.13;p=0.077)。在第2阶段中,自基线的平均(SD)变化为-1.3(5.10)(d6-DM/Q)和0.0(5.14)(安慰剂),导致LS平均治疗差为-1.40(95%CI-3.99,1.19;p=0.284)(表31)。使用SPCD ANCOVA(表37)和使用mITT 12周平行组群体(表35)的分析展示类似趋势,其中在使用mITT 12周平行组群体的12周分析(p=0.028)中发现有利于d6-DM/Q的统计显著差异。
3.4.1.2.1.7 PANSS兴奋成分
兴奋成分包含PANSS的5个项目并且评分在5至35范围内,其中评分越高指示症状的严重程度越高。通过SPCD MMRM分析、SPCD ANCOVA分析或使用mITT 12周平行组群体的12周分析,未观测到经d6-DM/Q治疗的患者与经安慰剂治疗的患者之间存在PANSS兴奋成分的显著差异。
3.4.1.2.1.8 PANSS应答者分析
借助于使用mITT群体的SPCD分析和使用mITT 12周平行组群体的GEE分析,通过分析PANSS总分相比基线降低20%的患者的比例来评估治疗作用。在这些分析中未观测到d6-DM/Q与安慰剂之间存在统计显著差异。
对于PANSS Marder阴性因子和PANSS阴性分量表,还使用相同阈值(相比基线降低20%)进行事后分析(数据未显示)。
在两个阶段中,与安慰剂相比,d6-DM/Q组中的PANSS Marder阴性因子相比基线降低20%的患者的比例以统计方式显著更高(第1阶段:21.3%对比16.3%;第2阶段:27.3%对比3.3%;SPCD p=0.012)。
在两个阶段中,与安慰剂相比,d6-DM/Q组中的PANSS阴性分量表相比基线降低20%的患者的比例更高(第1阶段:23.4%对比13.8%;第2阶段:21.2%对比10%;SPCD p=0.054)。
3.4.1.2.2 NSA-16:总体阴性症状、总体功能水平、5因子领域和NSA-4
3.4.1.2.2.1总体阴性症状评级
NSA-16中的总体阴性症状评级是单一评分,其是基于将阴性症状的严重程度的整体印象分为1至7级,其中评分越高指示严重程度越高。在mITT群体中,通过主要SPCD MMRM分析(SPCD加权Z统计=-2.23,p=0.026,表52),在NSA-16总体阴性症状评级中观测到有利于d6-DM/Q的d6-DM/Q与安慰剂之间的统计显著差异。在第1阶段中,NSA-16总体阴性症状评级的自基线的平均(SD)变化为-0.4(0.68)(d6-DM/Q)和-0.2(0.65)(安慰剂),导致LS平均治疗差为-0.17(95%CI-0.40,0.07;p=0.167)。在仅包括随机分配至d6-DM/Q或安慰剂组的安慰剂无应答者的第2阶段中,NSA-16总体阴性症状评级的自基线的平均(SD)变化为-0.5(0.76)(d6-DM/Q)和-0.1(0.52)(安慰剂),导致LS平均治疗差为-0.29(95%CI-0.61,0.03;p=0.079;图9)。通过SPCD OLS ANCOVA观测到类似结果(p=0.016,表53)。
使用mITT 12周平行组群体的NSA-16总体阴性症状评级的自基线的变化的分析概述于表54(MMRM分析)和表55(ANCOVA)中。
3.4.1.2.2.2总体功能水平
总体功能水平是分为1至7级的单一评分,其提供患者的功能水平的整体评估,其中较高的评分指示严重的功能缺损。通过SPCD MMRM分析、SPCD ANCOVA分析或使用mITT 12周平行组群体的12周分析,未观测到经d6-DM/Q治疗的患者与经安慰剂治疗的患者之间存在NSA-16总体功能水平评分的显著差异。
3.4.1.2.2.3 NSA-4总分
NSA-4总分包含NSA-16的项目2、5、8和13,其使得临床医师能够快速确定精神分裂症的阴性症状的严重程度。这些项目关注以下行为:言语量受限(2)、情绪范围缩窄(5)、社交动力下降(8)和兴趣减少(13)。通过SPCD MMRM分析、mITT群体的SPCD ANCOVA分析或使用mITT 12周平行组群体的12周分析,未观测到经d6-DM/Q治疗的患者与经安慰剂治疗的患者之间存在NSA-4总分的显著差异。
3.4.1.2.2.4 NSA-16因子领域
将NSA-16中的项目分组以使用5因子模型描述阴性症状,其包括以下领域:交流(项目1、2、3和4)、情绪/反应(项目5、6和7)、社会参与(项目8、9和10)、动机(项目11、12、13和14)和迟缓(项目15和16)。通过使用mITT群体的SPCD分析或使用mITT 12周平行组群体的12周分析,在NSA-16的5因子领域中的任一者中都未观测到d6-DM/Q与安慰剂之间存在显著差异。
如下测定各领域的结果:
·交流:mITT群体的SPCD MMRM分析、SPCD ANCOVA分析;使用mITT 12周平行组群体的12周分析
·情绪/反应:mITT群体的SPCD MMRM分析、SPCD ANCOVA分析;使用mITT 12周平行组群体的12周分析
·社会参与:mITT群体的SPCD MMRM分析、SPCD ANCOVA分析;使用mITT 12周平行组群体的12周分析
·动机:mITT群体的SPCD MMRM分析、SPCD ANCOVA分析;使用mITT 12周平行组群体的12周分析
·迟缓:mITT群体的SPCD MMRM分析、SPCD ANCOVA分析;使用mITT 12周平行组群体的12周分析
3.4.1.2.3患者总体印象-变化(PGI-C)
使用PGI-C在第6周(第1阶段)相对于基线并且在第12周(第2阶段)相对于第4次访视(第1阶段结束)评估患者的治疗应答印象,其中1=极显著改善至7=极显著恶化。测定mITT群体的各阶段的类别应答的概述。通过对所观测的数据进行SPCD ANCOVA分析(SPCD p=0.170),在d6-DM/Q与安慰剂组之间未观测到PGI-C评分的统计显著差异。在第1阶段中,27.7%的用d6-DM/Q治疗的患者(相对于24%的用安慰剂治疗的患者)在其症状变化中被评级为“显著改善”或“极显著改善”。在第2阶段中,34.4%的用d6-DM/Q治疗的患者(相对于13.3%的用安慰剂治疗的患者)在第12周时报告其症状得到“显著改善”或“极显著改善”(图10,SPCD p=0.170)。概述mITT群体的各阶段的使用比例优势回归的PGI-C的分析。
PGI-C评分的使用mITT 12周平行组群体的12周分析呈现于表21中并且各阶段的比例优势回归呈现于表22中。在mITT 12周平行组群体中,在第12周相对于基线被评级为‘显著改善’或‘极显著改善’的患者的比例为33.3%(14/42)(d6-DM/Q/d6-DM/Q组)和18.8%(6/32)(安慰剂/安慰剂组)(p=0.158,通过比例优势回归)。
3.4.1.2.4临床总体印象-变化(CGI-S)
CGI-S评分在1至7范围内,其中评分越高表示疾病的严重程度越高。在mITT群体中,d6-DM/Q和安慰剂组在基线时的平均(SD)CGI-S评分分别为3.7(0.74)和3.9(0.74)。通过SPCD ANCOVA分析或使用mITT 12周平行组群体的12周分析,在d6-DM/Q与安慰剂之间未观测到CGI-S评分的显著差异。
3.4.1.2.5临床总体印象-变化(CGI-C)
使用CGI-C评估在第6周(第1阶段)时相对于基线以及在第12周(第2阶段)时相对于第4次访视(第1阶段结束)的精神疾病的变化的总体印象,其中1=极显著改善至7=极显著恶化。测定mITT群体的各阶段的类别应答的概述。通过对所观测的数据进行SPCD ANCOVA分析或mITT群体的各阶段的比例优势回归,在d6-DM/Q与安慰剂组之间未观测到统计显著差异。在第1阶段中,d6-DM/Q组中的48.9%(23/47)的患者和安慰剂组中的35.9%(28/78)的患者在CGI-C评级中具有‘极小改善’、‘显著改善’或‘极显著改善’,并且在第2阶段中,d6-DM/Q组中的43.7%(14/32)的患者和安慰剂组中的36.7%(11/30)的患者在CGI-C评级中具有‘显著改善’或‘极显著改善’(SPCD p=0.057)。
mITT 12周平行组群体和PP 12周平行组群体的分析结果类似,各组的CGI-C评分之间不存在统计显著差异,然而,在使用PP群体的SPCD ANCOVA中观测到各组之间的统计显著差异(SPCD p=0.044)。
3.4.1.2.6卡尔加里精神分裂症抑郁量表(CDSS)
CDSS评分在范围0至27内,其中较高评分指示抑郁症的严重症状。在此研究中,仅包括评分<6的患者。总体而言,参与研究的患者在基线时具有低CDSS评分;在第1阶段中,平均(SD)评分为1.1(1.34)(随机分配至d6-DM/Q组的患者)和0.9(1.31)(随机分配至安慰剂组的患者)。通过SPCD MMRM分析(表56)、SPCD ANCOVA分析(表57)或使用mITT 12周平行组群体的12周分析(表58),在经d6-DM/Q治疗的患者与经安慰剂治疗的患者之间未观测到CDSS评分的显著差异。在mITT 12周平行组群体中,在第12周时,CDSS评分的自基线的平均(SD)变化为-0.2(1.27)(d6-DM/Q/d6-DM/Q组)和-0.4(0.99)(安慰剂/安慰剂组)(表58)。
3.4.1.2.7 MATRICS认知功能成套测验(MCCB)
MCCB综合评分在0至70范围内,其中评分越高指示认知症状的严重程度越低。通过使用mITT群体的SPCD ANCOVA分析,与安慰剂相比,观测到d6-DM/Q的MCCB综合评分的自基线的变化的数值上的有利改善(SPCD加权OLS Z统计=1.78,p=0.074,表63)。在第1阶段中,MCCB综合评分的自基线的平均(SD)变化为1.2(5.11)(d6-DM/Q)和1.6(4.55)(安慰剂),导致LS平均治疗差为-0.12(95%CI-1.88,1.64;p=0.893)。在第2阶段中,MCCB综合评分的自基线的平均(SD)变化为1.6(3.71)(d6-DM/Q)和-1.6(4.06)(安慰剂),导致LS平均治疗差为3.21(95%CI 1.11,5.30;p=0.003)。使用PP群体的类似分析展示有利于d6-DM/Q的统计显著差异(p=0.046,表64)。
MCCB综合评分的自基线的变化的SPCD ANCOVA分析的结果呈现于表65(使用mITT12周平行组群体)和表66(使用PP 12周平行组群体)中。在这些分析中未观测到d6-DM/Q与安慰剂治疗之间存在统计显著差异。
3.4.1.2.8奖励与努力付出任务(EEfRT)
分析以下8种变量的EEfRT评分:
·基线按压评级,首先促使受试者用其非惯用小指尽可能快地按压用于困难任务的按键持续21秒。将值编码为平均按压次数/秒。
·选择RT 1st 50,在第一个50次试验期间进行选择所花费的平均反应时间(毫秒)。仅受试者作出选择的试验。
·完成,在第一个50次试验期间所完成的任务(简单或困难)的比例。
·12%机率-机率高努力选择-1st 50,在任务的第一个50次试验期间,在低(12%)机率条件下作出困难任务选择的比例。
·50%机率-机率高努力选择-1st 50,在任务的第一个50次试验期间,在中等(50%)机率条件下作出困难任务选择的比例。
·88%机率-机率高努力选择-1st 50,在任务的第一个50次试验期间,在高(88%)机率条件下作出困难任务选择的比例。
·所有比例高努力选择-1st 50,在任务的第一个50次试验中作出困难任务选择的整体比例。
·差异比例高努力选择-1st 50,在任务的第一个50次试验期间,在高机率条件下作出困难任务选择的比例之间的差异。
通过SPCD ANCOVA分析或使用mITT 12周平行组群体的12周分析,在经d6-DM/Q治疗的患者与经安慰剂治疗的患者之间未观测到显著差异。
3.4.1.2.9戒烟
评估当前吸烟者和mITT 12周平行组群体的每天吸食的香烟的数目的自基线的变化,并且在戒烟方面,经d6-DM/Q治疗的患者与经安慰剂治疗的患者之间不存在有意义的差异。
3.4.1.3探索性生物标记物
在已抽取血液的任何访视时收集全血样品以用于探索性生物标记物分析。在可用时,在单独报告中呈现生物标记物分析的结果。
3.4.1.4事后分析
3.4.1.4.1在整个研究中由单个评级者进行的评估
尽管进行训练工作,但来自指定锚点的评级者差异和偏移对可靠评级的不一致性具有显著影响。在研究过程期间,研究中的约三分的一的患者具有超过一位评级者评估主要和次要终点。进行事后分析(排除来自安慰剂和d6-DM/Q治疗组的所有具有超过一位评级者的患者)以评估在整个研究期间,使用单个评级者评估相同终点的影响。这些发现结果概述于表12中。
即使患者群组较小,但在NSA-16和PANSS的多个终点中,对阴性症状的积极治疗作用增强。在SPCD组合第1阶段和第2阶段NSA-16总分(p=0.004)、NSA-16总体阴性症状评分(p=0.010)和NSA-16交流领域(p<0.001)中观测到统计显著治疗益处。
SPCD组合第1阶段和第2阶段总分(p=0.008)、阴性分量表(p=0.009)、PANSSMarder阴性因子(p=0.007)、一般精神病理学评分(p=0.049)和亲社会因子评分(p=0.009)的单个评级者分析还一致地表明在主要数据分析中发现的积极治疗作用的增强。
3.4.1.4.2对交流和表达的评估
患有精神分裂症的个体通常在语言和非语言交流方面具有严重表达缺陷。这种交流能力的缺损引起严重功能缺陷,导致适应性亲社会行为减少、社交孤立和退缩。进行PANSS以及NSA-16和MCCB中的测量交流和表达领域的特定因子的事后分析。研究中使用的仪器捕获由参与者使用的语言和非语言交流(Axelrod等人J Psychiatr Res.1993;27(3):253-8)。
在用d6-DM/Q治疗的患者中,存在关注交流和互动的子领域得到改善的强烈趋势。相信此变化的整体模式反映患者与他人互动和接洽的能力的改善,所述能力的缺损是精神分裂症的阴性症状的标志之一以及这些患者的长期社交退缩的关键原因。还在MCCB的注意力/警惕性子领域和NSA-16交流因子领域中显而易见对这些交流领域的改善的支持。这些结果呈现于表13中。
3.4.2个体应答数据的列表
测定以下的应答数据和其他相关研究信息的患者列表:
·随机化流程
·中止的患者
·方案偏差
·从分析排除的患者
·人口统计和基线特征数据
·并用药物
·依从性/药物浓度
·患者功效应答
3.4.3药物剂量、药物浓度以及与应答的关系
3.4.3.1药物剂量
用于随机分配或再随机分配至d6-DM/Q组的患者的剂量是基于d6-DM/Q-24/4.9QD保持1周、d6-DM/Q-24/4.9 BID保持1周和d6-DM/Q-34/4.9 BID保持4或10周的固定滴定流程。安全性群体中d6-DM/Q暴露的持续时间呈现于表14中。
表14.暴露持续时间-安全性群体
患者数目-年等于所有患者暴露持续时间(天数)的总和除以365.25。
SD=标准偏差。
3.4.3.2药物浓度和PK参数估算:d6-DM/Q
在基线(第1天)、第4次访视(第6周)和第7次访视(第12周)时测量d6-DM、d3-DX和Q的血浆浓度并且按代谢者子群和所有代谢者类型进行概述。所有患者的d6-DM的平均浓度为49.7ng/mL(d6-DM/Q/d6-DM/Q组,第4次访视[第6周]时)、53.2ng/mL(d6-DM/Q/d6-DM/Q组,第7次访视[第12周]时)和54.0ng/mL(安慰剂/d6-DM/Q组,第7次访视[第12周]时)。所有患者的d3-DX的平均浓度为101.2ng/mL(d6-DM/Q/d6-DM/Q组,第4次访视[第6周]时)、111.6ng/mL(d6-DM/Q/d6-DM/Q组,第7次访视[第12周]时)和124.5ng/mL(安慰剂/d6-DM/Q组,第7次访视[第12周]时)。所有患者的d3-3-MM的平均浓度为20.5ng/mL(d6-DM/Q/d6-DM/Q组,第4次访视[第6周]时)、19.4ng/mL(d6-DM/Q/d6-DM/Q组,第7次访视[第12周]时)和23.4ng/mL(安慰剂/d6-DM/Q组,第7次访视[第12周]时)。d6-DM、d3-DX和d3-3-MM的平均值随代谢者类型而变化。所有患者的Q的平均浓度为17.9ng/mL(d6-DM/Q/d6-DM/Q组,第4次访视[第6周]时)、20.1ng/mL(d6-DM/Q/d6-DM/Q组,第7次访视[第12周]时)和21.2ng/mL(安慰剂/d6-DM/Q组,第7次访视[第12周]时)。
在第4次访视(第6周)和第7次访视(第12周)时评估所有经随机分配以接受d6-DM/Q的患者的d6-DM、d3-DX和Q的C
3.4.3.3 d6-DM/Q PK参数与应答的关系
对于mITT群体,在第4次访视(第6周;C
3.4.4药物浓度:第二代抗精神病药(SGA)
根据研究参与标准,患者在基线时正在使用至少1种SGA。分析以下SGA的血浆浓度:阿立哌唑、鲁拉西酮、奥氮平、帕潘立酮、喹硫平、利培酮和齐拉西酮(ziprasidone)。对于经随机分配以在第1阶段和第2阶段中接受d6-DM/Q(d6-DM/Q/d6-DM/Q)、在第1阶段和第2阶段中接受安慰剂(d6-DM/Q/d6-DM/Q)或仅在第2阶段中接受d6-DM/Q(安慰剂/d6-DM/Q)的患者,按代谢者子群概述结果。在基线时使用每种SGA的患者的数目如下:阿立哌唑32名患者、鲁拉西酮6名患者、奥氮平36名患者、帕潘立酮18名患者、喹硫平23名患者、利培酮31名患者和齐拉西酮3名患者。
3.4.5药物-药物和药物-疾病相互作用
对mITT群体中的在基线时并用苯二氮
还对在基线时,MCCB综合评分<30的患者(表59)相对于MCCB综合评分≥30的患者(表60)进行主要终点的分析,以评估认知水平对患者NSA-16总分的影响。尽管任一个子群中安慰剂与d6-DM/Q之间都没有统计显著差异,但在基线时的MCCB评分≥30的子群中观测到NSA-16总分出现数值上更高的改善。在基线时的MCCB评分≥30的子群的标准效应值为-0.477(第1阶段)和-0.527(第2阶段),并且在基线时的MCCB评分≤30的子群的标准效应值为-0.226(第1阶段)和0.059(第2阶段)。
3.4.6患者显示
此研究中未报告患者显示数据,因为组平均值数据代表主要分析。
3.4.7功效
·通过SPCD分析,与安慰剂相比,在主要功效终点、NSA-16总分的自基线的变化中观测到d6-DM/Q的数值上更高的改善(SPCD加权Z统计=-1.79,p=0.073)。d6-DM/Q与安慰剂之间的LS平均治疗差为-1.79(95%CI-3.86,0.29)(第1阶段)和-1.28(95%CI-4.39,1.83)(第2阶段)。通过分析的SUR方法(p=0.048)和SPCD OLS ANCOVA LOCF数据(p=0.042)方法,在主要终点的敏感性分析中发现显著治疗作用。
·在PANSS总分(p=0.025)、PANSS阴性分量表(p=0.027)、PANSS Marder阴性因子(p=0.024)和PANSS亲社会因子(p=0.009)的SPCD分析中观测到与安慰剂相比,有利于d6-DM/Q的统计显著差异。在PANSS的阳性分量表(p=0.700)、兴奋成分(p=0.723)和一般精神病理学分量表(p=0.054)中观测到2个组之间的统计显著差异。
·还在NSA-16总体阴性症状评级的SPCD分析中观测到与安慰剂相比,有利于d6-DM/Q的统计显著差异(p=0.026)。d6-DM/Q与安慰剂之间的LS平均治疗差为-0.17(95%CI-0.40,0.07)(第1阶段)和-0.29(95%CI-0.61,0.03)(第2阶段)。
·在PGI-C量表中,在第1阶段中,27.7%的用d6-DM/Q治疗的患者(相对于24%的用安慰剂治疗的患者)在其症状变化中被评级为“显著改善”或“极显著改善”。在第2阶段中,34.4%的用d6-DM/Q治疗的患者(相对于13.3%的用安慰剂治疗的患者)在第12周时报告其症状得到“显著改善”或“极显著改善”(SPCD p=0.170)。在12周平行组mITT群体中,在第12周相对于基线被评级为‘显著改善’或‘极显著改善’的患者的比例为33.3%(14/42)(d6-DM/Q/d6-DM/Q组)和18.8%(6/32)(安慰剂/安慰剂组)(p=0.158,通过比例优势回归)。
·通过其他次要终点(包括CGI-S、CGI-C、CDSS总分、MCCB综合评分和NSA-4总分)的SPCD分析,在d6-DM/Q与安慰剂组之间未观测到统计显著差异,然而,观测到CGI-C评分和MCCB综合评分的有利于d6-DM/Q的数值上更高的改善。
·所有患者的d6-DM的平均浓度为49.7ng/mL(d6-DM/Q/d6-DM/Q组,第4次访视[第6周]时)、53.2ng/mL(d6-DM/Q/d6-DM/Q组,第7次访视[第12周]时)和54.0ng/mL(安慰剂/d6-DM/Q组,第7次访视[第12周]时)。d6-DM、d3-DX和d3-3MM的浓度随代谢者类型而变化。估算d6-DM、d3-DX和Q的PK参数。在使用这些所估算的PK参数的PK/药效学(PD)分析中,皮尔森相关系数值通常指示NSA-16总分的自基线的变化的弱相关性。
4安全性评估
4.1生命体征、体检结果以及其他与安全性相关的观测结果
4.1.1生命体征
测定生命体征实际值、自基线的变化和自基线的变化百分比。未观测到表明d6-DM/Q对所测量的生命体征值具有影响的趋势。
测定潜在临床上显著(PCS)生命体征异常的概述。各治疗组之间不存在PCS生命体征异常的发生率的有意义的差异。
4.1.2身体和神经检查
仅在筛选访视时计划对所有患者进行全面身体和神经检查。报告3名患者的体检的临床显著异常并且记录于患者的医疗史中。
4.1.3心电图
确定各治疗组的ECG参数以及这些参数的自基线的变化和自基线的变化百分比的概述。此外,测定在第1天和第6周时,ECG参数从给药前至给药后的变化。总体而言,ECG参数的自基线的变化较小并且不存在表明d6-DM/Q对ECG结果具有影响的趋势。
在所有安慰剂组或所有d6-DM/Q组中,不存在自基线的QTcF变化≥60msec的患者并且不存在PR间期>200msec的患者。总体而言,2名女性(1名在所有d6-DM/Q组中并且1名在所有安慰剂组中)符合QTcF间期中的ECG异常的PCS标准,其中基线后值在>460至≤485msec范围内,并且3名男性(1名在所有d6-DM/Q组中并且2名在所有安慰剂组中)符合QTcF间期中的ECG异常的PCS标准,其中基线后值在>450至≤480msec范围内。
两名患者在心脏病症SOC中报告AE;所有d6-DM/Q组中的1名患者(120-003)报告心动过速并且所有安慰剂组中的1名患者(119-004)报告心房纤颤。
4.1.4其他安全性观测结果
4.1.4.1哥伦比亚自杀严重程度评级量表(C-SSRS)
测定各次访视时的C-SSRS意念严重程度和各次访视时的C-SSRS自杀行为类型的概述。测定最严重的意念的强度。此外,测定各次访视时的致死性最高的尝试的实际和潜在致死性。总体而言,在安慰剂或d6-DM/Q治疗情况下都未观测到与自杀意念、严重程度和致死性相关的临床上有意义的信号。
4.1.4.2辛普森安格斯锥体外系症状量表(SAS)
各性别以及男性和女性组合的SAS的总分和个别项目评分的概述提供于表61中。此外,安全性群体以及女性和男性子群的SAS的个别项目的偏移表呈现于表62中。
总体而言,不存在指示用d6-DM/Q进行的治疗于在基线时患有锥体外系症状的患者中起到使此类症状恶化的作用的信号或趋势。
4.1.4.3巴恩斯静坐不能量表(BAS)
测定各性别以及男性和女性组合的BAS的总分和个别项目评分的概述。此外,测定安全性群体以及女性和男性子群的BAS的总体临床评估和个别项目的偏移表。
总体而言,不存在指示用d6-DM/Q进行的治疗于在基线时患有坐立不安症状的患者中起到使此类症状恶化的作用的信号或趋势。
4.1.4.4异常不自主运动量表(AIMS)
测定各性别以及男性和女性组合的AIMS的总分和分量表评分的概述。此外,测定安全性群体以及女性和男性子群的AIMS的个别项目的偏移表。
总体而言,不存在指示用d6-DM/Q进行的治疗于在基线时患有运动障碍症状的患者中起到使此类症状恶化的作用的信号或趋势。在用d6-DM/Q治疗的患者中未报告运动障碍的AE。所有安慰剂组中的一名患者报告运动障碍作为第2阶段中的AE。研究者认为所述AE为轻度并且可能与研究药物相关。
4.2安全性
·在此研究期间未报告死亡。
·总体而言,所有d6-DM/Q治疗组的30名(34.1%)患者和所有安慰剂治疗组中的39名(40.6%)患者报告≥1TEAE。TEAE的严重程度是轻度至中度。
·所有d6-DM/Q组或所有安慰剂组中最频繁(>2名患者)报告的TEAE分别为口干(4.5%对比1.0%)、腹泻(2.3%对比3.1%)、头晕(3.4%对比3.1%)、头痛(2.3%对比5.2%)、嗜睡(1.1%对比3.1%)和鼻咽炎(3.4%对比4.2%)。
·所有d6-DM/Q和所有安慰剂治疗组中所报告的治疗相关TEAE分别为13.6%和18.8%。所有d6-DM/Q组中的≥2名患者中报告的治疗相关TEAE包括口干(2.3%)和头痛(2.3%),并且在所有安慰剂组中,这些治疗相关TEAE包括头痛(3.1%)、头晕(2.1%)、嗜睡(3.1%)、腹泻(2.1%)和镇静(2.1%)。
·所有安慰剂组中的2名患者并且所有d6-DM/Q组中无患者报告SAE。2种SAE是精神分裂症恶化和泌尿道感染。在经历精神分裂症恶化的SAE的患者中,在其住院后中止研究药物并且不再恢复使用。在经历泌尿道感染的SAE的患者中将研究药物中断一天。此患者完成研究。认为两种SAE都与研究药物无关。
·总体而言,10名患者由于TEAE而中止研究;所有d6-DM/Q组中有2名患者并且所有安慰剂组中有8名患者。所有d6-DM/Q组中的引起研究中止的TEAE是视力模糊和皮疹,并且在所有安慰剂组中,引起研究中止的TEAE是眼痛、皮疹、心房纤颤、头晕、头痛、精神分裂症和勃起功能障碍。
·在所有安慰剂组或所有d6-DM/Q组中,不存在自基线的QTcF变化≥60msec的患者并且不存在PR间期>200msec的患者。
·患者在基线时,在SAS(锥体外系症状)、BAS(坐立不安)和AIMS(运动障碍)中具有低评分,并且在研究过程期间不存在指示用d6-DM/Q进行的治疗起到使此类症状恶化的作用的AE事件或趋势。
5结果
在具有精神分裂症的阴性症状的患者中进行的此研究中,在历史上认可用于评估精神分裂症的阴性症状的多种经良好验证且可靠的仪器中观测到d6-DM/Q的恒定功效信号。尽管主要终点,NSA-16总分的变化未达到统计显著性,但存在改善的趋势(p=0.073)。在特异性地评估阴性症状的若干功效评级量表的组合(第1阶段和第2阶段)SPCD mITT分析中,与安慰剂相比,用d6-DM/Q治疗的患者经历显著更大的自基线的改善。这些改善包括NSA-16总体阴性症状评分(p=0.026)、PANSS Marder阴性因子(p=0.024)和PANSS阴性因子评分(p=0.027)。在PANSS表达缺陷领域(p=0.034)和PANSS经验缺陷领域(p=0.022)、PANSS亲社会因子评分(p=0.009)中存在与安慰剂治疗相比有利于d6-DM/Q的显著改善,以及对PANSS一般精神病理学分量表评分(p=0.054)具有积极作用的趋势。PANSS总分的改善是由阴性症状和一般精神病理学分量表的改善促成。患者具有低基线PANSS阳性评分并且在研究期间显示极小的变化,因此所述发现结果不具有假特异性。
对于NSA-16总分、PANSS总分、PANSS Marder和PANSS阴性,各阶段的标准化效应值(绝对值)在0.30至0.75范围内(数据在档案中),表明即使在一个结果不统计显著时,在与其他经认可的神经科学化合物相比时仍观测到有意义的治疗作用。
为了测定评级者变化性对功效终点的影响,针对NSA-16和PANSS进行在整个研究期间始终由同一名评级者进行评级的患者的事后分析。此事后分析的结果具有高显著性并且使若干结果量度增强,包括NSA-16总分(p=0.004)、NSA-16总体阴性症状评分(p=0.010)以及PANSS Marder阴性因子(p=0.007)和阴性因子评分(p=0.009)。此分析包括mITT群体中的总共87名患者。
在共识声明中,Schooler和同事(Schooler等人Schizophr Res.2015;162(1-3):169-174)提出对精神分裂症的阴性症状的有意义的治疗益处将是“对评估阴性症状的有效且可靠的量度的显著改善,大于或等于总分的20%”。在此研究(实施例1)中,使用此20%或更大改善的阈值进行PANSS阴性分量表和PANSS Marder阴性因子评分的分析。
与安慰剂相比,显著更高的百分比的经d6-DM/Q治疗的患者显示PANSS Marder阴性因子评分的20%或更大的自基线的改善(第1阶段:21.3%对比16.3%;第2阶段:27.3%对比3.3%;p=0.012)。在两个阶段中,与安慰剂相比,d6-DM/Q组中的PANSS阴性分量表相比基线降低20%的患者的比例还较高(第1阶段:23.4%对比13.8%;第2阶段:21.2%对比10%;SPCD p=0.054)。
使用患者报告的结果量度,PGI-C,在第1阶段结束时,在PGI-C中具有‘显著改善’或‘极显著改善’评级的患者的比例为27.7%(用d6-DM/Q治疗的患者)和24%(用安慰剂治疗的患者)(在第2阶段,相对于基线的第12周)。在第2阶段中,与安慰剂相比,存在高超过2倍的百分比的使用d6-DM/Q的患者的从基线至研究结束时的症状评级为“极显著”或“显著”改善(34.4%对比13.3%)。在12周平行组mITT群体中,在第12周相对于基线被评级为‘显著改善’或‘极显著改善’的患者的比例为33.3%(14/42)(d6-DM/Q/d6-DM/Q组)和18.8%(6/32)(安慰剂/安慰剂组)(p=0.158,通过比例优势回归)。在处于疾病的残余状态并且使用稳定剂量的非典型抗精神病药(约3个月)的此稳定的患者群体中,PGI-C可以可靠的方式用于支持临床上有意义的信息。
经由NSA-16总体阴性症状严重程度评分评估临床医师对患者的阴性症状的总体严重程度的观测,而通过CGI-S和CGI-C评估疾病的整体水平。观测到对NSA-16总体阴性症状严重程度评分的统计显著益处(SPCD;p=0.026)。尽管通过CGI-C和CGI-S量度未观测到d6-DM/Q与安慰剂之间存在统计显著差异,但结果指示d6-DM/Q具有优于安慰剂的益处的趋势。与CGI-C/S相比,在NSA-16总体阴性症状评分中评估的不同锚点和疾病类型可说明通过NSA-16总体阴性症状评分而发现的更强的统计显著结果。与正常健康年轻人相比,NSA-16总体对阴性症状具有极高特异性,而CGI-C/S使用患者本身或其他患有精神分裂症的患者作为锚点并且对整体疾病而非阴性症状具有特异性。CGI-C/S的评估目标症状学的严重程度和变化的经修改的版本可提供更有意义的信息并且可在未来研究中考虑。
还存在认知改善的趋势,如通过MCCB综合评分和MCCB的注意力/警惕性领域评估,其中在研究的第2阶段发现显著益处。显示认知改善的能力已成为本领域中的另一困难障碍,并且在本文中,似乎存在值得在其他研究中探索的针对认知益处的潜力。
在此研究(实施例1)中,存在表明与安慰剂相比,在用d6-DM/Q治疗的患者中的关于PANSS的特定因子中的语言和非语言交流的方面的功效的信号。这些信号还在MCCB的注意力/警惕性子领域和NSA-16交流因子领域中显而易见。
患者具有低基线抑郁症症状(通过卡尔加里精神分裂症抑郁量表评估)和锥体外系症状并且在整个研究期间未经历这些症状的显著变化,证实研究结果关于阴性症状的特异性。通常,d6-DM/Q在此研究(实施例1)中是安全且良好耐受的,具有符合相同化合物的其他临床试验的安全性概况。所有d6-DM/Q组或所有安慰剂组中最频繁(>2名患者)报告的TEAE分别为口干(4.5%对比1.0%)、腹泻(2.3%对比3.1%)、头晕(3.4%对比3.1%)、头痛(2.3%对比5.2%)、嗜睡(1.1%对比3.1%)和鼻咽炎(3.4%对比4.2%)。在研究中未报告死亡并且所有d6-DM/Q组中未出现严重AE。
总体来说,来自此研究(实施例1)的结果证实广泛范围的可靠且经验证的量度的功效的恒定、积极信号,表明用d6-DM/Q进行的辅助治疗可提供用于表现出阴性症状的稳定的精神分裂症患者的安全、有效且临床上有意义的治疗选择方案。d6-DM/Q的安全性概况是有利的并且支持此适应症的进一步研究。
用于评估d6-DM/Q(氢溴酸氘化右美沙芬[d6-DM]/硫酸奎尼丁[Q])治疗精神分裂症的阴性症状的功效、安全性和耐受性的多中心、随机化、双盲、安慰剂对照、平行臂研究的示例性参数
在来自实施例1的研究中,与使用安慰剂的患者相比,用34/4.9mg每天两次(BID)的剂量的d6-DM/Q治疗的患者显示精神分裂症的阴性症状的改善。d6-DM/Q在该研究中是安全且良好耐受的。
为了评估较高剂量的d6-DM/Q(42.63/4.9mg)的功效、安全性和耐受性,在随机化治疗期的第一周期间,从每天一次至两次d6-DM28mg/Q 4.9mg(d6-DM/Q-28/4.9)的d6-DM/Q的初始剂量对随机分配至d6-DM/Q组的患者进行滴定;接着,患者在随机化治疗期的其余时间接受每天两次口服施用的d6-DM 42.63mg/Q 4.9mg(d6-DM/Q-42.63/4.9)。
d6-DM/Q-42.63/4.9 BID剂量与d6-DM暴露相关,所述暴露在d6-DM/Q的1期和2期研究中通常证实具有良好耐受性的范围内并且在其中受体结合足以测试有效性假设的范围内。跨越多种适应症研究此较高剂量的d6-DM/Q 42.63/4.9mg BID,迄今为止,d6-DM/Q临床开发程序中未出现新的安全性信号。
12周治疗持续时间确保暴露于d6-DM/Q的目标最佳剂量达到足以观测治疗应答以及评估应答的持续时间的时间段。包含国家心理卫生研究所(NIMH)、FDA、学院和行业的代表的共识组已鉴别最小12周治疗持续时间适于设计精神分裂症的阴性症状的研究(Laughren和Levin,Schizophr Bull.2006;32(2):220-222)。
1研究性计划
1.1整体研究设计
这是用于评估d6-DM/Q在具有精神分裂症的阴性症状的患者中的功效、安全性和耐受性的多中心、随机化、双盲、安慰剂对照研究的可能的设计。研究包括至多4周筛选期、12周双盲治疗期和30天随访期。多达370名患者参与。
筛选期(第-28天至第-1天)
筛选期是28天(4周)并且在获得同意书时开始。如果需要,则可由医学监测员请求延长至多额外14天(最多总共42天)以满足合格性要求。
双盲治疗期
患者以1∶1比率经随机分配以在过程中接受d6-DM/Q或匹配安慰剂胶囊。随机分配至d6-DM/Q组的患者开始一周滴定期,其中其在前3天每天上午(qam)接受d6-DM 28mg/Q4.9mg(d6-DM/Q-28/4.9)的给药并且在每天晚上(qpm)接受安慰剂的给药,接着在接下来的4天接受d6-DM/Q-28/4.9 BID。在此一周滴定期之后,患者在双盲治疗期中的其余11周接受每天两次口服施用的d6-DM 42.63mg/Q 4.9mg(d6-DM/Q-42.63/4.9)。
随访期
在最后一次访视(第5次访视[第12周]或提前终止[ET])后开始30天随访期。在第16周时通过电话联系患者以收集在最后一次施用研究药物之后的30天的不良事件(AE)和并用药物信息。
评估和访视:
患者在筛选、第1次访视(第1天(基线))以及在第3周、第6周、第9周和第12周或在ET时参加临床访视。在第1周、第4周、第7周、第10周以及在第12周之后的第30天或在ET访视时进行电话呼叫。
在每次访视时进行研究程序,如评估和访视时间表中所概述(表16)。主要功效量度是PANSS Marder阴性因子评分。次要功效量度包括:NSA-16总体阴性症状评分、患者总体印象-严重程度(PGI-S)和患者总体印象-变化(PGI-C)。其他结果量度包括:PANSS阳性分量表和卡尔加里精神分裂症抑郁量表(CDSS)。在所有访视时进行NSA-16,其中评估NSA-16总体,然而NSA-16并非研究中的功效量度。
由在第2次、第4次和第6次访视(或ET)时收集的样品测量d6-DM、Q和某些代谢物的血浆浓度的药物动力学(PK)测量值。
通过所报告的AE、体检、生命体征、临床实验室测量、12导联心电图(ECG)和以下量表评估d6-DM/Q的安全性和耐受性:哥伦比亚自杀严重程度评级量表(C-SSRS)、异常不自主运动量表(AIMS)、巴恩斯静坐不能量表(BAS)和辛普森安格斯锥体外系症状量表(SAS)。
1.2患者数目
多达370名患者参与。
1.3治疗分配
基于由赞助商提供的随机化流程随机分配(1∶1比率)治疗(12周、双盲、治疗期;d6-DM/Q或安慰剂)。
1.4研究评估和程序
研究评估和程序的说明提供于章节8中。
2患者的选择和退出
符合条件的患者是成年门诊患者、年龄在18与60岁之间、经诊断患有精神分裂症、展示精神分裂症的阴性症状的证据、临床上稳定并且符合以下章节提及的所有包含标准并且不符合所有排除标准。
由研究者使用6.0.0或7.0.2版简明国际神经精神访谈(M.I.N.I.)(分别为附录11A或11B),根据精神病症诊断与统计手册第5版(DSM-V)进行初始诊断评估以评估患者是否符合精神分裂症的诊断标准。
2.1患者包含标准
1.在签署知情同意书时年龄为18至60岁(包括端点)的男性和女性。
2.由7.0.2版M.I.N.I确认符合精神分裂症的DSM-V诊断标准的患者,在筛选之前至少1年发作并且已在临床上稳定至少6个月(未发生精神病院入院或急性加重)。在与医学监测员商讨后允许在筛选之前的最近6个月内由于社交原因而住院的患者,但排除当前住院患者。
3.患者需要在筛选之前至少30天处于稳定生活状态。
4.患者在筛选之前应用除氟氮平以外的第二代非典型抗精神病药物(SGA),以任何经批准的剂型治疗至少3个月并且在筛选之前保持稳定剂量至少30天;患者必须保持临床稳定的(以主要研究者的观点)至第1次访视并且不可用超过一种SGA治疗(除用于失眠的低剂量喹硫平(至多在晚上使用50mg)以外)。
5.根据研究者的判断,阴性症状已存在至少6个月。
6.患者必须具有以下评分:
a.在PANSS项目P1:妄想;P3:幻觉;P6:猜疑/被害;和P7:敌对性上的评分≤4;和
b.在PANSS的以下项目中的任2项上的评分≥4(中度)或在任1项上的评分≥5:N1:情感迟钝;N2:情绪退缩;N4:被动/淡漠社交退缩;或N6:交谈缺乏自发性/流畅性。
7.性活跃的具有生育潜力的女性在试验过程期间必须使用有效避孕方法并且在研究药物的最后一次给药之后保持至少30天。为了最小化一种避孕方法失败的风险,必须使用以下防护措施中的两者:输精管结扎、输卵管结扎、阴道子宫帽、子宫内装置、避孕丸、避孕储槽式注射剂、避孕植入物或具有杀精子剂的保险套或具有杀精子剂的海绵。周期性禁欲(例如日历法、排卵法、症状体温法、排卵后方法)、宣称在暴露于研究药物的持续时间内禁欲或停药并非可接受的避孕方法。不育(即,卵巢切除和/或子宫切除)、停经(连续12个月无月经,无替代性医学原因)或实践真实禁欲(当此方法符合患者的优选和常用生活方式时)免除此要求。
8.允许并用抗抑郁剂,诸如选择性血清素再吸收抑制剂(SSRI;例如氟西汀、舍曲林、西酞普兰)和血清素-去甲肾上腺素再吸收抑制剂(SNRI;例如文拉法辛、去甲文拉法辛、度洛西汀、沃托西汀、维拉唑酮),只要在筛选之前剂量已保持稳定3个月(90天)、在整个研究期间保持稳定并且所用剂量在该药物的美国说明书的指导范围内即可。允许使用帕罗西汀,一种细胞色素P450(CYP)2D6底物,前提条件是剂量不超过10毫克/天。
9.允许在就寝时并用安眠药(例如5至10mg剂量的唑吡坦或等效物)以用于失眠的夜间治疗,前提条件是在第1次访视之前剂量已保持稳定至少1个月(30天)并且在整个研究期间保持稳定。
10.使用多达每天2mg的总剂量的劳拉西泮以用于治疗失眠、焦虑症、坐立不安或躁动的患者。患者应在第1次访视之前保持稳定剂量达至少1个月(30天)并且剂量应在研究持续时间内保持稳定。
11.在已完全说明参与研究的性质和风险之后,愿意签署患者知情同意书(ICF)并且接收其复本。
12.充分理解和合作以实现遵守所有程序和评估。
13.患者必须具有可靠的信息提供者(例如病例管理人员、社会工作者、家庭成员)。以研究者的观点,信息提供者应与患者一起度过足够长的时间以能够处理行为、活动和症状。研究者(或指定人员)应在其初始患者评估时(或在筛选周期期间)处理此关系。
2.2患者排除标准
如果患者符合以下标准中的任一者,则其不参与研究:
1.当前患有重性抑郁障碍(MDD)和/或在筛选访视时CDSS评分≥6的患者。
2.诊断患有分裂情感性障碍或双相障碍的患者。
3.在第1次访视之前的3个月(90天)内具有氯氮平使用史的患者。在研究期间不允许使用氯氮平。
4.基于研究者判断,具有继发于正在使用的抗精神病药物的假性帕金森症的患者。
5.在筛选期间,在辛普森-安格斯量表(SAS)的前八个项目的总和上的评分>3的患者。
6.在第1次访视之前的3个月(90天)内或在研究持续时间期间用任何典型抗精神病药(例如氟哌啶醇、氯丙嗪等)治疗的患者。
7.在第1次访视之前的1个月(30天)内使用抗胆碱能药物治疗与抗精神病药物相关的AE的患者。
8.使用左旋多巴的患者。
9.在筛选时具有不可检测的水平的SGA的患者。在筛选时具有次治疗水平的SGA的患者由医学监测员审查合格性。
10.使用除劳拉西泮以外的任何苯二氮
11.在筛选或第1次访视时具有以下心血管病史或结果中的任一者的患者:
a.完全性心脏传导阻滞、心室性心动过速、如由中央读取器评估的临床上显著的心室性早期收缩(PVC)的存在、QTc延长或尖端扭转型心室心动过速的病史或证据。
b.基于筛选访视时的中央审查,在筛选时使用弗氏公式的QTc(QTcF)对于男性而言>450msec并且对于女性而言>470msec,除非归因于心室起搏。
c.先天性QT间期延长综合征的任何家族病史。
d.临床上显著的晕厥、直立性低血压或体位性心动过速的病史或存在。
12.患者当前患有临床上显著的神经、肝、肾、代谢、血液学、免疫、心血管、肺或胃肠道疾病,诸如心肌梗塞、充血性心脏衰竭、HIV血清反应呈阳性状态/获得性免疫缺陷综合征或慢性乙型或丙型肝炎的任何病史。较小或良好控制的医学疾患可视为可接受的,前提条件是所述疾患在试验过程期间不会使患者暴露于显著不良事件的不当风险或者干扰安全性或功效的评估。在其中研究者不确定患者的医学疾患的稳定性以及所述疾患在参与试验时的潜在影响的任何情况下应联系医学监测员。
13.对DM、d6-DM、Q、鸦片药物(可待因等)或研究药物的任何其他成分具有已知的超敏反应的患者。
14.在第1次访视之前的3个月(90天)内接受与Q共同施用的DM的患者。
15.曾经接受与Q共同施用的d6-DM或参与来自实施例1的研究的患者。
16.患有重症肌无力(Q的禁忌症)的患者。
17.在第1次访视之前的2周内用单胺氧化酶抑制剂(MAOI)治疗的患者。在整个研究期间禁止MAOI直至研究药物中止后的第2周。
18.在第1次访视之前的2周或5个半衰期(以较长者为准)内使用禁止并用药物的患者。
19.使用休闲或药用大麻的患者,如由筛选时的大麻尿液药检呈阳性所证明。
20.在C-SSRS自杀意念项目4(有某些行动意图但无具体计划的主动自杀意念)上回答“是”并且最近一次符合此C-SSRS项目4的标准的发作在最近6个月内发生的患者,或者
在C-SSRS自杀意念项目5(有具体计划和意图的主动自杀意念)上回答“是”并且最近一次符合此C-SSRS项目5的标准的发作在最近6个月内发生的患者,或者
在5个C-SSRS自杀行为项目(实际尝试、被中断的尝试、放弃的尝试、预备的行动或行为)中的任一项上回答“是”并且最近一次符合这5个C-SSRS自杀行为项目中的任一项的标准的发作在最近2年内发生的患者,或者
以研究者的观点,具有严重的自杀或杀人风险的患者。
21.在筛选访视时,具有临床上显著的实验室异常(血液学、化学和尿分析)或者具有临床潜在关注的安全性值或者天冬氨酸转氨酶(AST)或丙氨酸转氨酶(ALT)>2x正常值上限(ULN)的患者。
22.以研究者的观点,不愿意或不能遵守研究指示。
23.在筛选之前的6个月内有物质和/或酒精滥用史的患者。允许使用所有烟草和烟碱产品。
24.乙型肝炎表面抗原、丙型肝炎抗体或HIV抗体测试呈阳性的患者。
25.在筛选之前的一年内接受电痉挛治疗(ECT)、重复经颅磁刺激(rTMS)或深部脑刺激(DBS)的患者。
26.处于哺乳期、怀孕或计划怀孕的女性。
27.在临床试验受试者数据库(CTS数据库)中发现与在第1次访视之前的30天内参与另一项介入性药物或装置研究的患者“几乎确定”匹配的患者。
2.3任选的患者包含和排除标准
在以上包含标准1中,6个月未住院周期可缩短至4个月周期。
以上包含标准6(a)可由以下包含标准替换:患者在PANSS项目P1:妄想;P3:幻觉;和P7:敌对性上的评分必须≤4。
以下其他包含标准可适用:患者在筛选和第1次访视时的PANSS Marder阴性因子评分必须≥20并且2次访视之间的绝对差<4分。PANSS Marder阴性因子:N1:情感迟钝;N2:情绪退缩;N3:交流障碍;N4:被动/淡漠社交退缩;N6:交谈缺乏自发性/流畅性;G7:行动迟缓;G16:主动回避社交。
以上包含标准9和10可由以下包含标准替换:允许在就寝时并用安眠药(例如右佐匹克隆、嗦吡旦(solpidem)、扎来普隆、曲唑酮[多达100毫克/天])以用于失眠的夜间治疗,前提条件是在基线之前剂量已保持稳定至少1个月(30天)并且在整个研究期间保持稳定。在参与研究之前使用劳拉西泮治疗焦虑症、坐立不安或躁动的患者在研究持续时间应保持相同治疗方案。除用于失眠和行为障碍的短期或必要治疗的劳拉西泮以外,不允许使用任何其他苯二氮
以上排除标准12可由以下排除标准替换:患有可混淆研究的安全性结果的说明的并发性的临床上显著或不稳定的全身性疾病(例如恶性疾病[除皮肤基底细胞癌或未经治疗的前列腺癌以外]、控制不佳的糖尿病、控制不佳的高血压、肺不稳定、肾或肝疾病、不稳定的局部缺血性心脏疾病、扩张型心肌病或不稳定的心脏瓣膜病)、认知和其他神经退化性病症的患者。可由研究者和医学监测员个别地评估一些情况。
以下其他排除标准可适用:当前正在参与或在基线之前的30天内参与其他介入性(药物或装置)临床研究的患者。
2.4患者信息提供者
应认识到,具有阴性症状的患者由于其疾病而通常难以发展与他人的关系。然而,关于数据准确性,重要的是,信息提供者与患者足够熟悉以能够报告其行为、活动和症状以及这些项目的任何变化。信息提供者每周应与患者一起度过足够长的时间以能够准确地报告这些项目。尽管无特定需求,但要求研究者记录信息提供者与患者之间的关系(即,家庭成员、社会工作者或病例管理人员)并且指定关系长度。此数据由医学监测员审查并且在研究的合格性过程中考虑信息提供者关系。
2.5患者退出标准
口头并且在书面ICF中告知患者和照护者,他们具有在任何时间退出研究的权力而不会遭受偏见或损失其在其他方面获取的权益。研究者或赞助商可在以下情况下中止患者参与研究:间发病、不良事件、其他与患者的健康或康乐有关的原因、患者的影响其继续参与研究的能力的理解或认知功能的降低,或在缺乏合作、无依从性、违反协议或其他行政原因的情况下。如果患者未返回进行预定访视,则应尽最大努力联系患者/照护者。与情况无关,应尽最大努力记录患者结果。研究者应询问退出原因,要求患者/照护者返还所有未使用的研究药物并且针对任何未解决的不良事件随访患者/照护者。
此外,在随机化之后的任何时间呈现QTc间期(QTcF)>500msec(除非归因于心室起搏)或相比给药前基线ECG的QTcF间期变化>60msec的患者退出研究。针对临床显著性评估并且记录QTcF值。
3患者的治疗
3.1研究药物的说明
d6-DM/Q以含有活性成分d6-DM和Q的立即释放、蓝色、不透明、经印刷的硬明胶胶囊(3号尺寸)形式提供。非活性成分是交联羧甲纤维素钠、微晶纤维素、胶态二氧化硅和硬脂酸镁。研究药物的每个胶囊含有以下中的一者:
·d6-DM/Q-28/4.9:28mg d6-DM和4.9mg Q
·d6-DM/Q-42.63/4.9:42.63mg d6-DM和4.9mg Q
·匹配安慰剂:外观与d6-DM/Q胶囊相同;仅含有非活性成分
以盲式(双盲)、单独、预先标记的泡壳包装卡形式向研究点提供药物供应器。
根据良好生产规范(GMP)指南、国际协调理事会(International Council onHarmonisation;ICH)、良好临床实验规范(Good Clinical Practice;GCP)指南以及适用的法律和法规制备、封装和标记研究药物。
3.2研究药物的施用
研究药物的施用如下。
双盲、随机化治疗期:
患者以1∶1比率随机分配(按安慰剂-应答者状态和中心分级)以接受口服施用的d6-DM/Q或匹配安慰剂胶囊。
随机分配至d6-DM/Q的患者具有以下固定滴定时间表:前3天d6-DM/Q-28/4.9qam,随后的4天d6-DM/Q-28/4.9 BID,并且接着在第八天开始d6-DM/Q-42.63/4.9 BID至剩余11周。
在双盲治疗期期间的研究药物的施用如下:
·第1天-第3天:每天上午d6-DM/Q-28/4.9胶囊;每天晚上施用匹配安慰剂
·第4天-第7天:d6-DM/Q-28/4.9胶囊,BID
·第8天-第85天:d6-DM/Q-42.63/4.9胶囊,BID
·第22天-第106天:匹配d6-DM/Q安慰剂胶囊,BID
研究药物的封装描述于章节4.2中。
3.3并用药物
患者在研究期间或在第1天开始给药之前的2周或5个半衰期(以较长者为准)内不可使用章节3.3.2中列举的禁止药物中的任一者。在每次访视时,询问患者是否使用任何并用药物,如果使用,研究者记录所使用的药物及其使用原因。
d6-DM/Q含有硫酸奎尼丁(Q),其是P-糖蛋白抑制剂。与地高辛(digoxin)(一种P-糖蛋白底物)一起用于心脏适应症的以较高剂量进行的Q的并用施用导致血浆或血清地高辛浓度最高可翻倍;应密切监测并用地高辛的患者中的地高辛浓度并且根据需要降低剂量。
在前药的作用是由细胞色素P450同功酶2D6(CYP2D6产生的代谢物,例如可待因和氢可酮,其镇痛和止咳作用似乎分别由吗啡和氢吗啡酮介导)介导的情况下,由于Q介导的CYP2D6抑制,在存在d6-DM/Q的情况下有可能无法实现所需临床益处。应考虑替代性治疗。
3.3.1允许的并用药物,苯二氮
允许的并用药物由研究者评估并且根据需要由医学监测员论述以确定在研究期间使用是否会存在任何问题。
允许在就寝时并用安眠药(例如5至10mg剂量的唑吡坦或等效物)以用于失眠的夜间治疗,前提条件是在第1次访视之前剂量已保持稳定至少1个月并且在整个研究期间保持稳定。使用多达每天2mg的总剂量的劳拉西泮以用于治疗失眠、焦虑症、坐立不安或躁动的患者应在第1次访视之前保持稳定剂量达1个月(30天)并且在研究持续时间内保持相同治疗方案。
除用于焦虑症、失眠和/或行为障碍的短期或“根据需要”治疗的劳拉西泮以外,不允许使用任何其他苯二氮
在任何计划功效或安全性量表评估(包括锥体外系症状(EPS)量表)之前的8小时内不应施用苯二氮
允许并用抗抑郁剂,诸如SSRI(例如氟西汀、舍曲林、西酞普兰)和SNRI(例如文拉法辛、去甲文拉法辛、度洛西汀、沃托西汀、维拉唑酮),只要在筛选之前剂量已保持稳定3个月(90天)、在整个研究期间保持稳定并且所使用的剂量在该药物的美国说明书的指导范围内即可。允许使用帕罗西汀,一种CYP2D6底物,前提条件是剂量不超过10毫克/天。
3.3.2禁止药物
禁止并用药物包括可潜在地改变Q和/或d6-DM的血浆水平的药物,或与Q相关的药物。禁止药物的实例的列表提供于以上表3中。
在研究期间或在研究药物给药(第1天)的2周或5个半衰期(以较长者为准)内,患者不可使用表3中列举的禁止药物中的任一者。在整个研究期间禁止使用单胺氧化酶抑制剂(MAOI)。使用MAOI的患者应在研究药物的第一次给药之前的至少14天以及在研究药物的最后一次剂量之后的至少14天内不使用MAOI。
研究中不允许使用左旋多巴。在研究期间不允许使用休闲和/或药用大麻。
3.4治疗依从性
在治疗期间的每次研究访视期间指示每位患者返还未使用的研究药物和空的药物泡壳包装卡。研究点工作人员进行胶囊计数并在药物责任日志中记录依从性并且将信息输入eCRF中。
在胶囊计数和患者访谈之后评估研究药物依从性;依从性定义为摄取研究药物的计划剂量的至少80%(依从性范围:80%至120%)。
使用另一探索工具(AiCure)帮助确保患者依从性;然而,测量依从性的主要方法是由研究点工作人员进行的胶囊计数。
3.5随机化和盲式处理
在进入研究后(在筛选时签署ICF之后),对每位患者分配9位数患者编号。前6个数字由国家代码中心编号组成;最后3个数字由交互式网络应答系统(IWRS)以001开始依序分配。此9位数编号是每位患者的主要标识符。
患者以1∶1比率经随机分配以接受d6-DM/Q胶囊或匹配安慰剂胶囊(双盲方式)。由赞助商或指定人员设计随机化流程并且在IWRS内管理。每位患者有50%的机率接受d6-DM/Q。
4研究药物材料和管理
4.1研究药物
d6-DM/Q以含有活性成分d6-DM(28mg或42.63mg)和Q(4.9mg)的立即释放、蓝色、不透明、经印刷的硬明胶胶囊(3号尺寸)形式提供。d6-DM/Q和匹配安慰剂胶囊的组成描述于章节3.1中。
4.2研究药物封装和标记
封装
每个研究药物药盒是单独标记的泡壳包装卡,其预先封装用于3周治疗加额外一周供应。在泡壳包装卡内存在四块板,每块板由2列泡壳条组成,一列是上午剂量(如AM所指示),一列是晚上剂量(如PM所指示)以用于1周供应。
4.3研究药物储存
临床供应器必须按照标记要求储存在安全位置并且保持在室温下;25℃(77°F),允许偏移15℃至30℃(59°F至86°F)。
4.4研究药物施周
每位患者根据其由IWRS随机化流程分配的药品标识号(随机化编号)接受研究药物。每个研究点的指定工作人员分配研究药物泡壳包装卡。在适用研究访视当天,患者在存在研究点人员的情况下在诊所使用其研究药物的上午剂量(与当天时间无关),除此以外患者自行施用研究药物。
指示每位患者每天两次与水一起口服摄取1粒研究药物胶囊,约每12(±4)小时一次(上午和晚上)(每天2粒胶囊)。对于每位患者,在整个双盲治疗期间,每次摄取研究药物的剂量的时间应保持一致。在AiCure药物依从性监测平台中记录研究药物剂量。
所有研究药物以双盲方式供应和施用。
5功效评估
功效评估包括作为主要评估的PANSS Marder阴性因子评分以及作为次要终点的NSA总体阴性症状评分、患者总体印象-严重程度(PGI-S)和患者总体印象-变化(PGI-C)。其他结果量度包括PANSS阳性分量表和卡尔加里精神分裂症抑郁量表(CDSS)。这些量表描述于以下章节中。
5.1阳性和阴性综合征量表(PANSS)Marder阴性因子评分
PANSS(附录2)是经验证的临床量表,其已广泛用作精神分裂症的阴性和阳性症状的可靠和有效量度(Daniel,Schizophr Res.2013;150(2-3):343-5)。所述量表包含30个不同的项目,这些项目共同评估精神分裂症的阳性和阴性综合征,包括其彼此之间的关系以及与总体精神病理学的关系。各项目以“1”(无)至“7”(极重度)评分。
PANSS的现有心理测量特性使得能够评估阳性、阴性和一般精神病理学作为精神分裂症的类别或维度视角的一部分(Kay等人Schizophr Bull.1987;13(2):261-276;Kumari等人J Addict Res Ther.2017;8(3))。因此,通常以因子结构形式分析项目的不同组合以对精神分裂症的阴性综合征的特定方面进行评分。
PANSS的五因子解决方案的概念已成功地用于临床试验中,并且鉴别精神分裂症的五个因子或维度:1)阴性症状;2)阳性症状;3)思维混乱;4)不受控的敌对性/兴奋;和5)焦虑症/抑郁症(Lindenmayer等人Psychopathology.1995;28(1):22-31;Muder等人J ClinPsychiatry.1997;58(12):538-546)。Marder在两项受控试验中研究五因子解决方案并且发现与氟哌啶醇相比,利培酮对所有五个维度产生显著更大的改善,其中与氟哌啶醇相比,利培酮对因子1(阴性症状)具有尤其强效的作用。因子1,即,PANSS Marder阴性因子,相对于阴性分量表具有若干个经改善的内容效度的方面,更一致地符合在2006 NIMH-MATRICS共识声明中鉴别的领域(Kirkpatrick等人Schizophr Bull.2006;32(2):214-219)。
PANNS Marder阴性因子评分
PANSS Marder阴性因子评分是精神分裂症的阴性症状的可靠且经验证的量度,并且包含30项目型PANSS中的以下7个项目:
Marder阴性因子:
1.N1:情感迟钝
2.N2:情绪退缩
3.N3:交流障碍
4.N4:被动/淡漠社交退缩
5.N6:交谈缺乏自发性/流畅性
6.G7:行动迟缓
7.G16:主动回避社交
PANSS Marder阴性因子中的每一者与阴性症状的五个主要领域中的一者相关(Kirkpatrick等人Schizophr Bull.2006;32(2):214-219)。PANSS项目N1:情感迟钝,与情感迟钝相关;N6:交谈缺乏自发性/流畅性,与失语症相关;而N4:被动/淡漠社交退缩;G16:主动回避社交;和N3:交流障碍,是与无社会性相关的因子。PANSS项目N2:情绪退缩,与快感缺乏相关;并且G7:行动迟缓,与快感缺乏和无意志相关(Daniel,Schizophr Res.2013;150(2-3):343-5)。
PANSS Marder阴性因子评分中的两个项目(N4和G16)仅基于从信息提供者获得的信息。患者需要鉴别可靠的信息提供者(例如病例管理人员、社会工作者、家庭成员),其与患者一起度过足够长的时间以能够向PANSS评级者提供信息。
PANSS阳性分量表(P1-P7)还评估精神病症状的变化,并且包括P1:妄想;P2:概念紊乱;P3:幻觉行为;P4:兴奋;P5:夸大;P6:猜疑/被害;和P7:敌对性。
在筛选(第-28天至第-1天)、第1次访视(第1天)、第2次访视(第22天)、第3次访视(第43天)、第4次访视(第64天)和第5次访视/ET访视(第85天)时进行PANSS评估。
5.2阴性症状评估-16(NSA-16)总体阴性症状评分
认为NSA-16(附录3A或3B)是与精神分裂症相关的阴性症状的存在、严重程度和范围的有效且可靠的量度;其跨越语言和文化具有高评级者间和测试-再测试可靠性(Daniel,Schizophr Res.2013;150(2-3):343-345;Axelrod等人J Psychiatr Res.1993;27(3):253-258)。NSA-16使用5因子模型描述阴性症状:1)交流,2)情绪/反应,3)社会参与,4)动机,和5)迟缓。由结构式访谈评估的这些因子是全面的并且经明确定义以帮助标准化评估。作为25项目型NSA的截短版本,NSA-16仍捕获阴性症状的多维性,但可在约15至20分钟内完成。与受试者进行比较的“正常”参考群体是二十多岁的健康年轻人。
NSA总体阴性症状评分对定义为通常存在于健康年轻人中的行为消失或减少的阴性症状的整体严重程度评级。评级不应取决于来自NSA或任何其他类似工具的任何特定项目。反而应衡量评级者的访谈的完形并且在NSA-16访谈完成之后评估(Alphs等人Int JMethods Psychiatr Res.2011;20(2):e31-37)。
在第1次访视(第1天)、第2次访视(第22天)、第3次访视(第43天)、第4次访视(第64天)和第5次访视/ET访视(第85天)时进行NSA-16和NSA总体阴性症状评分评估。
5.3患者总体印象-严重程度(PGI-S)
PGI-S(附录4)是7点(1-7)患者评级量表,其用于如下评估患者的精神分裂症的严重程度:1)正常,完全无疾病;2)处于疾病边缘;3)轻度疾病;4)中度疾病;5)明显疾病;6)重度疾病;7)极重度疾病。
在第1次访视(第1天)、第2次访视(第22天)、第3次访视(第43天)、第4次访视(第64天)和第5次访视/ET(第85天)时进行PGI-S评估并且集中于精神分裂症的阴性症状。
5.4患者总体印象-变化(PGI-C)
PGI-C(附录5A或5B)是7点(1-7)患者评级量表,其用于如下评估与患者的精神分裂症有关的治疗应答:极显著改善、显著改善、极小改善、无变化、极小恶化、显著恶化或极显著恶化。
在第2次访视(第22天)、第3次访视(第43天)、第4次访视(第64天)和第5次访视/ET访视(第856天)时进行PGI-C评估并且集中于精神分裂症的阴性症状。
5.5卡尔加里精神分裂症抑郁量表(CDSS)
CDSS(附录6)是来源于汉密尔顿抑郁量表(Ham-D)的9项目型量表,其被设计用于特异性地评估患有精神分裂症的患者中的抑郁症(Addington等人Schizophr Res.1996;19(2-3):205-212)。与Ham-D不同,CDSS不含与精神分裂症的阴性症状重叠的抑郁症状,诸如快感缺乏和社交退缩。CDSS展示极佳的心理测量特性。量表中的每个项目评分为0,无;1,轻度;2,中度;或3,重度。通过将每个项目评分相加来获得CDSS评分。对于预测存在重性抑郁发作,高于6的评分具有82%特异性和85%敏感性。
在筛选(第-28天至第-1天)、第1次访视(第1天)、第3次访视(第43天)和第5次访视/ET访视(第85天)时进行CDSS评估。
6药物动力学的评估
由在第1天(第0周)给药后、第43天(第6周)给药前和给药后以及第85天(第12周)给药前收集的样品测量d6-DM、其代谢物(d3-DX和d3-3-MM)和Q的血浆浓度。按照赞助商提供的指示收集这些样品。
用eCRF记录每次样品收集的日期和时间以及在样品收集之前研究药物的最后一次给药的日期和时间。
通过离心分离血液样品并且接着在-20℃下冷冻,直至在分析单元中进行分析。由赞助商在研究开始时向研究点提供分析样品的收集、储存和装运的程序。
以描述方式概述d6-DM、d3-DX、d3-3-MM和Q的血浆浓度并且可用于未来的群体PK分析中。
7安全性评估
7.1不良事件
AE是从签署ICF时发生的任何不当的医学事件或不希望的变化(包括身体、精神[例如抑郁症]或行为),包括在开始治疗之后的临床试验的过程期间发生的间发性疾病,无论是否视为与治疗相关。因此,AE可以是在时间上与药品的使用相关的任何不利的和不希望的体征(包括例如异常实验室结果)、症状或疾病,无论是否视为与药品相关。与临床上通常预期的发生率或量值相比没有差异的与正常生长和发育相关的变化不是AE(例如在生理学上合适的时间发生的月经)。
在可能时,应通过诊断而非症状描述临床AE(例如感冒、季节性过敏,代替“流鼻涕”)。
过度剂量是故意或无意地以高于方案中指定的剂量且高于已知治疗剂量的剂量施用治疗。必须无关结果报告过度剂量,即使未观测到毒性作用。
在3点量表中对AE进行分级并且在eCRF中如所指示进行详细报告:
每种AE与研究药物的关系应由研究者使用以下说明确定:
7.2严重不良事件
严重不良事件(SAE)是在任何剂量下发生的引起以下结果中的任一者的任何AE:
·死亡
·危及生命的经历(在初始报告者看来,从AE出现时便使受试者具有立即死亡的风险的经历;即,其不包括在以更严重的形式出现时才可能引起死亡的AE)
·持续性或显著功能障碍/失能(功能障碍是受试者进行正常生活功能的能力的实质性破坏)
·住院或住院时间延长
·先天性异常/出生缺陷
当基于合适的医学判断,可能不引起死亡、不会危及生命或无需住院的重要医学事件会危害患者或需要医学或手术介入以防止所列举的结果中的一者时,可将其视为SAE。
怀孕不视为AE或SAE,除非发生符合AE或SAE要求的并发症,但必须在怀孕报告表中报告。此研究排除怀孕或可能有孕的女性。在患者在研究期间有孕的情况下,必须中止研究药物,必须完成怀孕报告表以捕获在怀孕期间的潜在药物暴露,并且必须在发现后24小时内报告怀孕。必须随访任何怀孕患者直至已知其怀孕结果(即,正常分娩、异常分娩、自然/自发性/治疗性流产)。必须随访孕妇(即,母体和胎儿)直至分娩以获知结果。
在男性患者的女性伴侣在研究药物的最后一次给药或研究完成之后的30天内(以较长者为准)有孕的情况下,还必须完成妊娠报告表。
术语“重度”是强度的量度;因此,重度AE未必严重。举例来说,持续若干小时的恶心可评级为重度,但可能在临床上并非严重的。
7.3报告不良事件
在所有筛选后访视时针对AE询问患者。研究者评估和记录所有所报告的AE。跟踪在患者接受研究药物的最后一次给药之后并且直至研究结束访视时新近报告的任何AE直至消退(患者的健康恢复至其基线状态或者所有变量恢复至正常值)或直至所发生的事件稳定(研究者不预期事件的任何进一步改善或恶化)。
必须向赞助商报告在研究期间发生或在停止研究药物之后30天内引起研究者注意的死亡(无论是否视为与治疗相关)。
对于所有SAE,包括评估为临床上显著的异常实验室测试值,研究者应在知晓事件之后的24小时内与赞助商的医学监测员或指定代表(根据需要)进行协商并且以传真/电子邮件形式报告任何SAE,如下文详细描述。接着,必须评估SAE的以下细节:事件严重性、开始日期、停止日期、强度、发生率、与研究药物的关系、关于测试药物采取的行动、所需治疗和迄今为止的结果。
7.4身体和神经检查
在筛选(第-28天至第-1天)和最终临床访视(第5次访视[第12周])时进行身体和神经检查。身体体检包括评估头部、眼睛、耳朵、鼻子、咽喉、淋巴结、皮肤、四肢、呼吸道、胃肠道、肌肉骨胳、心血管和神经系统。神经检查包括评估精神状态、颅脑神经、运动系统、反射、协调性、步态和站姿以及感觉系统。在可能时,每次应由同一个人进行身体和神经检查。
在筛选时由研究者测定为临床上显著的身体和神经检查异常应记录为医疗史。与筛选检查相比,身体和神经检查结果的任何临床上显著的变化应记录为AE。
7.5生命体征测量
在所有临床访视时进行(重复两次)血压(BP)和心率(HR)测量。仅在筛选访视时测量直立性BP和HR并且描述于下文中;在患者坐立/半卧时进行所有其他BP和HR测量。
在筛选、第1天(给药之前)和最终临床访视(第5次访视[第12周])时评估呼吸率(呼吸/分钟)和体温(°F)。
在筛选、第1天(第1次访视)和第85天(第5次访视)时记录体重和身高。
7.6心电图(ECG)
在筛选和每次临床访视(第1次、第2次、第3次、第4次、第5次和第6次访视)时进行静息12导联ECG;筛选ECG重复进行三次(1分钟间隔)。在第1次访视和第2次访视时,在给药前和给药后1-2小时(±15分钟)进行ECG;在所有其他访视时,仅在给药前进行ECG(第3次、第4次和第5次访视)。
通过中央读取器向研究中心提供ECG设备。在研究中心记录ECG评估并且其包括一般结果,包括心率(跳动/分钟)、综合QRS以及PR和QTc间期。通过中央读取器在72小时内向研究者提供结果。在筛选时存在的任何ECG异常记录为医疗史。由研究者视为临床上显著的相比筛选时的ECG状态的任何变化应捕获为AE。
应与医学监测员一起论述任何临床上显著的异常ECG,并且如果有必要,应在约1周内重复ECG。此外,在随机化之后的任何时间呈现QTcF间期>500msec(除非归因于心室起搏)或相比给药前基线ECG的QTcF间期增加(第22天/第3周)>60msec的任何患者退出研究。
7.7安全性量表
出于安全性目的评估AIMS、BAS、SAS和C-SSRS量表并且简单描述于以下章节中。
7.7.1异常不自主运动量表(AIMS)
AIMS(附录7A或7B)由12个项目构成并且用于评估运动障碍。使用5点量表(0=无至4=重度)对与口颌面、四肢和躯干运动的严重程度、关于失能的总体判断以及患者意识相关的项目进行评级。使用“是”或“否”应答对与牙齿状态相关的两个项目进行评分。
在第1次访视(第1天)、第3次访视(第43天)和第5次访视/ET访视(第85天)时进行AIMS评估。
7.7.2巴恩斯静坐不能量表(BAS)
BAS(附录8)由评估静坐不能的客观存在和发生率、个体主观意识和苦恼程度以及总体严重程度的项目组成。
BAS评分如下:
客观静坐不能、坐立不安的主观意识以及与坐立不安相关的主观苦恼在4点量表中以0-3进行评级并且相加得到在0至9范围内的总分。静坐不能的总体临床评估使用在0-4范围内的5点量表。
在第1次访视(第1天)、第3次访视(第43天)和第5次访视/ET访视(第85天)时进行BAS评估。
7.7.3辛普森安格斯锥体外系症状量表(SAS)
SAS(附录9A或9B)由10个项目构成并且用于评估假性帕金森症。使用5点量表对每个项目的严重程度的级别进行评级。SAS评分可在0至40范围内。所评估的体征包括步态、落臂、摇肩、肘强直、腕强直、腿摆动、头部旋转、眉间轻敲、震颤和流涎。
在筛选(第-28天至第-1天)、第1次访视(第1天)、第3次访视(第43天)和第5次访视/ET访视(第85天)时进行SAS评估。
7.7.4哥伦比亚自杀严重程度评级量表(C-SSRS)
C-SSRS(附录10)是提供意念和行为的概述的临床访谈,其可在任何评估或风险评估期间施用以鉴别所存在的自杀倾向的程度和类型。C-SSRS还可在治疗期间使用以监测临床恶化。
在研究期间使用C-SSRS量表的“基线/筛选”和“自上次访视后”版本监测自杀倾向。在筛选时对所有患者完成“基线/筛选”版本以测定合格性,所述版本评估具有自杀事件和自杀意念的患者的生命经历,以及在进入研究之前,在指定时间段内发生的自杀事件或意念。应从研究排除任何在最近6个月内具有主动自杀意念、在最近2年内具有自杀行为或在研究者的临床判断中呈现严重自杀风险的患者(参见章节2.2,排除标准)。还在筛选之后的访视时完成“自上次访视以后”C-SSRS表格。
在筛选(第-28天至第-1天)、第1次访视(第1天)、第2次访视(第22天)、第3次访视(第43天)、第4次访视(第64天)和第5次访视/ET访视(第85天)时进行C-SSRS评估。
8评估和程序的时间表
评估和程序的时间表提供于表16中。
研究程序的说明
在每次研究访视时,要求研究工作人员的成员将关于患者数据和预定研究评估结果的信息输入IWRS数据库中。在IRT站点手册中提供其他指示。
下文列举的给药前和给药后程序通常在合理的灵活程度内以其执行顺序列举。NSA-16、NSA-16总体、PANSS、PGI-S和PGI-C评估都在给药前进行并且应在可适用的访视的研究程序期间尽早完成。任何给药前ECG和给药前抽血应仅在每次访视时完成以上量表之后进行。在筛选时的用于双重入选的CTS数据库检验应作为第一批签署知情同意书后程序中的一者进行,使得研究点在进行更复杂的任务之前了解潜在的双重入选。
筛选访视(第-28至-1天)
在第1天之前的28天内,在筛选时进行以下程序。
1.研究者或受委托的研究点工作人员向患者提供知情同意书文件并且说明研究的基本原理,向患者提供足够的时间以用于咨询问题。在进行任何研究相关程序之前,患者必须提供书面知情同意书。
2.完成授权表格并且将患者信息输入CTS数据库。
3.合格且经认证的评级者施用PANSS。
4.记录精神病史并且进行M.I.N.I.检查。
5.经充分培训且经认证的评级者施用CDSS以评估抑郁症的症状。
6.经充分培训的研究者(或合格的指定人员)完成“基线/筛选”C-SSRS表格以排除具有显著自杀行为风险的患者。
7.审查并且记录医疗史,包括患者人口统计数据,和任何并用药物使用(包括非处方[OTC]药物、维生素和补充剂)。
8.审查包含和排除标准(合格性表格)。
9.收集并且记录关于患者与其信息提供者的关系的详细信息。
10.重复两次测量并且记录生命体征(直立性BP和HR(仅筛选时)[参见章节7.5]、呼吸率和体温);还记录体重和身高。
11.进行身体和神经检查。
12.经充分培训且有经验的临床医师施用SAS以评估EPS。
13.重复三次进行静息12导联ECG(1分钟间隔)。
14.收集血液和尿液样本以用于临床实验室评估(化学,包括空腹葡萄糖、脂质、TSH和HbA1c;血液学;和尿分析)。
15.收集血液样本以用于评估血浆抗精神病药水平。
16.对所有具有生育潜力的女性进行血清妊娠测试。
17.收集血液样本以用于乙型和丙型肝炎(HBsAg和HCAb)以及HIV抗体筛选。
18.收集尿液样本以用于药物筛选。
19.审查评估以验证患者符合继续参与研究的条件。
在用于评估包含和排除标准的筛选程序之后,向医学监测员提交方案合格性表格以进行审批。由研究者和医学监测员认为符合条件的患者返回进行第1次访视(第1天)。中止(不随机分配)具有研究者视为临床上显著的不属于参考正常值范围内并且可能使患者具有较高研究参与风险的ECG结果或实验室测试结果的患者。
第1次访视(基线,随机分配;第1天/第0周±3天窗口)
在双盲治疗期期间,将患者随机分配(1∶1比率)以接受d6-DM/Q或匹配安慰剂胶囊保持12周。在第1天,在诊所施用双盲治疗期的研究药物的第一次给药。
在完成所有第2次访视给药前程序之后进行随机分配;此访视应在3天窗口内。在第1次访视(第1天±3天窗口)时进行以下程序:
1.合格的且经认证的评级者施用NSA-16、NSA-16总体、PANSS、PGI-S和PGI-C。
2.经充分培训且经认证的评级者施用CDSS以评估抑郁症的症状。
3.经充分培训的研究者(或合格的指定人员)完成“自上次访视以后”C-SSRS表格。
4.测量和记录生命体征(坐立/半卧BP和HR[重复两次])。
5.进行静息12导联ECG(给药前,在已施用以上量表之后)。
6.询问患者关于AE和并用药物使用(包括OTC药物、维生素和补充剂)情况。
7.收集血液和/或尿液样本以用于安全性实验室评估(化学[包括空腹葡萄糖和脂质]、血液学、尿分析)-所有抽血应在施用以上量表之后进行。
8.收集血液样本以用于评估血浆抗精神病药水平。
9.对所有具有生育潜力的女性进行尿液妊娠测试。
10.经充分培训且有经验的临床医师施用AIMS、SAS和BAS以评估EPS。
11.由研究点工作人员进行所有未使用的研究药物的审查。分配足够的研究药物以用于每天两次给药直至下一次访视。
在确定患者满足所有包含标准并且不满足所有排除标准后(基于上文所描述的筛选和第1次访视评估),将患者随机分配并且经由IWRS分配研究药物药盒编号。
在诊所从研究药物泡罩包装卡的AM条施用研究药物的第一次给药,与当天时间无关。
1.收集血液样本以用于血浆d6-DM、Q和d6-DM代谢物的PK评估(在给药后1至3小时)。
2.在给药后1至2小时(±15分钟)进行静息12导联ECG。
患者指示
指示患者每天两次使用研究药物(上午使用1粒来自标记有AM的泡壳封装的胶囊并且晚上使用1粒来自标记有PM的泡壳封装的胶囊;约每12小时一次)直至下一次访视。还指示患者在每次研究访视时带回任何未使用的研究药物。
提醒患者使用AiCure以监测诊所外的研究药物依从性。
研究者和/或研究协调员向患者提供关于研究程序的详细指示。还指示患者在使用任何非研究药物之前与研究点进行协商。当面审查这些要求并且还可根据研究者判断向患者提供除知情同意书以外的书面说明书。
研究者在每次访视结束时询问患者以确定患者理解对其的要求。
12周,双盲治疗期
8.1.3.1安全性电话联系(第8天/第1周±3天窗口)
患者在第1周(第8天±3天)接受电话访问以评估AE并且询问并用药物情况。还询问患者是否依照指导使用其研究药物。
第2次访视(第22天/第3周±6天窗口)
在第22天(±6天窗口)的上午进行以下程序:
1.合格的且经认证的评级者施用NSA-16、NSA-16总体、PANSS、PGI-S和PGI-C。
2.进行静息12导联ECG(给药前,在已施用以上量表之后)。
在完成以上程序之后,在诊所从新近分配的泡壳封装的AM条施用研究药物。
1.询问患者关于AE和并用药物使用(包括OTC药物、维生素和补充剂)情况。
2.经充分培训的研究者(或合格的指定人员)完成“自上次访视以后”C-SSRS表格。
3.重复两次测量并且记录生命体征(坐立/半卧BP和HR)。
4.对所有具有生育潜力的女性进行尿液妊娠测试。
5.由研究点工作人员进行所有未使用的研究药物的审查。分配足够的研究药物以用于每天两次给药直至下一次访视。
6.在给药后1至2小时(±15分钟)进行静息12导联ECG。
患者指示
指示患者每天两次使用研究药物(上午使用1粒来自标记有AM的泡壳封装的胶囊并且晚上使用1粒来自标记有PM的泡壳封装的胶囊;约每12小时一次)直至下一次访视。还指示患者在每次研究访视时带回任何未使用的研究药物。
提醒患者使用AiCure以监测诊所外的研究药物依从性。
研究者和/或研究协调员向患者提供关于研究程序的详细指示。还指示患者在使用任何非研究药物之前与研究点进行协商。当面审查这些要求并且还可根据研究者判断向患者提供除知情同意书以外的书面说明书。
研究者在每次访视结束时询问患者以确定患者理解对其的要求。
安全性电话联系(第29天/第4周±3天窗口)
患者在第4周(第29天±3天)接受电话访问以评估AE并且询问并用药物情况。还询问患者是否依照指导使用其研究药物。
第3次访视(第43天/第6周±6天窗口)
在第64天(±6天窗口)的上午进行以下程序:
1.合格的且经认证的评级者施用NSA-16、NSA-16总体、PANSS、PGI-S和PGI-C。
2.进行静息12导联ECG(给药前,在已施用以上量表之后)。
3.收集血液样本以用于血浆d6-DM、Q和代谢物的PK评估(给药前,在已施用以上量表之后)。
在诊所从新近分配的泡壳封装的AM条施用研究药物,与当天时间无关。
1.询问患者关于AE和并用药物使用(包括OTC药物、维生素和补充剂)情况。
2.经充分培训且有经验的临床医师施用AIMS、SAS和BAS以评估EPS。
3.经充分培训且经认证的评级者施用CDSS以评估抑郁症的症状。
4.经充分培训的研究者(或合格的指定人员)完成“自上次访视以后”C-SSRS表格。
5.对所有具有生育潜力的女性进行尿液妊娠测试。
6.收集血液和/或尿液样本以用于安全性实验室评估(化学[包括空腹葡萄糖和脂质]、血液学、尿分析)。
7.收集血液样本以用于评估血浆抗精神病药水平。
8.收集血液样本以用于血浆d6-DM、Q和代谢物的PK评估(在给药后1至3小时)。
9.由研究点工作人员进行所有未使用的研究药物的审查。分配足够的研究药物以用于每天两次给药直至下一次访视。
患者指示
指示患者每天两次使用研究药物(上午使用1粒来自标记有AM的泡壳封装的胶囊并且晚上使用1粒来自标记有PM的泡壳封装的胶囊;约每12小时一次)直至下一次访视。还指示患者在每次研究访视时带回任何未使用的研究药物。
提醒患者使用AiCure以监测诊所外的研究药物依从性。
研究者和/或研究协调员向患者提供关于研究程序的详细指示。还指示患者在使用任何非研究药物之前与研究点进行协商。当面审查这些要求并且还可根据研究者判断向患者提供除知情同意书以外的书面说明书。
研究者在每次访视结束时询问患者以确定患者理解对其的要求。
安全性电话联系(第50天/第7周±3天窗口)
患者在第7周(第50天±3天)接受电话访问以评估AE并且询问并用药物情况。还询问患者是否依照指导使用其研究药物。
第4次访视(第64天/第9周±6天窗口)
在第85天(±6天窗口)的上午进行以下程序。
1.合格的且经认证的评级者施用NSA-16、NSA-16总体、PANSS、PGI-S和PGI-C。
2.进行静息12导联ECG(给药前,在已施用以上量表之后)。
在诊所从新近分配的泡壳封装的AM条施用研究药物,与当天时间无关。
1.重复两次测量并且记录生命体征(坐立/半卧BP和HR)。
2.询问患者关于AE和并用药物使用(包括OTC药物、维生素和补充剂)情况。
3.经充分培训的研究者(或合格的指定人员)完成“自上次访视以后”C-SSRS表格。
4.对所有具有生育潜力的女性进行尿液妊娠测试。
5.由研究点工作人员进行所有未使用的研究药物的审查。分配足够的研究药物以用于每天两次给药直至下一次访视。
患者指示
指示患者每天两次使用研究药物(上午使用1粒来自标记有AM的泡壳封装的胶囊并且晚上使用1粒来自标记有PM的泡壳封装的胶囊;约每12小时一次)直至下一次访视(第6次访视;第106天)。还指示患者在每次研究访视时带回任何未使用的研究药物。
提醒患者使用AiCure以监测诊所外的研究药物依从性。
研究者和/或研究协调员向患者提供关于研究程序的详细指示。还指示患者在使用任何非研究药物之前与研究点进行协商。当面审查这些要求并且还可根据研究者判断向患者提供除知情同意书以外的书面说明书。
研究者在每次访视结束时询问患者以确定患者理解对其的要求。
安全性电话联系(第71天/第10周±3天窗口)
患者在第10周(第71天±3天)接受电话访问以评估AE并且询问并用药物情况。还询问患者是否依照指导使用其研究药物。
第5次访视(第85天/第12周±6天窗口)/提前终止访视在第85天(±6天窗口)或ET访视时对在第12周之前退出的患者进行以下程序:
·合格的且经认证的评级者施用NSA-16、NSA-16总体、PANSS、PGI-S和PGI-C。
·进行静息12导联ECG(给药前,在已施用以上量表之后)。
·收集血液样本以用于血浆d6-DM、Q和代谢物的PK评估(给药前,在已施用以上量表之后)。
对于完成双盲治疗的患者(未提前停止的患者),从患者带回的研究药物泡壳包装卡的AM条施用研究药物的最后一次给药,与当天时间无关。
·重复两次测量并且记录生命体征(坐立/半卧BP和HR、呼吸率和体温);还记录体重和身高。
·进行身体和神经检查。
·询问患者关于AE和并用药物使用(包括OTC药物、维生素和补充剂)情况。
·经充分培训且有经验的临床医师施用AIMS、SAS和BAS以评估EPS。
·经充分培训且经认证的评级者施用CDSS以评估抑郁症的症状。
·经充分培训的研究者(或合格的指定人员)完成“自上次访视以后”C-SSRS表格。
·收集血液和/或尿液样本以用于安全性实验室评估(化学[包括空腹葡萄糖、脂质和HbA1c]、血液学、尿分析)。
·收集血液样本以用于评估血浆抗精神病药水平。
·对所有具有生育潜力的女性进行尿液妊娠测试。
·患者返还所有未使用的研究药物;由研究点工作人员进行所有未使用的研究药物的审查。
在最后一次联系患者时,研究点工作人员访问CTS数据库,输入患者研究ID和最后一次联系的性质(即,完成者或ET)。
提前终止的程序:
所有患者在第5次访视/ET时经历完成评估。此外,对任何在随机分配至试验中之后的任何时间退出的患者完成第5次访视/ET评估;在可能时,应在研究药物的最后一次给药的48小时内完成评估。对于施用任何新的精神药物之前的第5次访视/ET访视,应进行尝试以完成所有评估,特别是功效评估。然而,如果患者在V5/ET程序之前因精神分裂症症状恶化而接受新的急救药品,则不应进行功效评估。
对于停止治疗的患者,当天在诊所不施用研究药物,因此,不存在12导联ECG和血液/尿液样本收集(临床实验室测试或药物动力学)的特定时间范围。
安全性随访电话联系(第16周/第115天±3天窗口)
在第16周(第115天±3天)时电话联系患者并且询问患者从最后一次访视开始是否经历任何AE或其药物的变化。经历AE的患者可能需要返回诊所以进行用于安全性评估的非预定访视。
随访任何先前报告的并且在此电话联系时尚未解决的AE直至解决(患者的健康恢复至其基线状态或者所有变量恢复至正常值)或直至所发生的事件稳定(研究者不预期事件的任何进一步改善或恶化)。
随访在接受研究药物的最后一次给药之后并且直至在接受研究药物的最后一次给药之后30天的任何新近报告的AE直至解决(患者的健康恢复至其基线状态或者所有变量恢复至正常值)或直至所发生的事件稳定(研究者不预期事件的任何进一步改善或恶化)。
9统计
用于此研究的计划统计方法的概述提供于下文中。在研究揭盲之前,在统计分析计划(SAP)中提供完全统计方法。
9.1分析群体
此研究的分析群体定义如下:
安全性群体:经随机分配并且进行研究药物的至少一次给药的所有患者。所有安全性分析都是基于安全性群体。
调整意向治疗(mITT):安全性群体中具有基线和至少一次基线后PANSS测量的所有患者。所有功效分析都是基于mITT群体。
9.2人口统计数据和基线特征
使用描述性统计概述各治疗组的人口统计数据和基线特征。
9.3功效分析
9.3.1研究终点
9.3.1.1主要功效终点
主要功效终点是PANSS Marder阴性因子评分的从基线至第5次访视(第12周)的变化。
9.3.1.2次要功效终点
次要功效终点包括以下结果量度的从基线至第5次访视(第12周)的变化(PGI-C原始评分量度变化):
·NSA-16总体阴性症状评分
·PGI-S
·PGI-C
9.3.1.3其他结果量度
其他结果量度包括以下量度的从基线至第5次访视(第12周)的变化:
·PANSS阳性分量表
·卡尔加里精神分裂症抑郁量表(CDSS)
9.3.2主要功效分析
主要功效终点是PANSS Marder阴性因子评分的从基线至第12周的变化。通过对所观测的数据使用基于可能性的线性混合效应模型重复测量(MMRM)来分析主要功效终点。模型包括治疗、试验中心、访视、治疗与访视相互相用以及基线与访视相互相用的固定效应。使用非结构化协方差模型。在数据库揭盲之前根据区域和实践汇集小型试验中心用于分析。
此研究的目的是评估与安慰剂相比,由开始使用d6-DM/Q的患者产生的未来群体中的预期药物作用。主要兴趣的被估目标定义如下:
·群体:mITT
·变量:PANSS Marder阴性因子评分的从基线至第12周的变化。
·间发事件:遵守“治疗方针”策略,由此使用相关变量的值而与对随机分配的治疗的依从性和/或禁止药物的起始无关。
·群体水平概述:PANSS Marder阴性因子评分的从基线至第12周的平均变化的治疗间差异。
在统计分析中使用在研究期间收集的所有数据。关于主要功效分析,使用上文所描述的MMRM方法估计治疗作用。在随机缺失(MAR)假设下,MMRM提供对治疗期的治疗作用的无偏估计。进行在非随机缺失(MNAR)下通过多重设算(MI)来设算的缺失值的分析和其他方法作为敏感性分析。
9.3.3次要功效和其他结果量度分析
在合适时,以与主要功效分析类似的方式分析次要功效终点和其他结果量度。其他细节和分析方法描述于SAP中。
9.4药物动力学分析
以描述方式概述d6-DM、其代谢物d3-DX和d3-3-MM以及Q的血浆浓度。
9.5安全性分析
安全性分析基于安全性群体,所述安全性群体定义为经随机分配并且进行研究药物的至少一次给药的所有患者。该分析由生物学参数和AE的数据概述组成。按治疗对安全性分析进行列表。
9.5.1不良事件
使用监管活动医学词典(MedDRA)对AE进行编码。按治疗、系统器官类别(SOC)、死亡、非致死性SAE、AE、引起研究中止的AE和治疗引发的AE(TEAE)概述经历1种或多种AE的患者的百分比。
9.5.2生命体征和心电图
提供BP(舒张和收缩)、心率、呼吸率、体重和ECG参数的绝对值和自基线(第1次访视;第1天/第0周)的变化百分比的概述统计。在个体患者数据列表中突出显示在预定正常值范围以外的所有值。
9.5.3临床实验室量度
经由描述性统计并且经由在第1次访视(第0周)与治疗结束之间的结果相对于正常值范围的偏移(如增加、降低或无变化)来概述实验室参数。
9.5.4安全性量表
经由描述性统计并且经由从基线(第1次访视;第1天/第0周)至基线后访视和治疗结束时,每个评分的患者的数目和百分比的偏移表来概述C-SSRS、SAS、BAS和AIMS。