基于LncRNA的mLs模型的构建方法及其应用
文献发布时间:2024-04-18 19:58:26
技术领域
本发明属于生物医药领域,涉及一种基于LncRNA的mLs模型的构建方法及其应用,特别涉及一种基于LncRNA的用于前列腺癌辅助诊断和预后评估的mLs模型的构建方法及其应用。
背景技术
前列腺癌(prostate cancer,PCa)是男性第2大常见恶性肿瘤,据统计,前列腺癌占所有肿瘤新发病率的7.3%和死亡率的3.8%。它不仅对男性健康产生了严重威胁,而且给社会造成了巨大的经济负担。虽然目前临床上前列腺癌的治疗方式较多,如前列腺根治疗法(RP)、雄激素剥夺疗法、放射治疗、内分泌治疗和免疫治疗等,但由于前列腺癌的高度异质性,当前的临床指标如血清前列腺特异性抗原(PSA)、格林森评分(Gleason Score)和肿瘤分期等难以满足现代医学中个体化治疗的需要,这往往容易导致治疗不足或过度治疗。因此,迫切需要对前列腺癌异质性进行更深入地研究。
肿瘤异质性是肿瘤发生发展过程中的关键特征。肿瘤的表型异质性不仅建立在慢性或偶发性基因组不稳定性和由此产生的肿瘤细胞遗传异质性的基础上,而且还与非突变表观遗传异质性有关.非突变表观遗传被认为是肿瘤进展中的又一重要特征,它主要包括DNA甲基化、组蛋白修饰、RNA修饰和染色质重塑等,其中RNA修饰是目前研究较多的一个新兴领域。而在种类繁多的RNA修饰中,N6-methyladenosine(m6A)是真核生物中最常见、最丰富的转录后水平RNA修饰,约占总甲基化核糖核苷酸的50%和细胞总RNA中所有腺苷的0.1-0.4%。m6A修饰是一个动态可逆过程,主要由m6A甲基化酶、去甲基化酶和结合蛋白组成。m6A修饰具有广泛的生物学功能,它参与调控RNA的转录、剪接、降解和翻译等过程,在RNA代谢中起着重要作用。m6A修饰不仅在mRNA中发挥关键作用,而且也参与调节non-codingRNAs(ncRNAs)的生成和功能。ncRNAs是一类缺乏蛋白质编码功能的RNA,主要包括microRNAs(miRNAs)、long noncoding RNAs(LncRNAs)和circular RNAs(circRNAs)。其中LncRNAs中的m6A修饰水平较高,并且LncRNAs中的m6A修饰相关研究已在多种肿瘤中被广泛报道,例如有研究人员通过m6A-seq发现与正常脑组织相比,胶质母细胞瘤组织中m6A修饰的LncRNA的丰度明显更高,且根据实验发现m6A修饰的LncRNA WEE2-AS1通过阻止cul2介导的RPN2 K322泛素化促进RPN2蛋白稳定,从而通过激活AKT信号通路促进胶质母细胞瘤恶性进展。然而前列腺癌中相关研究数量有限,仍需要进一步探索。
发明内容
本发明的目的在于解决现有技术中所存在的前列腺癌患者预后及生存情况难以精准预测、临床应用繁琐的技术问题,从而提供了一种基于LncRNA的新的评价指标即mLs,用于对前列腺癌进行辅助诊断和预后评估。通过对前列腺癌患者体内特定生物标志物表达水平进行检测并分析,从而对患者生化复发风险、预后及生存情况等进行有效预测,可作为一种风险评估工具筛选出高风险的前列腺癌患者,以进行合理的提前预防和治疗,指导前列腺癌患者的个体化精准治疗,进一步优化前列腺癌患者的管理,避免因治疗不及时而导致的疾病进展或因治疗过度导致的医疗资料浪费。
为了解决上述技术问题,本发明是通过如下技术方案得以实现的。
本发明第一方面提供了一种基于LncRNA的用于前列腺癌辅助诊断和/或预后评估的mLs模型的构建方法,包括如下步骤:
(1)获取原发性(primary)前列腺癌患者肿瘤组织样本和转移性(metastatic)前列腺癌患者肿瘤组织样本,分别利用MeRIP-seq和RNA-seq分析对其主要成分进行分析,获得各组中的m6A峰及LncRNA数量;
(2)对原发性前列腺癌患者肿瘤组织样本组、转移性前列腺癌患者肿瘤组织样本组中重叠的m6A峰和LncRNA进行分析,并按照如下条件对所获得的重叠的m6A峰及LncRNA进行差异表达识别,以获得两组中具有显著差异丰度的m6A峰和差异表达的LncRNA:
p<0.05且|fold change|>2;
(3)对步骤(2)所获得的原发性前列腺癌患者肿瘤组织样本组、转移性前列腺癌患者肿瘤组织样本组中具有差异丰度的m6A峰和差异表达的LncRNA进行交叉分析(cross-analysis),获得同时具有甲基化差异以及表达水平差异的LncRNA;
(4)利用PCaDB工具对步骤(3)获得的同时具有甲基化差异以及表达水平差异的LncRNA进行分析,筛选得到与前列腺癌具有显著相关性的LncRNA;
(5)基于步骤(4)中所述与前列腺癌具有显著相关性的LncRNA按照如下公式进行mLs的计算:
其中N代表所检测的LncRNA的数量,Coef
应理解的是,在没有特别说明的情况下,在本发明上下文中mLs、mLs Score、mLs-Score、mLs Scr、mLs-Scr、mLs评分、mLs得分等表述,均为同一意思的表达,即为m6A-modified LncRNAs Score的简化表述,理解为对前列腺癌患者预后状况的一种量化评估方式,进而对前列腺癌患者生化复发风险、生存率、预后等进行合理预测。
作为优选地,步骤(4)中所述与前列腺癌具有显著相关性的LncRNA由COL1A1、MIR210HG、AL158071.5、SNHG8、AL390719.2、LINC00920、PCA3、LINC01088、AF165147.1组成。
作为优选地,步骤(5)中各LncRNA的Coef
作为优选地,步骤(5)中N=9。
本发明第二方面提供了根据上述构建方法得到的基于LncRNA的用于前列腺癌辅助诊断和/或预后评估的mLs模型。
本发明第三方面提供了用于评价前列腺癌患者mLs的试剂盒,包括用于检测标志物表达水平的试剂,所述标志物由COL1A1、MIR210HG、AL158071.5、SNHG8、AL390719.2、LINC00920、PCA3、LINC01088、AF165147.1组成。
作为优选地,所述检测标志物表达水平的试剂包括用于检测标志物基因表达水平的引物对和/或检测标志物基因编码的蛋白含量的试剂。
应理解的是,在没有特别说明的情况下,在本发明上下文中,所述引物和/或引物对是指用于在PCR中合成各标志物基因cDNA链的PCR引物,从而用于检测各标志物基因的表达水平。除本发明所列出的引物和/或引物对外,本领域技术人员完全有能力根据各标志物的基因序列采用包括但不限于分子生物学等本领域的常规方法手段进行相应引物和/或引物对的设计,并通过常规实验手段对所设计的引物和/或引物对进行筛选,只要能够实现特异性检测各标志物表达水平即可。同时,对于所述用于检测各标志物基因编码的蛋白含量的抗体试剂,本领域技术人员可以通过市售获得,也可以根据各标志物基因序列和/或蛋白结构自行设计完成。
作为优选地,所述试剂盒还包括PCR酶、PCR缓冲液、dNTPs、荧光底物中的一种或多种。
作为优选地,所述荧光底物选自Syber Green或荧光标记的探针。
本发明第四方面提供了上述试剂盒在制备对癌症患者进行辅助诊断和/或预后评估的产品中的应用。
作为优选地,所述癌症选自前列腺癌、间皮瘤(MESO)、胰腺癌(PAAD)、乳腺浸润性癌(BRCA)、胆管癌(CHOL)、卵巢浆液性囊腺癌(OV)、脑低度胶质瘤(LGG)、葡萄膜黑色素瘤(UVM)、胶质母细胞瘤细胞瘤(GBM)、肝癌(LIHC)、脑低度胶质瘤(LGG)、胶质母细胞瘤细胞瘤(GBM)、肺鳞状细胞癌(LUSC)、肾乳头状细胞癌(KIRP)、肾透明细胞癌(KIRC)、肾嫌色细胞癌(KICH)、宫颈鳞状细胞癌和宫颈内腺癌(CESC)、肾上腺皮质癌(ACC)、葡萄膜黑色素瘤(UVM)、淋巴样肿瘤弥漫性大b细胞淋巴瘤(DLBC)中的一种或多种。
本发明第五方面提供了一种用于对前列腺癌患者辅助诊断和/或预后评估的系统,包括:
输入单元,用于输入待评估前列腺癌患者的数据;
分析单元,用于对待评估前列腺癌患者的样本中的标志物表达水平进行检测,所述标志物由COL1A1、MIR210HG、AL158071.5、SNHG8、AL390719.2、LINC00920、PCA3、LINC01088、AF165147.1组成;
评估单元,用于对分析单元分析得到的数据按照如下公式进行计算,以获得前列腺癌患者的mLs值,并根据mLs值将前列腺癌患者分为mLs高得分组或mLs低得分组,最后对前列腺癌患者的预后和总生存率作出评估:
其中N代表所检测的标志物的数量,Coef
作为优选地,输入单元中所述数据包括但不限于前列腺癌患者的肿瘤组织样本数据、血液样本数据、癌旁组织样本数据中的一种或多种。
作为优选地,所述评估单元中按照如下标准对前列腺癌患者进行评估:
(a)mLs得分与总生存率呈现负相关;mLs得分越高,总生存率相对越低,mLs得分越低,总生存率相对越高;
以及(b)mLs得分与生化复发率呈现正相关;mLs得分越高,生化复发率相对越高,mLs得分越低,生化复发率相对越低;
以及(c)mLs得分与远处转移率呈现正相关;mLs得分越高,远处转移率相对越高,mLs得分越低,远处转移率相对越低。
本发明第六方面提供了一种计算机可读的存储介质,其中存储有处理器可执行的指令,所述处理器可执行的指令在处理器执行时执行上述用于对前列腺癌患者辅助诊断和/或预后预测进行评估的系统。
应理解的是,在没有特别说明的情况下,本发明上下文中所述的“模型”即为临床预测模型,本领域技术人员能够理解所述的“模型”为一种产品。临床上通过使用参数/半参数/非参数数学模型来评估受试者当前患有某种疾病的概率或将来发生某种结局的可能性。通过该模型,利用已知特征来计算未知结局发生的概率。临床预测模型一般采用各种回归分析方法建模,回归分析的统计本质是寻找一种定量因果关系,可用于对疾病进行诊断和/或预后评估,从而在诊断治疗决策、患者预后管理、公共卫生资源配置等方面发挥作用。
与许多其他类型的肿瘤不同,前列腺癌尤其是局限性前列腺癌大多数进展较为缓慢,很多患者仅表现为前列腺增生甚至无明显症状,部分患者的10年癌症特异性生存率甚至超过94%。目前,临床上主要依赖PSA、Gleason Score、Surgical margin和淋巴结转移与否等指标来前列腺癌患者进行预后预测和生存评估,但有研究表明即使是采用这些指标的组合,其预测准确率仍然只有75-85%,即现有的临床参数已经难以满足对前列腺癌患者进行精准的危险分层以及当下个体化治疗的需要。现有技术中已经公开了部分能够实现对前列腺癌进行预后预测的相关指标,然而这些指标存在准确性差、缺乏稳定性等缺陷,同时大多数指标所需要检测的标志物数量过多,例如Decipher需要检测22个标志物表达水平、Prolaris需要检测46个标志物表达水平,Oncotype_DX_GPS需要检测17个标志物表达水平,更有甚者需要检测高达200余种的标志物表达水平,其大大增加了临床检测的时间和成本,难以大规模推广应用。
由于前列腺癌的高度异质性和非突变表观遗传机制,过度治疗或治疗不足仍是前列腺癌个体化治疗中普遍存在的问题。然而,作为一种最常见的RNA表观遗传修饰方式,m6A修饰在前列腺癌的发生和进展中的作用仍有待阐明。在本发明中,通过收集了来自患者的原发性PCa和转移性PCa样本,并利用MeRIP-seq来定位m6a甲基化的LncRNAs。随后于3个多中心队列中构建并验证了一种基于9个甲基化差异LncRNAs的前列腺癌预后指标,并将它命名为前列腺癌m6A-modified LncRNA score(mLs)。结果表明,m6A修饰的差异与m6A修饰的Lncrna表达的改变呈正相关,mLs高得分组患者的生化复发发生时间更早,且mLs的预测性能优于5个已知的预测指标。此外,本发明的结果表明,mLs与脂代谢之间呈负相关,且mLs也是部分其他类型肿瘤预后的保护或危险因素。通过选择其中的高风险因素之一MIR210HG进行实验验证发现,其高表达可促进PCa增殖和侵袭。总之,本发明基于对前列腺癌患者体内多项LncRNA的分析,构建了能够用于系统评价前列腺癌患者辅助诊断和预后评估的可量化模型-mLs,其能够更加精准地对前列腺癌患者进行危险分层和预后预测,以便于精准识别出需要积极干预的高风险前列腺癌患者,避免因判断错误而导致的治疗不足或过度治疗;同时本发明所提供的mLs需要检测的标志物数量显著减少,大大简化了临床检测的过程,可有效避免医疗资源的浪费。
本发明相对于现有技术具有如下技术效果:
(1)本发明对前列腺癌的发病、发展机制进行了深入研究,发现准确预测前列腺癌患者生化复发状况对于评估前列腺癌患者的生存率和预后具有关键意义,并基于对前列腺癌患者体内多项LncRNA的分析,构建了能够用于系统评价前列腺癌患者预测的可量化模型-mLs,从而能够通过对受试者体内特定LncRNA表达水平的分析,更加准确地对前列腺癌患者预后及生存率状态进行合理预测和评估。
(2)本发明揭示了mLs与前列腺癌生化复发之间的关联性,相对于现有技术中常用预后指标来说具有更高的预测准确率,能够更加精准地对前列腺癌患者进行危险分层和预后预测,以便于精准识别出需要积极干预的高风险前列腺患者,避免因判断错误而导致的治疗不足或过度治疗;同时本发明所提供的mLs需要检测的基因数量显著减少,大大简化了临床检测的过程,可有效避免医疗资源的浪费,且相对于现有的预测模型具有更加稳定可靠的预测性能。本发明的mLs对于填补实现前列腺癌精准预后评估空白,更好地实现前列腺癌患者的个体化精准治疗具有重要的现实意义,具有极高的社会价值和市场应用前景。
附图说明
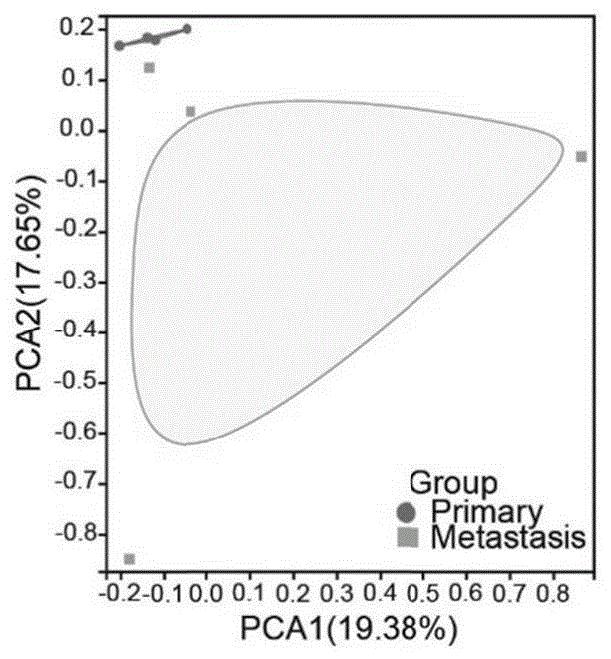
图1为原发性前列腺癌和转移性前列腺癌样本之间的PCA分析结果示意图。
图2为原发性前列腺癌和转移性前列腺癌样本之间m6A peaks和LncRNA情况分析结果示意图。
图3为对原发性前列腺癌和转移性前列腺癌样本之间具有显著差异丰度的m6A峰分析结果示意图。
图4为对原发性前列腺癌和转移性前列腺癌样本之间存在差异表达的LncRNA分析结果示意图。
图5为对原发性前列腺癌和转移性前列腺癌样本之间所存在的同时具有甲基化差异以及表达水平差异的LncRNA分析结果示意图。
图6为对m6A峰差异丰度与LncRNA差异表达量之间相关性分析结果示意图。
图7为在3个不同队列中mLs得分对前列腺癌患者生存率影响结果示意图。
图8为在3个不同队列中mLs得分对前列腺癌患者BCR影响结果示意图。
图9为对3个不同队列进行单变量和多变量Cox回归分析结果示意图。
图10为在3个不同队列中将mLs与其他临床指标的预后预测性能进行比较的结果示意图。
图11为在3个不同队列中mLs与其他5个已知模型的预测性能进行比较的结果示意图。
图12为对前列腺癌患者3年、5年以及8年无生化复发生存期预测列线图。
图13为所预测的3年、5年以及8年无生化复发生存期与实际情况相比较结果示意图。
图14为列线图中3年、5年以及8年无生化复发生存期的AUC情况结果示意图。
图15为mLs对脂肪酸降解和脂肪酸延长的影响结果示意图。
图16为mLs与脂肪酸降解和脂肪酸延长之间相关性分析结果示意图。
图17为在mLs在不同癌症中的预测情况结果示意图。
图18为mLs对相关存活率(DSS)、总体生存率(OS)和无进展间隔(PFI)的影响结果示意图。
图19为在不同癌症中mLs的Cox单因素分析以明确其风险情况结果示意图。
图20为不同前列腺癌细胞中MIR210HG表达情况结果示意图。
图21为前列腺癌患者肿瘤组织FISH实验结果示意图。
图22为对MIR210HG与m6A修饰情况分析结果示意图。
图23为不同siRNA对前列腺癌细胞中MIR210HG抑制情况结果示意图。
图24为siRNA对前列腺癌细胞增殖情况影响结果示意图。
图25为siRNA对前列腺癌细胞克隆形成影响结果示意图。
图26为前列腺癌细胞Transwell实验结果示意图。
具体实施方式
为使本发明的目的、技术方案及效果更加清楚、明确,以下参照实施例对本发明作进一步详细说明。应当理解,此处所描述的具体实施例仅用以解释本发明,并不用于限定本发明。
在无特别说明的情况下,本发明上下文中所使用的试剂中,均通过市售获得。前列腺癌细胞系PC-3、22Rv1、C4-2和C4-2B均购自于BeNa Culture Collection。22Rv1、C4-2和C4-2B在RPMI-1640培养基中培养;PC-3在DMEM培养基中培养。两种培养基中均添加了10%胎牛血清和1%双抗生素(青霉素和链霉素)。所有细胞系均在37℃恒温和5%CO
生物学实验重复中选择具有代表性的结果呈现在上下文附图中,数据按照图示中规定的以mean±SD和mean±SEM展示。所有实验至少重复三次。数据的统计分析和可视化由R软件(version 4.1.0)和GraphPad Prism 8.0完成。Pearson和Spearman相关性分析用于计算相关系数。使用Wilcoxon秩和检验或Student’s t-tests比较分析两个连续变量之间的差异,卡方检验或Fisher精确检验用于分类变量间的比较。p<0.05被认为是一个显著的差异。
实施例1
首先对用于前列腺癌辅助诊断和预后评估的mLs模型进行构建,具体步骤如下:
(1)获取原发性前列腺癌患者肿瘤组织样本(primary组)和转移性前列腺癌患者肿瘤组织样本(metastatic组),使用Trizol从样品中提取总RNA,并使用NanoDrop ND-1000(Thermo Fisher Scientific)评估质量,随后分别利用MeRIP-seq和RNA-seq分析对其主要成分(PCA)进行分析,并获得各组中的m6A峰及LncRNA数量。其中RNA-seq、MeRIP-seq均按照本领域的常规方法进行,具体地,对于RNA-seq,首先使用
结果如图1-2所示。结果显示,primary组和metastasis组在PCA方面基本没有重叠(参见图1)。在primary组中,共鉴定出11313个m6A峰,对应7931个LncRNA;在metastasis组中,共鉴定出10971个m6A峰,对应7802个LncRNA。在这两组中,重叠的m6A peaks和LncRNA数量分别为7696和6013,而primary组特有的m6A峰和LncRNA数量分别为3617和1918,metastasis组特有的m6Apeaks和LncRNA数量分别为3275,1789(参见图2)。
(2)对primary组和metastasis组中重叠的m6A峰和LncRNA进行分析,并按照如下条件对所获得的重叠的m6A峰及LncRNA进行差异表达识别,以获得两组中具有显著差异丰度的m6A峰和差异表达的LncRNA:
p<0.05且|fold change|>2。
分析结果如图3-4所示。结果显示,primary组和metastasis组的7696个m6A重叠峰中有3712个具有显著差异丰度,其中有1764个显著低甲基化(hypo-methylated)的m6A峰和1948个显著高甲基化(hyper-methylated)的m6A峰(参见图3)。此外,两组共有的6013个LncRNA中有92个存在差异表达,其中包括54个表达下调的LncRNAs和38个表达上调的LncRNAs(参见图4)。
(3)为了研究m6A methylation与lncRNA表达量之间的关系,进一步对步骤(2)所获得的primary组和metastasis组中具有差异丰度的m6A峰和差异表达的LncRNA进行交叉分析(cross-analysis),获得同时具有甲基化差异以及表达水平差异的LncRNA。
结果如图5-6所示。结果显示,primary组和metastasis组之间存在21个同时具有甲基化差异以及表达水平差异的LncRNA,其中10个高甲基化的LncRNA显著上调,11个低甲基化的LncRNA显著下调(参见图5)。通过对MeRIP-seq和RNA-seq数据的相关性分析进一步证明了m6A峰差异丰度与LncRNA差异表达量之间呈现明显的正相关性(参见图6)。
(4)利用PCaDB工具(http://bioinfo.jialab-ucr.org/PCaDB/)中“Transcriptome Analysis-Prognostic Model”模块的“CoxLasso”方法的对步骤(3)获得的同时具有甲基化差异以及表达水平差异的LncRNA进行分析,筛选得到9个与前列腺癌具有显著相关性的LncRNA,分别为COL1A1、MIR210HG、AL158071.5、SNHG8、AL390719.2、LINC00920、PCA3、LINC01088和AF165147.1。
(5)基于步骤(4)中9个与前列腺癌具有显著相关性的LncRNA按照如下公式进行mLs的计算:
其中N代表所检测的标志物的数量,本实施例中N=9;Coef
本实施例中,各标志物的Coef
表1各LncRNACoef
实施例2
为了评估mLs的预后价值,以TCGA-PRAD作为训练集,以Taylor和GSE54460作为验证集,采用Kaplan-Meier进行生存分析。分析结果如图7-8所示。结果显示,在TCGA-PRAD训练集和2个验证集中,mLs高得分组患者的生存率状况明显不如mLs低得分组患者,且mLs高得分组患者的BCR发生时间均明显早于mLs低得分组患者。
此外,在包括mLs、前列腺癌格利森评分(Gleason Score)、年龄、TNM分期和前列腺特异性抗原(Prostate-Specific Antigen(PSA))等临床因素在内的单、多因素Cox回归分析表明,在3个长期队列中,mLs均可作为前列腺癌BCR的独立危险因素(p<0.05,HR>1)(参见图9)。通过在3个队列中对各个临床因素变量的C指数进行计算比较发现,mLs的表现与其他几个临床指标相当甚至优于其他临床指标,尤其在TCGA-PRAD队列中(参见图10)。这表明mLs在大多数长期队列中具有可靠稳定的预测性能。
随后,选择现有技术中的5个基于LncRNA表达量构建前列腺癌预测模型,分别于TCGA-PRA、Taylor和GSE54460这3个队列中进行C指数的计算并与mLs相比较,所述5个预测模型的PMID分别为35850679(Liu.5.BC)、31281469(Shao.5.JC)、30860081(Shao.7.AJA)、35860569(Tang.10.FIO)、35731271(Liu.5.JCRCO)。分析结果如图11所示,结果显示,在个长期队列中,本发明的mLs的C指数均明显优于其他预测模型。这表明mLs在相对于现有的前列腺癌预测模型具有更加可靠稳定的预测性能。
进一步地,根据包括mLs、前列腺癌格利森评分(Gleason Score)、年龄、TNM分期和前列腺特异性抗原(Prostate-Specific Antigen(PSA))等在内的临床指标采用rms包(version 6.5-0)分别对前列腺癌患者3年、5年以及8年无生化复发(BCR-free)生存期进行列线图绘制(参见图12)。通过校准分析发现,图12列线图所预测的3年、5年以及8年无生化复发生存期与实际情况基本一致(参见图13),列线图中3年、5年以及8年无生化复发生存期的AUC值分别为0.795、0.836和0.822(参见图14),表明本发明所构建的列线图是准确可靠的。综合上述结果进一步证实了本发明所构建的mLs具有稳定可靠的预测价值。
实施例3
为了进一步研究mLs相关的功能通路,从MSigDB(https://www.gsea-msigdb.org/gsea/msigdb/)数据库获取h.all.v7.4.symbols.gmt基因集。使用Spearman相关性分析分别评估mLs与所有mRNA之间的相关性。然后,将所有mRNA按照相关系数降序排列,排序的基因列表进一步使用clusterProfiler包(version 4.2.2)进行GSEA(Gene set enrichmentanalysis)分析。其中,FDR(false Discovery Rate)<0.05的基因集被认为是显著富集。分析结果显示,mLs高得分组主要富集了许多与肿瘤进展和转移相关的通路,例如mitotic、UVresponse、TGFβsignaling、KRAS signaling和G2M checkpoint等;而mLs低得分组则主要与能量代谢和脂代谢有关,如氧化磷酸化、脂肪酸代谢和胆固醇顺势疗法等。
为了探讨mLs中的代谢重编程谱,我们利用KEGG数据库中的metabolic pathways进行了GSVA分析,具体如下:从KEGG(https://www.genome.jp/kegg/)数据库获取metabolic pathways并进行GSVA。limma包(version 3.50.3)用于识别高mLs组和低mLs组之间显著变化的通路,p<0.05被认为具有显著性差异。结果显示,mLs与能量代谢和脂质代谢等呈负相关。其中,mLs低得分组中脂肪酸降解和脂肪酸延长的激活程度显著增高(参见图15),且mLs与脂肪酸降解和脂肪酸延长呈现明显的负相关(参见图16)。
前述研究结果表明了mLs在前列腺癌预后预测中具有稳健优异的预测能力。为了进一步明确mLs在其他类型癌症中是否具有同样的预测性能,采用mLs进行了泛癌分析。结果表明,间皮瘤(MESO)、胰腺癌(PAAD)、乳腺浸润性癌(BRCA)、胆管癌(CHOL)、卵巢浆液性囊腺癌(OV)等肿瘤中mLs的得分较高,而脑低度胶质瘤(LGG)、葡萄膜黑色素瘤(UVM)、胶质母细胞瘤细胞瘤(GBM)、肝癌(LIHC)等肿瘤中mLs的得分较低(参见图17)。Kaplan-Meier生存分析表明,mLs高得分组患者的疾病相关存活率(DSS)、总体生存率(OS)和无进展间隔(PFI)均明显劣于mLs低得分组(参见图18)。
进一步地单因素Cox分析结果表明,mLs是脑低度胶质瘤(LGG)、胶质母细胞瘤细胞瘤(GBM)、肺鳞状细胞癌(LUSC)、肾乳头状细胞癌(KIRP)、肾透明细胞癌(KIRC)、肾嫌色细胞癌(KICH)、宫颈鳞状细胞癌和宫颈内腺癌(CESC)、肾上腺皮质癌(ACC)和葡萄膜黑色素瘤(UVM)的危险因素,是淋巴样肿瘤弥漫性大b细胞淋巴瘤(DLBC)的保护性因素(参见图19)。综合上述结果可以明确,mLs不仅可作为前列腺癌预后的特异性指标,同时在其他癌种中具备同样稳健的预测性能和外部泛化能力,因此在其他肿瘤中的应用仍需进一步研究。
实施例4
前述实施例中明确了mLs模型构建所基于的LncRNA标志物,对此,选取其中风险因素最高的标志物之一MIR210HG通过功能性实验对mLs模型的预测功能进行进一步验证。
首先,选择前列腺癌细胞,采用RT-qPCR对其中MIR210HG的表达水平进行检测,具体步骤如下:
(1)将前列腺癌细胞(PC-3、22Rv1、C4-2和C4-2B)分别培养于6孔板中,待细胞生长至对数期时分别收集1×10
(2)向细胞液中加入1mL Trizol溶液,吹打混匀使细胞充分裂解,静置5min;加入200μL氯仿,剧烈振荡混匀30s,使水相和有机相充分解除,室温静置2min。
(3)4℃下12000g离心10min,可见分为三层,其中RNA在上层水相中,转移至新的RNase-free EP管中;加入等体积异丙醇,轻柔地充分混匀,于-20℃静置10min。
(4)4℃下12000g离心10min,收集RNA沉淀,取上清,用75%乙醇洗涤两次,超净台风干,加入20-60μL DEPC水溶解沉淀;总RNA的纯度控制OD
(5)取1μg总RNA作为模板RNA,采用EasyScript First-Strand cDNA SynthesisSuperMix(TransGen,货号AE301-02)进行逆转录,逆转录反应体系按照试剂盒说明书或者本领域的常规体系进行,反应条件为:25℃保温10min;50℃保温30min;85℃保温5min。
(6)采用RT-qPCR检测前列腺癌细胞中MIR210HG表达水平,所采用的序列如下:MIR210HG-F:GGCAGATTTAGTGGACGCCT;MIR210HG-R:GTTAGCTCTGCAGGTGTGGA;qRT-PCR反应过程按照Power SYBR Green PCR Master Mix说明书操作;通过融解曲线分析和电泳确定目的条带,ΔΔCT法进行相对定量。
检测结果如图20所示。结果显示,在前列腺癌细胞中MIR210HG表达水平明显升高(*p<0.05,**p<0.01,***p<0.001,****p<0.0001,以正常前列腺上皮细胞系中MIR210HG表达水平作为对照)。
随后,收集前列腺癌患者的肿瘤组织进行FISH分析,具体步骤如下:将前列腺癌细胞于15mm的共聚焦培养皿(JET BIOFIL)中进行培养,针对homo-MIR210HG进行探针序列设计(Ribo Bio)(建议提供具体的序列),按照Ribo
考虑到MIR210HG的表达与mLs模型中的m6A修饰密切相关,随后进行了MeRIP-qPCRassay,具体步骤如下:收集前列腺癌细胞(22Rv1、PC-3),使用Trizol从样品中提取总RNA,并使用NanoDrop ND-1000(Thermo Fisher Scientific)评估质量,将RNA片段化并按一定比例作为输入对照。针对MeRIP-seq结果鉴定的甲基化位点设计引物(MIR210HG-F:GGCAGATTTAGTGGACGCCT;MIR210HG-R:GTTAGCTCTGCAGGTGTGGA),利用上述引物进行qPCR,进一步分析m6A的富集情况。通过将扩增周期值归一化到相应的输入部分,计算MIR210HG基因上m6A的相对富集程度。分析结果如图22所示,结果显示,在PC-3和22Rv1细胞系中抗m6A抗体可以显著富集MIR210HG,这表明MIR210HG可受到m6A直接修饰。
为了验证MIR210HG对前列腺癌的作用,基于MIR210HG序列设计并合成了三种siRNA,其序列分别为:si-1:CAACACAGTTCACAATATA;si-2:GAGCTAACTTACTGCCAGA;si-3:GAAATAACCAAGCCGAGTT)。将其分别转染至PC-3和22Rv1细胞中,通过上述RT-qPCR对其中MIR210HG表达情况进行检测。结果显示,si-1和si-3均能够对MIR210HG的表达产生有效抑制作用,显著下调PC-3和22Rv1细胞中MIR210HG的表达水平(参见图23)。
进一步地,通过细胞增殖实验、平板集落形成实验以及Transwell实验发现,在利用si-1和si-3对MIR210HG的表达水平进行抑制后,能够显著降低前列腺癌细胞的增殖能力、集落形成能力以及侵袭能力(参见图24-26,*p<0.05,***p<0.001),更进一步验证了本发明mLs对前列腺癌预测的准确性和可靠性。
近年来,随着高通量测序等新兴技术的飞速发展,研究人员越来越多地关注到RNA修饰等表观遗传机制。大量研究表明,RNA修饰等表观遗传参与调控细胞分化、个体发育以及肿瘤发生、发展等众多生物学过程。在众多RNA修饰类型中,m6A甲基化是最常见和最丰富的RNA表观遗传修饰,m6A修饰是通过信使核糖核酸加工中多个步骤的修饰来调节基因表达的重要途径。然而,随着转录组学研究的不断深入,研究人员发现m6A修饰也以多种方式影响着ncRNA,其中LncRNA是丰度较高的ncRNA。目前越来越多的研究集中在分析m6A修饰和LncRNA在肿瘤进展中的作用,然而,涉及m6A修饰LncRNA在前列腺癌中的研究数量有限,这些LncRNA在前列腺癌中的作用仍有待阐明。
本发明通过收集前列腺癌组织标本进行MeRIP-seq和RNA-seq分析,明确了原发性前列腺癌和转移性前列腺癌中的m6A修饰方式,初步分析了m6A修饰LncRNA在前列腺癌预后中的作用,发现原发性前列腺癌样本和转移性前列腺癌样本中的m6A修饰方式存在差异,其中原发性前列腺癌组中的总m6A水平更高,并且比转移性前列腺癌组多342个m6A峰。根据MeRIP-seq和RNA-seq的分析结果,筛选出了21个同时具有差异甲基化和差异表达水平的LncRNA。为了进一步分析这些LncRNA在前列腺癌中的作用,利用3个多中心队列构建了一种包含9个m6A修饰LncRNAs的前列腺癌预后模型(mLs)。通过验证发现,无论是在3个临床队列(TCGA-PRAD、Taylor还是GSE54460)中还是与其他5个已知的前列腺癌预后指标进行外部相比,mLs均表现出了稳定的预测性能。在这9个LncRNA中,通过选择较高丰度的MIR210HG进行了进一步的验证,发现其作为一个促癌因子,能够显著促进前列腺癌的增殖和侵袭,由此进一步证实了本发明mLs准确可靠的预测性能。
以上具体实施方式部分对本发明所涉及的分析方法进行了具体的介绍。应当注意的是,上述介绍仅是为了帮助本领域技术人员更好地理解本发明的方法及思路,而不是对相关内容的限制。在不脱离本发明原理的情况下,本领域技术人员还可以对本发明进行适当的调整或修改,上述调整和修改也应当属于本发明的保护范围。
- 一种微流控芯片和基于该芯片的肿瘤转移模型及模型构建方法和应用
- 基于类器官方法构建正常免疫小鼠人胃癌移植瘤模型的方法和应用
- 基于类器官方法的患者来源的肝癌正常免疫小鼠移植瘤模型的构建方法及其应用
- 基于RNA结合蛋白相关lncRNA组合构建的模型在制备预测浸润性乳腺癌预后产品中的应用
- 基于m6A相关lncRNA的浆液性卵巢癌预后模型的构建及其临床应用