一种麦角甾醇化学对照品的制备方法
文献发布时间:2023-06-19 11:40:48
技术领域
一种麦角甾醇化学对照品的制备方法,尤其涉及一种西南手参内生菌Tulasnellaceae sp.中麦角甾醇化学对照品的分离制备方法属于技术领域。
背景技术
麦角甾醇,别名麦角固醇,平面结构图见图7,为白色或无色光亮的小叶晶或白色结晶粉末。麦角甾醇不但具有独特的生理作用,还被广泛应用到药物的开发中。麦角甾醇作为真菌细胞膜的重要组成成分,结构稳定,专一性强,对测定生物量来说,它比葡糖胺更具代表性,所以可以通过检测麦角甾醇的含量来测量真菌的生物量。麦角甾醇的来源主要通过微生物发酵合成得到,近年来也有学者从一些菌丝体中提取。
西南手参(Gymnadenia orchidis)系属手参属(Gymnadenia)、兰科(Orchidaceae)植物。现代研究表明手参属植物中的化学成分主要包括天麻素类。现代药理学的研究表明,手参属植物(包含西南手参)化学成分具有多方面的药理作用,如益智、镇静、助眠等功效。目前,并未有西南手参引种栽培成功范例,为了加快西南手参可持续利用和相关新药的研发步伐,从中分离培养其所含的内生菌并发展内生菌中高纯度化学对照品的高效制备方法,尤其规模化制备技术显得尤为重要。
目前,尚未见有关从西南手参中分离培养内生菌及从中分离化学对照品的报道,已有的研究也未做到系统化的研究。因此,亟需建立一种工艺简单、规模化从西南手参内生菌Tulasnellaceae sp中快速制备化学对照品的方法。
发明内容
本发明所要解决的技术问题是:克服现有技术的不足,提供一种高效、高纯度的从西南手参内生菌Tulasnellaceae sp中快速制备化学对照品的方法。
本发明解决其技术问题所采用的技术方案是:一种麦角甾醇化学对照品的制备方法,其特征在于:包括以下步骤:
1)西南手参内生菌Tulasnellaceae sp.粉碎后加入其质量6~10倍的乙酸乙酯,在室温下提取、过滤,得滤液A,滤液A减压干燥,得西南手参内生菌Tulasnellaceae sp.乙酸乙酯提取物样品;
2)西南手参内生菌Tulasnellaceae sp.乙酸乙酯提取物样品用二氯甲烷溶解,配制样品浓度为100.0~400.0 mg/mL;该样品经中压色谱塔分离,经检测波长为270 nm的紫外检测器检测,收集制备色谱图中17~22分钟的色谱峰对应馏分,该馏分经减压干燥即得含有目标成分的组分;
3)所述含有目标成分的组分用95~100%乙醇水溶液溶解,配制样品浓度为10.0~20.0 mg/mL,经0.45 μm微孔滤膜过滤,得到滤液B,滤液B经反相C18柱分离,经检测波长为270 nm的紫外检测器检测,收集制备色谱图中25~28分钟色谱峰对应的馏分,该色谱峰馏分经减压干燥得到麦角甾醇化学对照品。
西南手参内生菌
优选的,步骤1)、2)和3)中所述的减压干燥条件为:真空度50~250 mbar,温度40~60℃。
优选的,步骤2)所述的中压色谱塔中装填有不定形硅胶材料。
优选的,步骤2)所述的中压色谱塔工作条件为:硅胶柱分离的工作参数为色谱柱柱长460 mm、直径15~49 mm,流动相A为二氯甲烷,B为甲醇,色谱分离条件为:0~30min,0~35%B,进样量为2.2~10 g,流速为10~59 mL/min。
优选的,步骤3)所述的反相C18柱工作条件为:色谱柱柱长250 mm、直径10~50 mm,反相C18柱为5 μm的耐纯水C18或5 μm的常规C18,流动相为体积分数75~95%的乙醇-水溶液,进样体积为0.5~10 mL,流速为5~80 mL/min。
优选的,步骤1)所述的西南手参内生菌Tulasnellaceae sp.粉碎前经过阴干。阴干后降低水分再提取,更易于乙酸乙酯将有效成分溶出,提高提取率。
优选的,步骤1)所述的提取为:提取3次,每次加入西南手参内生菌Tulasnellaceae sp.质量6~10倍的乙酸乙酯。分次提取,能够充分将有效成分溶出,提高提取率。
优选的,每次提取2~4h。保证有效成分充分溶出。
与现有技术相比,本发明所具有的有益效果是:所用的原料西南手参内生菌Tulasnellaceae sp.易于培养、成本低,易于规模化;使用的提取剂、硅胶柱富集溶剂、反向C18柱分离溶剂均可回收利用,不定性硅胶与反向C18材料均可重复利用,大规模实施成本低;采用一步中压色谱富集与一步高压色谱分离结合,能够保证产品纯度大于98%;分离材料可以装于中压柱系统并接在制备液相色谱上,并且使用的反相C18柱,为快速的等度方法,均非常适宜大规模生产。
附图说明
图1为实施例1硅胶柱富集色谱图;
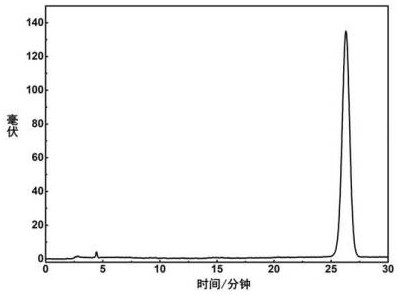
图2为实施例1反相C18柱分离色谱图;
图3为本发明西南手参内生菌
图4为本发明分离得到的麦角甾醇化学对照品ESI+分子离子峰质谱图;
图5为本发明分离得到的麦角甾醇化学对照品
图6为本发明分离得到的麦角甾醇化学对照品
图7为本发明分离得到的麦角甾醇平面结构图。
具体实施方式
下面结合实施例对本发明做进一步说明,实施例1是本发明的最佳实施例。
实施例1
一种麦角甾醇化学对照品的制备方法,包括以下步骤:
1)将阴干的西南手参内生菌Tulasnellaceae sp.32.5g粉碎后加入其质量6倍的乙酸乙酯,在室温下提取3次,每次3h,过滤,合并滤液,得滤液A,滤液A减压干燥,得西南手参内生菌Tulasnellaceae sp.乙酸乙酯提取物样品2.2g;减压干燥的条件为真空度50mbar,温度40℃。
2)西南手参内生菌Tulasnellaceae sp.乙酸乙酯提取物样品用二氯甲烷溶解,配制样品浓度为366.7mg/mL;该样品经中压色谱塔分离,经检测波长为270 nm的紫外检测器检测,收集制备色谱图中17.2~21.3分钟(参照图1)的色谱峰馏分,该馏分经减压干燥即得含有目标成分的组分586mg;减压干燥的条件为真空度50 mbar,温度40℃;硅胶柱分离的工作参数为色谱柱柱长460 mm、直径10 mm,流动相A为二氯甲烷,B为甲醇,色谱分离条件为:0~30 min,0~35%B,进样量为2 g,流速为10 mL/min。
3)所述含有目标成分的组分用95%体积浓度乙醇溶液溶解,配制样品浓度为10.0mg/mL,经0.45 μm微孔滤膜过滤,得到滤液B,滤液B经反相C18柱分离,经检测波长为270 nm的紫外检测器检测,收集制备色谱图中25~27.9分钟(参照图2)色谱峰馏分,该色谱峰馏分经减压干燥得到纯度大于98%麦角甾醇化学对照品287mg;减压干燥的条件为真空度50 mbar,温度40℃;反相C18柱分离的工作参数是指色谱柱柱长250 mm、直径10 mm,反相C18柱为5 μm的耐纯水C18(Reprosil C18),流动相为体积分数85%的乙醇-水溶液,进样体积为0.5 mL,流速为5 mL/min。
实施例2
一种麦角甾醇化学对照品的制备方法,包括以下步骤:
1)将阴干的西南手参内生菌Tulasnellaceae sp.300g粉碎后加入其质量10倍的乙酸乙酯,在室温下提取3次,每次2h,过滤,合并滤液,得滤液A,滤液A减压干燥,得西南手参内生菌Tulasnellaceae sp.乙酸乙酯提取物样品20.5g;减压干燥的条件为真空度250mbar,温度60℃。
2)西南手参内生菌Tulasnellaceae sp.乙酸乙酯提取物样品用二氯甲烷溶解,配制样品浓度为400mg/mL;该样品经中压色谱塔分离,经检测波长为270 nm的紫外检测器检测,收集制备色谱图中17.2~21.3分钟的色谱峰馏分,该馏分经减压干燥即得含有目标成分的组分5.3g;减压干燥的条件为真空度250 mbar,温度60℃;硅胶柱分离的工作参数为色谱柱柱长460 mm、直径49mm,流动相A为二氯甲烷,B为甲醇,色谱分离条件为:0~30 min,0~35%B,进样量为10 g,流速为59mL/min。
3)所述含有目标成分的组分用95%体积浓度乙醇溶液溶解,配制样品浓度为20.0mg/mL,经0.45 μm微孔滤膜过滤,得到滤液B,滤液B经反相C18柱分离,经检测波长为270 nm的紫外检测器检测,收集制备色谱图中25~27.9分钟色谱峰馏分,该色谱峰馏分经减压干燥得到纯度大于98%麦角甾醇化学对照品2.7g;减压干燥的条件为真空度250 mbar,温度60℃;反相C18柱分离的工作参数是指色谱柱柱长250 mm、直径50 mm,反相C18柱为5 μm的常规C18(Odyssil C18),流动相为体积分数95%的乙醇-水溶液,进样体积为10 mL,流速为80mL/min。
实施例3
一种麦角甾醇化学对照品的制备方法,包括以下步骤:
1)将阴干的西南手参内生菌Tulasnellaceae sp.161.8g粉碎后加入其质量6倍的乙酸乙酯,在室温下提取3次,每次4h,过滤,合并滤液,得滤液A,滤液A减压干燥,得西南手参内生菌Tulasnellaceae sp.乙酸乙酯提取物样品10.6g;减压干燥的条件为真空度150mbar,温度50℃。
2)西南手参内生菌Tulasnellaceae sp.乙酸乙酯提取物样品用二氯甲烷溶解,配制样品浓度为100mg/mL;该样品经中压色谱塔分离,经检测波长为270 nm的紫外检测器检测,收集制备色谱图中17.2~21.3分钟的色谱峰馏分,该馏分经减压干燥即得含有目标成分的组分2.8g;减压干燥的条件为真空度150 mbar,温度50℃;硅胶柱分离的工作参数为色谱柱柱长460 mm、直径36 mm,流动相A为二氯甲烷,B为甲醇,色谱分离条件为:0~30 min,0~35%B,进样量为5 g,流速为30 mL/min。
3)所述含有目标成分的组分用95%体积浓度乙醇溶液溶解,配制样品浓度为15.0mg/mL,经0.45 μm微孔滤膜过滤,得到滤液B,滤液B经反相C18柱分离,经检测波长为270 nm的紫外检测器检测,收集制备色谱图中25~27.9分钟色谱峰馏分,该色谱峰馏分经减压干燥得到纯度大于98%麦角甾醇化学对照品1.3g;减压干燥的条件为真空度150 mbar,温度50℃;反相C18柱分离的工作参数是指色谱柱柱长250 mm、直径20 mm,反相C18柱为5 μm的耐纯水C18(Reprosil C18),流动相为体积分数75%的乙醇-水溶液,进样体积为5 mL,流速为20 mL/min。
对比例1
一种麦角甾醇化学对照品的制备方法,在实施例1的基础上,步骤3)配置样品浓度为8mg/mL,其他条件相同。最终获得纯度大于98%麦角甾醇化学对照品197mg。
对比例2
一种麦角甾醇化学对照品的制备方法,在实施例1的基础上,步骤3)配置样品浓度为23mg/mL,其他条件相同。最终获得纯度88%麦角甾醇化学对照品302mg。
性能测试
对实施例2步骤2)所得“含有目标成分的组分2.8g”经色谱检测,检测结果见图3,可证明其纯度大于98%。
对实施例3步骤3)所得纯度大于98%的麦角甾醇化学对照品做核磁共振检测,总终检测所得质谱图、氢图、碳图分别见图4~6,可证明所得产物为麦角甾醇。
以上所述,仅是本发明的较佳实施例而已,并非是对本发明作其它形式的限制,任何熟悉本专业的技术人员可能利用上述揭示的技术内容加以变更或改型为等同变化的等效实施例。但是凡是未脱离本发明技术方案内容,依据本发明的技术实质对以上实施例所作的任何简单修改、等同变化与改型,仍属于本发明技术方案的保护范围。
- 一种麦角甾醇化学对照品的制备方法
- 一种富含麦角甾醇的酵母菌株及麦角酵母粉的制备方法