一种养血清脑醇提浸膏中有关物质含量测定方法
文献发布时间:2024-04-18 20:00:50
技术领域
本发明属于药物检测领域,涉及一种养血清脑醇提浸膏中有关物质含量测定方法。
背景技术
养血清脑制剂是一种由当归、川芎、延胡索、决明子等11味中药作为原料药制备而成的中成药,具有养血平肝,活血通络作用,用于血虚肝旺所致头痛,眩晕眼花,心烦易怒,失眠多梦。养血清脑制剂在制备过程中部分中药采用醇提技术得到醇提浸膏,投入下一步制备,因此,醇提浸膏是养血清脑制剂的中间体,其制备方法如下:
当归、川芎、延胡索、决明子加70%乙醇回流提取二次,第一次2小时,第二次1小时,滤过,回收乙醇并浓缩至适量,即得。
因此,养血清脑制剂醇提浸膏中,含有当归、川芎、延胡索、决明子中的药物成分。
现有技术中尚无对当归、川芎、延胡索、决明子醇提物中阿魏酸、洋川芎内酯A、藁本内酯、橙黄决明素、大黄素和大黄酚的含量测定方法。
文献《UPLC-Q-TOF/MS
随着中药现代化、国际化工作的深入开展,国内外药品监管机构均对中药质量控制提出了更严格的要求。为了提高中药产品质量一致性,加强对整个生产过程的质量控制,亟须开发一种准确和方便的养血清脑醇提浸膏中有关物质的质量检测和控制方法。
发明内容
本发明提供一种检测养血清脑醇提浸膏中阿魏酸、洋川芎内酯A、藁本内酯、橙黄决明素、大黄素、大黄酚含量的方法,首先通过固相萃取柱预处理的方式,得到供试品溶液,再通过UPLC技术对样品液进行分析,得到以上各组分的含量。另外的,本发明还提供了检测养血清脑醇提浸膏中阿魏酸、洋川芎内酯A、藁本内酯、橙黄决明素、大黄素、大黄酚以及生物碱,包括延胡索乙素、延胡索甲素、四氢非洲防己碱、四氢黄连碱和四氢小檗碱的含量的方法。首先通过固相萃取柱预处理的方式,采用不同的洗脱液洗脱固相萃取柱,从一份样品中先后获得两份供试品溶液,再通过UPLC技术对样品液进行分析,得到上述11种相关组分的含量。本发明公开的测定方法具有精密度、重复性、样品稳定性和加样回收率良好的优点,结果符合含量测定方法学要求,且从一份样品中得到两份供试品溶液的方法有简化了检测步骤,缩短了检测时间的优点。
本发明要求保护以下技术方案:
一种养血清脑醇提浸膏中有关物质含量测定方法,包括以下步骤:
(1)供试品溶液1的配制:养血清脑醇提浸膏0.06-0.14g用含0.5-5%乙酸的60-70%甲醇水溶液溶解;上样至阳离子交换固相萃取柱,用洗脱液A洗脱得到供试品溶液1;其中,所述洗脱液A为含0.5-5%乙酸的60-70%甲醇水溶液;
任选的步骤(2),供试品溶液2的配制:萃取柱继续用洗脱液B洗脱得到供试品溶液2;其中,所述洗脱液B为1-15%氨水甲醇溶液;所述1-15%氨水甲醇溶液是指1-15%氨水80-100%甲醇溶液;
(3)含量测定:将供试品溶液1和/或供试品溶液2分别注入超高效液相色谱仪,得到色谱图,根据色谱图计算供试品溶液中有关物质的含量。
其中,对于供试品溶液1的测定,色谱条件如下:
色谱柱类型:适用于UPLC的十八烷基硅烷键合硅胶柱;
流动相:流动相A为0.2%甲酸水溶液,流动相B为乙腈;
梯度洗脱:流速:0.35-0.45mL/min;检测波长:265-290nm;所述梯度洗脱程序如下表所示。
进样量在1-3μL,优选为2μL。
优选的,梯度洗脱流速为0.4mL/min,检测波长为280nm。
所述适用于UPLC的十八烷基硅烷键合硅胶柱选自Waters HSS T3色谱柱(2.1x100mm),Kinetex(2.1x100mm)或ACE Excel C18(2.1x100mm)。优选的,所述适用于UPLC的十八烷基硅烷键合硅胶柱是Waters HSS T3色谱柱。
其中,对于供试品溶液2的测定,色谱条件如下:
色谱柱:ACQUITY UPLC BEH C18色谱柱或ACQUITY UPLC CSH C18色谱柱;
流动相:流动相A为0.05%乙酸铵水溶液,流动相B为乙腈;
梯度洗脱;流速:0.35-0.45mL/min;检测波长:270-290nm;所述梯度洗脱程序如下表。
进样量在1-3μL,优选为2μL。
优选的,梯度洗脱流速为0.4mL/min,检测波长为280nm。
根据需要,本发明的方法中还可以包括以下步骤:
对照品溶液1的制备:分别取阿魏酸、橙黄决明素、洋川芎内酯A、藁本内酯、大黄素和大黄酚对照品,加甲醇配制成每毫升含阿魏酸、橙黄决明素、洋川芎内酯A、藁本内酯、大黄素和大黄酚分别为0.01mg、0.04mg、1.00mg、0.15mg、0.01mg和0.005mg的混合溶液。对照品溶液1作为供试品溶液1的标准对照品,在测定供试品溶液1的含量时可以分别注入超高效液相色谱仪。
对照品溶液2的制备:分别取延胡索乙素、延胡索甲素、四氢非洲防己碱、四氢黄连碱和四氢小檗碱对照品,加甲醇配制成每毫升含延胡索乙素、延胡索甲素、四氢非洲防己碱、四氢黄连碱和四氢小檗碱分别为0.01mg、0.02mg、0.01mg、0.005mg和0.005mg的混合溶液。对照品溶液2作为供试品溶液2的标准对照品,在测定供试品溶液2的含量时可以分别注入超高效液相色谱仪。
优选的,在一些实施方案中,在步骤(1)中,所述养血清脑醇提浸膏溶液是由所述养血清脑醇提浸膏0.1g用8mL含1%乙酸的65%甲醇水溶液溶解制成。
优选的,在一些实施方案中,步骤(1)中,所述供试品溶液1的制备方法包含以下步骤,将所述养血清脑醇提浸膏溶液上样至阳离子交换固相萃取柱,用2mL洗脱液A洗脱,再用13ml甲醇作为洗涤液洗涤固相萃取柱,合并上样液,洗脱液和洗涤液,并用甲醇定容至25mL,得到供试品溶液1;所述洗脱液A为含1%乙酸的65%甲醇水溶液。
优选的,步骤(2)中,得到供试品溶液1后,再用8mL洗脱液B洗脱固相萃取柱,收集洗脱液,并用5%氨水甲醇溶液定容至10mL,得到供试品溶液2;所述洗脱液B为5%氨水甲醇溶液。
其中,步骤(1),(2)中所述阳离子交换固相萃取柱选自ProElut PXC固相萃取柱或Cleanert PCX固相萃取柱。优选的,所述阳离子交换固相萃取柱是ProElut PXC固相萃取柱。
本发明的测定方法,其中,任选的,包括的含义如下:
本发明只采用测定方法A,即对供试品溶液1的检测,检测成分为阿魏酸、橙黄决明素、洋川芎内酯A、藁本内酯、大黄素和大黄酚。
本发明只采用测定方法B,即对供试品溶液2的检测,检测成分为延胡索乙素、延胡索甲素、四氢非洲防己碱、四氢黄连碱和四氢小檗碱;也可以不包括。
本发明采用测定方法A和测定方法B的组合,即对供试品溶液1和供试品溶液2的检测。检测成分为阿魏酸、橙黄决明素、洋川芎内酯A、藁本内酯、大黄素和大黄酚,延胡索乙素、延胡索甲素、四氢非洲防己碱、四氢黄连碱和四氢小檗碱。本发明公开的测定养血清脑醇提浸膏中有关物质的方法,具有如下有益效果:
1.简化了检测步骤:本发明公开了用同一份样本检测阿魏酸、洋川芎内酯A、藁本内酯、橙黄决明素、大黄素、大黄酚,以及5种生物碱含量的方法。采用了固相萃取柱预处理的方式,通过采用不同的洗脱液洗脱,从一份样品中先后获得两份供试品溶液,可以检测一共11种成分含量,简化了检测步骤;
2.缩短了检测时间:本专利采用超高效液相色谱法进行测定,快速高效,供试品溶液1和供试品溶液2分别在17分钟和7分钟内实现6种和5种成分的有效分离,色谱稳定性良好;
3.线性关系好:本发明公开的检测方法养血清脑醇提浸膏中阿魏酸、洋川芎内酯A、藁本内酯、橙黄决明素、大黄素和大黄酚的含量等6种成分在相当大的浓度范围内,峰面积和浓度呈良好的线性关系;
4.进样精密度好:按照本发明的测试方法进行6次相同的实验,显示6次进样的峰面积RSD均≤2.0%,显示进样精密度良好;
5.重复性好:按照本发明的测试方法进行6次相同的实验,样品含量为0.01%时,测定结果的RSD≤4.0%,样品含量为0.001%时,测定结果的RSD应≤5.0%,结果显示本发明公开的测定方法的重复性良好;
6.加样回收率高:阿魏酸、橙黄决明素、洋川芎内酯A、藁本内酯、大黄素和大黄酚的平均加样回收率分别为99.4%、95.9%、95.6%、99.5%、98.0%和95.1%,均在合格范围内,RSD均≤5.0%,符合要求;
7.样品液稳定性好:取供试品溶液分别于0、2、4、6、8、12h进样,记录阿魏酸等6种成分的峰面积,计算相对标准偏差。结果显示,12h内样品峰面积的RSD均≤2.0%,显示供试品溶液稳定性良好。
附图说明
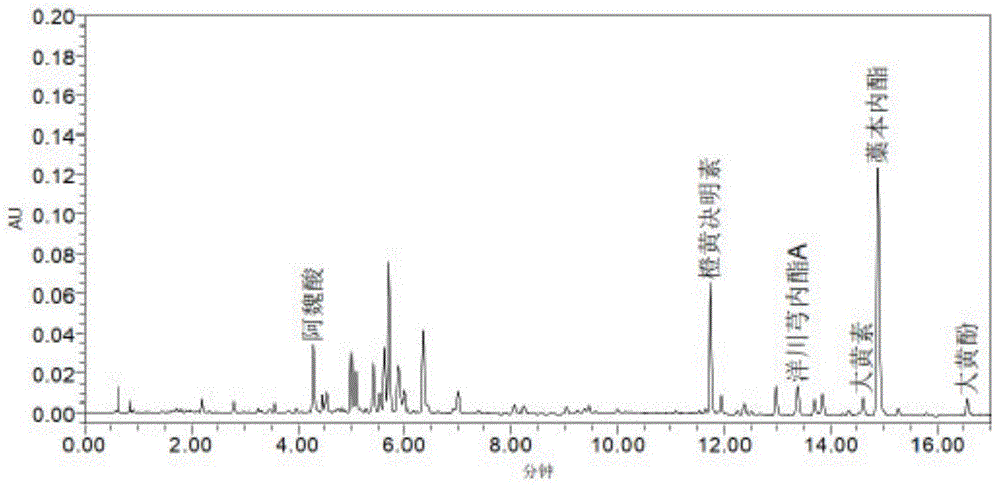
图1:测定方法A供试品溶液1色谱图;
图2:测定方法A对照品1色谱图;
图3:测定方法B供试品溶液2色谱图;
图4:测定方法B对照品2色谱图;
图5:对比例供试品溶液1色谱图。
具体实施方式
以下通过实施例进一步说明本发明,但不作为对本发明的限制。
实施例1:养血清脑醇提浸膏中相关化合物含量测定方法1
(1)供试品溶液的制备
取养血清脑醇提浸膏约0.1g,精密称定,加1%乙酸65%甲醇溶液8mL,超声溶解,上样至ProElut PXC(500mg/6mL)固相萃取柱。先用1%乙酸65%甲醇溶液2mL洗脱固相萃取柱,再用13ml甲醇作为洗涤液洗涤固相萃取柱,合并上样液、洗脱液和洗涤液至25ml量瓶中,加甲醇至刻度定容,摇匀,即得供试品溶液1;接着,用5%氨水甲醇8ml洗脱固相萃取柱,收集洗脱液至10ml量瓶中,加5%氨水甲醇定容,摇匀,得供试品溶液2。
(2)超高效液相色谱法分析
测定方法A
色谱条件:采用UPLC色谱系统,仪器为Waters Acquity超高效液相色谱仪。采用Waters HSS T3色谱柱,以0.2%甲酸水溶液为流动相A,乙腈为流动相B,按下表进行梯度洗脱,流速为0.4mL/min;检测波长:280nm。供试品溶液1色谱图见图1。
表1测定方法A梯度洗脱条件
对照品溶液配制:分别取阿魏酸、橙黄决明素、洋川芎内酯A、藁本内酯、大黄素和大黄酚对照品适量,加甲醇配制成每毫升含阿魏酸、橙黄决明素、洋川芎内酯A、藁本内酯、大黄素和大黄酚分别为0.01mg、0.04mg、1.00mg、0.15mg、0.01mg和0.005mg的混合溶液,即得对照品1。
测定法:分别精密吸取对照品1和供试品溶液1各2μl,注入液相色谱仪,测定,用外标法分别计算含量,即得。供试品溶液1色谱图见图1,对照品1的色谱图见图2。
测定方法B
采用ACQUITY UPLC BEH C18(2.1*100mm)色谱柱,以0.05%乙酸铵水溶液以流动相A,乙腈为流动相B,按下表中的规定进行梯度洗脱;流速为0.4mL/min,检测波长为280nm。
表2测定方法B梯度洗脱条件
对照品溶液的制备:
分别精密称取延胡索乙素、延胡索甲素、四氢非洲防己碱、四氢黄连碱和四氢小檗碱对照品适量,加甲醇溶解,制得每毫升含延胡索乙素和延胡索甲素分别为0.01mg、0.02mg、0.01mg、0.005mg和0.005mg的溶液,即得对照品2。
测定法:分别精密吸取对照品2和供试品溶液2各2μl,注入液相色谱仪,测定,用外标法分别计算含量,即得。供试品溶液2色谱图见图3,对照品2的色谱图见图4。
实施例2:养血清脑醇提浸膏中相关化合物含量测定方法2
(1)供试品溶液的制备
取养血清脑醇提浸膏约0.06g,精密称定,加0.5%乙酸70%甲醇溶液8mL,超声溶解,上样至Cleanert PCX(500mg/6mL))固相萃取柱。先用0.5%乙酸70%甲醇溶液2mL洗脱固相萃取柱,再用13ml甲醇作为洗涤液洗涤固相萃取柱,合并上样液、洗脱液和洗涤液至25ml量瓶中,加甲醇至刻度定容,摇匀,即得供试品溶液1;接着用1%氨水甲醇8ml洗脱固相萃取柱,收集洗脱液至10ml量瓶中,加1%氨水甲醇定容,摇匀,得供试品溶液2。
(2)超高效液相色谱法分析
测定方法A
色谱条件:采用UPLC色谱系统,仪器为Waters Acquity超高效液相色谱仪。采用Kinetex(2.1x100mm)色谱柱,以0.2%甲酸水溶液为流动相A,乙腈为流动相B,按表1进行梯度洗脱,流速为0.35mL/min;检测波长:265nm。
对照品溶液配制:同实施例1。
测定法:分别精密吸取对照品1和供试品溶液1各3μl,注入液相色谱仪,测定,用外标法分别计算含量,即得。
测定方法B
色谱条件:采用ACQUITY UPLC CSH C18(2.1*100mm)色谱柱,以0.05%乙酸铵水溶液以流动相A,乙腈为流动相B,按表2进行梯度洗脱;流速:0.35mL/min,检测波长为270nm。
对照品溶液的制备:同实施例1。
测定法:分别精密吸取对照品2和供试品溶液2各3μl,注入液相色谱仪,测定,用外标法分别计算含量,即得。
实施例3:养血清脑醇提浸膏中相关化合物含量测定方法3
(1)供试品溶液的制备
取养血清脑醇提浸膏约0.14g,精密称定,加5%乙酸60%甲醇溶液8mL,超声溶解,上样至ProElut PXC(500mg/6mL)固相萃取柱。先用5%乙酸60%甲醇溶液2mL洗脱固相萃取柱,再用13ml甲醇作为洗涤液洗涤固相萃取柱,合并上样液、洗脱液和洗涤液至25ml量瓶中,加甲醇至刻度定容,摇匀,即得供试品溶液1;接着,用15%氨水甲醇8ml洗脱至10ml量瓶中,加15%氨水甲醇至刻度,摇匀,得供试品溶液2。
(2)超高效液相色谱法分析
测定方法A
色谱条件:采用UPLC色谱系统,仪器为Waters Acquity超高效液相色谱仪。采用ACE Excel C18(2.1x100mm)色谱柱,以0.2%甲酸水溶液为流动相A,乙腈为流动相B,按表1进行梯度洗脱,流速为0.45mL/min;检测波长:290nm。
对照品溶液配制:同实施例1。
测定法:分别精密吸取对照品1和供试品溶液1各1μl,注入液相色谱仪,测定,用外标法分别计算含量,即得。
测定方法B
采用ACQUITY UPLC BEH C18(2.1*100mm)色谱柱,以0.05%乙酸铵水溶液以流动相A,乙腈为流动相B,按表2进行梯度洗脱;流速:0.45ml/min;检测波长为290nm。
对照品溶液的制备:同实施例1。
测定法:分别精密吸取对照品2和供试品溶液2各1μL,注入液相色谱仪,测定,用外标法分别计算含量,即得。
为了检验本发明检测养血清脑醇提浸膏中有关成分含量的方法是否准确和可靠,本发明对上述方法进行了方法学验证。
实验例:检测养血清脑醇提浸膏中有关成分含量测定的方法的方法学验证
1.线性和范围
精密称取经阿魏酸、橙黄决明素、芦荟大黄素、洋川芎内酯A、大黄素和大黄酚对照品适量,加甲醇配制成每毫升含阿魏酸、橙黄决明素、藁本内酯、洋川芎内酯A、大黄素和大黄酚分别为0.1mg、0.04mg、1.00mg、0.4mg、0.05mg和0.05mg的混合对照溶液,再分别取量取0.5、1、2、5、10ml混合对照溶液置于50ml量瓶中,加甲醇至刻度,混匀,制成系列溶液,按照实施例1测定方法A注入液相色谱仪,记录色谱图。以浓度对测得的峰面积进行线性回归,求得回归方程。
精密称取延胡索乙素、延胡索甲素、四氢非洲防己碱、四氢黄连碱和四氢小檗碱对照品适量,加甲醇配制成每毫升含延胡索乙素、延胡索甲素、四氢非洲防己碱、四氢黄连碱和四氢小檗碱分别约为0.1mg、0.2mg、0.1mg、0.05mg和0.05mg的混合对照储备溶液,再分别取量取0.5、1、2、5、10ml混合对照溶液置于25ml量瓶中,加甲醇至刻度,混匀,制成系列溶液,按照实施例1测定方法B注入液相色谱仪,测定,以浓度对测得的峰面积进行线性回归,求得回归方程,各成分的回归方程、相关系数及线性范围见表3。
表3回归方程、相关系数及线性范围
结果显示,阿魏酸等11种成分在上述浓度范围内,峰面积和浓度呈良好的线性关系。
2.进样精密度
分别取实施例1中供试品溶液1、2,按照实施例1测定方法A、B的色谱条件分别重复进样6次,记录阿魏酸等11种成分的峰面积,计算相对标准偏差。结果显示6次进样的峰面积RSD均≤2.0%,进样精密度良好,成分进样精密度结果见表4。
表4各相关成分进样精密度结果表
3.重复性试验
按照实施例1中供试品溶液1、2制备方法制备供试品溶液1、2各6份,按照实施例测定方法A和B的方法分别计算阿魏酸等11种成分的含量,并计算相对标准偏差。样品含量为0.01%时,测定结果的RSD应≤4.0%,样品含量为0.001%时,测定结果的RSD应≤5.0%,实验结果如表5所示,本测定方法的重复性符合验证要求。
表5各相关成分重复性试验结果表
4.回收率试验
取养血清脑醇提浸膏约0.05g,精密称定,加混合对照品溶液1ml(含阿魏酸、橙黄决明素、洋川芎内酯A、藁本内酯、大黄素、大黄酚、延胡索甲素、延胡索乙素、四氢非洲防己碱、四氢黄连碱和四氢小檗碱分别为0.075mg、0.070mg、0.108mg、0.725mg、0.025mg、0.035mg、0.031mg、0.019mg、0.012mg、0.004mg和0.078mg,加1%乙酸65%甲醇溶液7mL,超声溶解,上样至已处理好的ProElut PXC(500mg/6mL)固相萃取柱,参照实施例1中洗脱方法得到供试品溶液1和2,分别进样6次,以及参照实施例1的测定方法A和B进行色谱分析,计算回收率。
1)阿魏酸回收率(mg),结果见表6。
表6阿魏酸回收率(mg)
2)橙黄决明素回收率(mg)见表7。
表7橙黄决明素回收率(mg)
3)洋川芎内酯A回收率(mg)见表8。
表8洋川芎内酯A回收率(mg)
4)大黄素回收率(mg)见表9。
表9大黄素回收率(mg)
/>
5)藁本内酯回收率(mg)见表10。
表10藁本内酯回收率(mg)
6)大黄酚回收率(mg),见表11。
表11大黄酚回收率(mg)
7)四氢非洲防己碱回收率(mg)见表12。
表12四氢非洲防己碱回收率(mg)
8)延胡索乙素回收率(mg)见表13。
表13延胡索乙素回收率(mg)
9)四氢小檗碱回收率(mg)见表14。
表14四氢小檗碱回收率(mg)
10)延胡索甲素回收率(mg)见表15。
表15延胡索甲素回收率(mg)
11)四氢黄连碱回收率(mg)见表16。
表16四氢黄连碱回收率(mg)
结果显示,阿魏酸、橙黄决明素、洋川芎内酯A、大黄素、藁本内酯、大黄酚、延胡索甲素、延胡索乙素、四氢非洲防己碱、四氢黄连碱和四氢小檗碱的平均加样回收率分别为99.4%、95.9%、95.6%、99.5%、98.0%、95.1%、95.1%、96.6%、94.2%、95.4%和95.8%,均在合格范围内,RSD均≤5.0%,符合验证要求。
5.样品稳定性试验
按照实施例1的方法制备供试品溶液1和2,分别于0、2、4、6、8、12h进样,按照实施例1中的测定方法A和B进行检测,记录阿魏酸等11种成分的峰面积,计算相对标准偏差。结果显示,12h内样品峰面积的RSD均≤2.0%,样品稳定性良好。
表17样品稳定性试验结果
对比例1:采用现有技术(杨岱琳,佟玲,李晓稳,等.UPLC-Q-TOF/MS^E方法分析养血清脑颗粒的化学成分[J].药学学报,2016,51(5):9.)色谱条件测定养血清脑浸膏有关化合物的含量
试验方法:
(1)供试品溶液的制备方法
参照实施例1的方法制备得到的供试品溶液1。
(2)采用UPLC色谱系统,仪器为Waters Acquity超高效液相色谱仪。采用ACQUITYUPLC BEH C18(100mm×2.1mm,1.7μm)色谱柱(Waters公司),以0.1%甲酸水溶液为流动相A,乙腈为流动相B,按下表进行梯度洗脱,流速为0.25mL/min;检测波长:280nm。供试品溶液1色谱图见图5。
表18对比例洗脱梯度
由图5可知,采用上述色谱条件,对阿魏酸、橙黄决明素、洋川芎内酯A、藁本内酯、大黄素和大黄酚的分离度较差,杂质干扰较强,另外采用上述色谱条件分析时间较长。
实施例2和3检测结果和方法学验证与实施例1的检测结果和方法学验证具有类似的效果。
- 小程序的界面渲染方法、装置、电子设备和存储介质
- 渲染立体图形线框的方法、装置、计算机设备和存储介质
- 一种页面渲染方法、装置、设备和存储介质
- 页面渲染方法、装置、计算机设备及存储介质
- 在canvas中渲染数据的方法、装置、电子设备及存储介质
- 行驶轨迹渲染方法、装置、设备和存储介质
- 一种车辆行驶轨迹的渲染方法、装置、设备及存储介质