一种甲基化位点识别方法及装置
文献发布时间:2023-06-19 13:46:35
技术领域
本发明主要涉及基因数据处理技术领域,具体涉及一种甲基化位点识别方法及装置。
背景技术
DNA甲基化是一种重要的表观遗传修饰,与癌症的发生发展密切相关。DNA甲基化位点作为一种癌症相关的生物标志物,其有效识别对于了解癌症的发病机制、癌症分析判断和药物开发具有较高的指导意义。目前,在基因组、表观基因组、转录组等不同组学的研究中,利用计算机生物工具识别甲基化位点的研究成果不断涌现。但目前的处理模型存在偏差,将位点粗略地确定为甲基化位点,存在不够精确的情况。
发明内容
本发明所要解决的技术问题是针对现有技术的不足,提供一种甲基化位点识别方法及装置。
本发明解决上述技术问题的技术方案如下:一种甲基化位点识别方法,包括如下步骤:
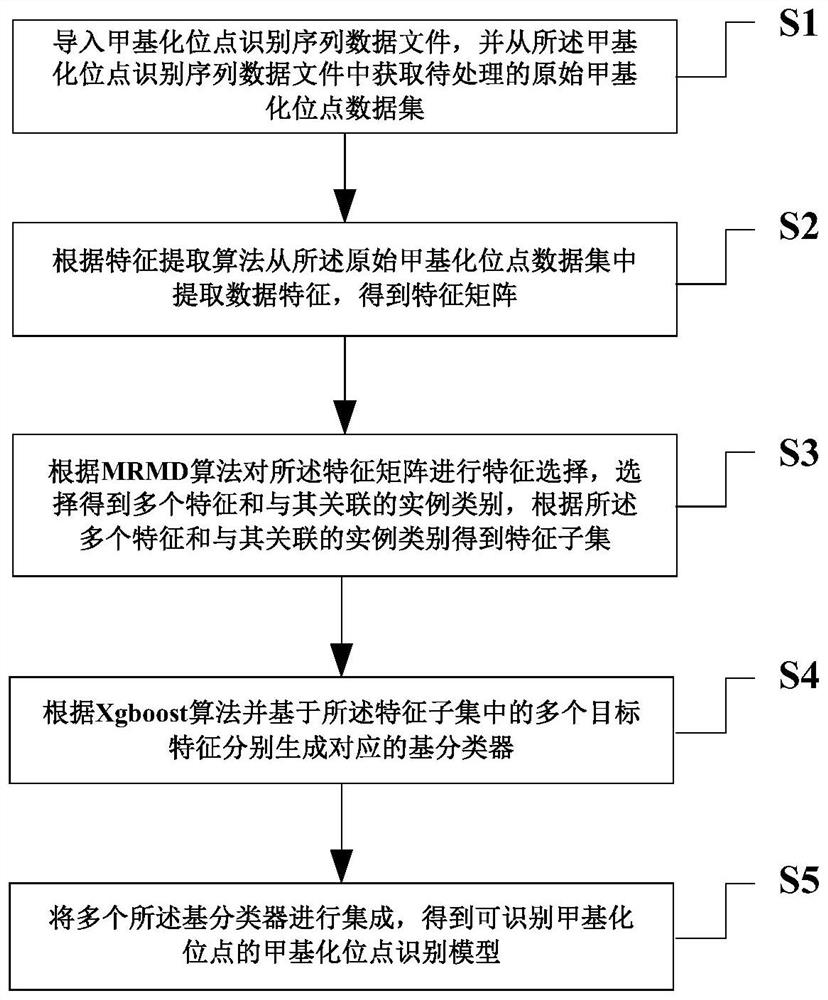
S1、导入甲基化位点识别序列数据文件,并从所述甲基化位点识别序列数据文件中获取待处理的原始甲基化位点数据集;
S2、根据特征提取算法从所述原始甲基化位点数据集中提取数据特征,得到特征矩阵;
S3、根据MRMD算法对所述特征矩阵进行特征选择,选择得到多个特征和与其关联的实例类别,根据所述多个特征和与其关联的实例类别得到特征子集;
S4、根据Xgboost算法并基于所述特征子集中的多个目标特征分别生成对应的基分类器;
S5、将多个所述基分类器进行集成,得到可识别甲基化位点的甲基化位点识别模型。
在上述技术方案的基础上,本发明还可以做如下改进。
进一步,所述特征提取算法包括基于序列特征的特征提取算法和基于物化性质的特征提取算法;
所述S2中,根据特征提取算法从所述原始甲基化位点数据集中提取数据特征,得到特征矩阵的过程包括:
基于序列特征的特征提取算法从所述原始甲基化位点数据集中提取Kmer,k间隔核苷酸对组成CKSNAP、核苷酸组成DNC和Mismatch组成,得到第一类数据特征;
基于物化性质的特征提取算法从所述原始甲基化位点数据集中提取并行相关伪三核苷酸组成PCPseTNC、系列相关伪二核苷酸组成SCPseDNC和序列相关伪三核苷酸组成SCPseTNC,得到第二类数据特征;
根据基因位点数据从所述原始甲基化位点数据集中提取基因衍生特征;
根据所述第一类数据特征、所述第二类数据特征和所述基因衍生特征得到特征矩阵。
采用上述进一步技术方案的有益效果是:利用核苷酸的组成来表达甲基化位点序列的特征,能够实现对甲基化位点的准确识别。
进一步,所述S3中,根据MRMD算法对所述特征矩阵进行特征选择的过程包括:
根据max(MR
并通过第二公式计算maxMD
其中,PCC(·)表示皮尔逊系数,F
采用上述进一步技术方案的有益效果是:通过MRMD算法对高维特征进行排序来过滤无信息数据,能够平衡特征排序和预测的准确性和稳定性。
进一步,还包括步骤,将所述原始甲基化位点数据集划分为正例数据集和反例数据集,所述正例数据集包括甲基化位点识别序列,所述反例数据集包括非甲基化位点识别序列。
进一步,所述将所述原始甲基化位点数据集划分为正例数据集和反例数据集的过程包括:
通过第三公式计算分类精度,所述第三公式为:
其中,ACC表示使用极限学习机算法对甲基化位点进行分类得到的分类精度,TP表示预测正确的甲基化位点数量,FP表示预测正确的非甲基化位点数量,TN表示预测错误的甲基化位点数量,FN表示预测错误的非甲基化位点数量;
本发明解决上述技术问题的另一技术方案如下:一种甲基化位点识别装置,包括:
获取模块,用于导入甲基化位点识别序列数据文件,并从所述甲基化位点识别序列数据文件中获取待处理的原始甲基化位点数据集;
处理模块,用于根据特征提取算法从所述原始甲基化位点数据集中提取数据特征,得到特征矩阵;
根据MRMD算法对所述特征矩阵进行特征选择,选择得到多个特征和与其关联的实例类别,根据所述多个特征和与其关联的实例类别得到特征子集;
根据Xgboost算法并基于所述特征子集中的多个目标特征分别生成对应的基分类器;
集成模块,用于将多个所述基分类器进行集成,得到可识别甲基化位点的甲基化位点识别模型。
本发明解决上述技术问题的另一技术方案如下:一种甲基化位点识别装置,包括存储器、处理器以及存储在所述存储器中并可在所述处理器上运行的计算机程序,当所述处理器执行所述计算机程序时,实现如上所述的甲基化位点识别方法。
本发明的有益效果是:本发明提出了一种全新的甲基化位点识别方法,从原始甲基化位点数据集中提取数据特征,对数据特征进行特征选择,得到多个特征和与其关联的实例类别,能够实现对甲基化位点的准确识别,为相应药物开发提供了理论基础,通过对基分类器进行集成进而构建甲基化位点识别模型,提升了甲基化位点识别精度。
附图说明
图1所示为本发明实施例提供的甲基化位点识别方法的流程示意图;
图2所示为本发明实施例提供的甲基化位点识别装置的功能模块示意图;
图3所示为本发明实施例提供的甲基化位点识别方法的数据性流程示意图。
具体实施方式
以下结合附图对本发明的原理和特征进行描述,所举实例只用于解释本发明,并非用于限定本发明的范围。
实施例1:
如图1所示,一种甲基化位点识别方法,包括如下步骤:
S1、导入甲基化位点识别序列数据文件,并从所述甲基化位点识别序列数据文件中获取待处理的原始甲基化位点数据集;
S2、根据特征提取算法从所述原始甲基化位点数据集中提取数据特征,得到特征矩阵;
S3、根据MRMD算法对所述特征矩阵进行特征选择,选择得到多个特征和与其关联的实例类别,根据所述多个特征和与其关联的实例类别得到特征子集;
S4、根据Xgboost算法并基于所述特征子集中的多个目标特征分别生成对应的基分类器;
S5、将多个所述基分类器进行集成,得到可识别甲基化位点的甲基化位点识别模型。
如图3所示,步骤S1中,原始甲基化位点数据集中分为两类数据,第一种来自于WHISLE,它包含两种模式(Full Transcript和mature MRNA)的数据。每一种模式包含6个训练数据集和6个独立数据集,每次取一个单基分辨率作为独立数据集,其余5个自动作为训练集。以A549为例进行详细说明。如果独立数据集为A549,则CD8T、HEK298_abacm、HEK298_sysy、HeLa和MOLM13构成训练数据集。所有完全转录的独立数据集和成熟mRNA统称为独立数据集1。所有训练数据集和独立数据集1的基本分辨率数据都直接从GEO(GeneExpression Omnibus)下载。根据不同的单基分辨率对同一地点的不同判断,对样本赋予不同的权重(负样本的权重为1,正样本的权重根据具体情况分为2、3、4、5),可以保证建立更可信的模型。在每次实验中,对负样本(原始正样本与负样本之比为1:10)进行相同程度的整合、聚类和随机抽样,这对提高模型的泛化能力非常有意义。第二类数据来自人类A549细胞,称为独立数据集2。
在获取待处理的原始甲基化位点数据集之前,需要对导入的甲基化位点识别序列数据文件进行格式判断和内容判断;格式判断的具体方法为:当读入的甲基化位点序列数据文件的行以字符串“>”为开头时,则取加一行的数据为序列文本数据;内容判断的具体方法为:读取的序列文本数据的内容是否由“A”、“U”、“C”或“G”四个字母组成,若有这四个字母之外的字母出现,则提示输入的文本有包括“A”、“U”、“C”和“G”之外的字母。
具体地,将所述原始甲基化位点数据集划分为正例数据集和反例数据集,所述正例数据集包括甲基化位点识别序列,所述反例数据集包括非甲基化位点识别序列。
具体地,所述将所述原始甲基化位点数据集划分为正例数据集和反例数据集的过程包括:
通过第三公式计算分类精度,所述第三公式为:
其中,ACC表示使用极限学习机算法对甲基化位点进行分类得到的分类精度,TP表示预测正确的甲基化位点数量,FP表示预测正确的非甲基化位点数量,TN表示预测错误的甲基化位点数量,FN表示预测错误的非甲基化位点数量;
上述实施例中,提出了一种全新的甲基化位点识别方法,从原始甲基化位点数据集中提取数据特征,对数据特征进行特征选择,得到多个特征和与其关联的实例类别,能够实现对甲基化位点的准确识别,为相应药物开发提供了理论基础,通过对基分类器进行集成进而构建甲基化位点识别模型,提升了甲基化位点识别精度。
具体地,所述特征提取算法包括基于序列特征的特征提取算法和基于物化性质的特征提取算法;
所述S2中,根据特征提取算法从所述原始甲基化位点数据集中提取数据特征,得到特征矩阵的过程包括:
基于序列特征的特征提取算法对所述原始甲基化位点数据集中的Kmer,k间隔核苷酸对组成CKSNAP、核苷酸组成DNC和Mismatch组成,得到第一类数据特征;
基于物化性质的特征提取算法从所述原始甲基化位点数据集中提取并行相关伪三核苷酸组成PCPseTNC、系列相关伪二核苷酸组成SCPseDNC和序列相关伪三核苷酸组成SCPseTNC,得到第二类数据特征;
根据物化性质的特征提取算法对所述原始甲基化位点数据集中的并行相关伪三核苷酸组成PCPseTNC、系列相关伪二核苷酸组成SCPseDNC和序列相关伪三核苷酸组成SCPseTNC,得到第二类数据特征;
根据基因位点数据从所述原始甲基化位点数据集中提取基因衍生特征;
根据所述第一类数据特征、所述第二类数据特征和所述基因衍生特征得到特征矩阵。
应理解地,考虑到单一的特征提取方法很难准确识别站点。从整个序列、理化性质和基因信息中提取特征,综合评价模型的性能。
如图3所示,核苷酸之间的差异可以通过序列直接反映出来。因此,有四种基于序列的特性:Kmer,组成的k间隔核苷酸对(CKSNAP),二核苷酸组成(DNC),Mismatch。Kmer通过表征k个相邻核酸的出现频率,生成一个255维的特征向量,CKSNAP通过计算任何k个核酸分隔的核酸对的频率来生成一个400维的特征向量,而Mismatch通过计算相邻的k个长度的核酸的出现频率来生成一个64维的特征向量,这些核酸最多相差m个错匹配。在这一过程中,AAAT、AACC、AACG、gac、TAGC、AG、CG、GA、GC、CG的间隙在确定位点的类别中起着至关重要的作用。
物化性质已广泛并成功地应用于DNA和RNA属性的预测任务中。如图3所示,本实验采用五种基于理化性质的特征来描述基因序列中核酸性质的整体组成:平行相关伪三核苷酸组成(PCPseTNC)、序列相关伪二核苷酸组成(SCPseDNC),和序列相关伪三核苷酸组成(SCPseTNC)。所有的特征编码方法根据具体的物理化学性质类型将4种主要的核酸分类为大类,分别根据不同的公式得到123-、18-、66-、28-、68维特征向量。NCP通过对腺嘌呤(a)、鸟嘌呤(G)、胞嘧啶(C)和尿嘧啶(U)设置不同的编码,生成123维特征向量,其中a、G、C、U四种不同类型的特征向量具有不同的化学结构和化学结合。根据化学性质,A、G、C、U分别表示为(1,1,1)、(0,1,0)、(1,0,0)、(0,0,1)。
几乎所有现有的预测算法都只包含序列派生的特征。在某种程度上,单序列特征几乎不可能捕获完整的站点信息。因此,本发明使用了14种基因特征提取方法来预测位点,这些位点可通过MRMD算法进行特征选择和排序。所选的64个特征表明,该位点是否与主RNA转录本的拓扑区域重叠,区域上的相对位置,区域长度(bp),与进化保守相关的得分,RNA二级结构,基因或转录本的属性,与生物学相关的RNA注释,rna结合蛋白注释、核苷酸距离剪接点或最近的核苷酸距离以及该位点是否为特殊基序对甲基化位点的识别有重要影响。
具体地,步骤S2中的特征提取算法包括基于序列特征的特征提取算法,基于物化性质的特征提取算法和基因衍生特征。所述的基于序列的特征提取算法包括但不仅限于Kmer,k间隔核苷酸对的组成(CKSNAP),Di核苷酸组成(DNC)和Mismatch。所述的基于物化性质的特征提取算法包括但不仅限于并行相关伪三核苷酸组成(PCPseTNC)系列相关伪二核苷酸组成(SCPseDNC)和序列相关伪三核苷酸组成(SCPseTNC)。所述的基因衍生特征为直接根据基因位点数据提取而得。
具体地,所述S3中,根据MRMD算法对所述特征矩阵进行特征选择的过程包括:
根据max(MR
并通过第二公式计算maxMD
其中,PCC(·)表示皮尔逊系数,F
应理解地,在MRMD算法中,特征与实例类别之间的相关性用皮尔逊系数表征,皮尔逊系数越大说明特征与实例类别之间的相关性越强,关系越紧密;特征之间的冗余性用欧式距离表征,欧式距离又与Euclidean距离ED,Cosine距离COS和Tanimoto系数TC相关,欧式距离越大说明特征之间的冗余性越低。
相对而言,特征选择的本质是用特定的评价标准来衡量给定特征子集的优势。表1为关键性特征示意,如表1所示,通过特征选择,去除原始集合中的名誉特征和无关特征,保留有用的特征。训练集的数量和特征提取方法的多样性,特征选择是必要的。在选择特征选择方法时,预测效果的稳定性是最重要的指标。综上所述,MRMD算法通过对高维特征进行排序来过滤无信息数据,能够平衡特征排序和预测的准确性和稳定性。与其他特征选择算法相比,MRMD算法最大的优点是可以兼顾特征选择和降维后的稳定性,可以保证降维后的特征仍然具有良好的性能。优秀的特征表示可以提升模型的性能,更容易理解数据的特征和底层结构,更方便地改进模型和算法。
表1
具体地,步骤S4引入了Xgboost算法。目前大多数用于站点识别的分类方法都是随机森林或者支持向量机(SVM),一个更强大的分类算法被期待。作为一个升序树模型,Xgboost算法是集成了许多树模型(树模型是CART回归树模型)的一个强大分类器。
作为一个升序树模型,Xgboost算法是集成了许多树模型(树模型是CART回归树模型)的一个强大分类器。Xgboost算法被设计为通过添加越来越多的树和分割特性来生长树。实际上,当添加一棵树时,就学习了一个新的函数,用来拟合上次预测的残差。训练完成后,得到K棵树。根据样本的特征,在每棵树中找到相应的叶节点和预测得分。最后,每棵树的预测得分之和就是样本的预测值。
F={f(x)=w
式中,w_q(x)是叶子节点q的得分,f(x)是一棵回归树。使用Xgboost进行站点识别有几个原因。
(1)Xgboost算法中使用了多种过拟合预防策略。如果模型过度学习了训练集的特征,很可能是模型将训练样本的某些特征作为了一般属性,导致泛化能力下降。对于机器学习算法来说,过拟合是不能完全避免的,这意味着使用防止过拟合策略在机器学习中具有重要意义。(2)通过对样本设置不同的权重,可以使重要的样本得到更多的关注。为了在训练数据集中获得更准确的模型,对不同的样本赋予不同的权值,进一步提高了效果。
将步骤S3生成的选择后的特征矩阵与Xgboost算法进行结合以生成基分类器。
步骤S5对步骤S4所生成的基分类器进行了集成,集成策略的选取应根据数据特征,物种特性和时间复杂度进行选取。
下面通过实验对本发明进行论述:
在本实验中,两种模式(full transcript和mature mRNA)的数据分别由6个训练数据集和6个独立数据集组成。如图3所示,模型的构建过程详细如下:数据处理。序列数据的生成。本实验从原始基因组坐标数据(原始数据中仅包含基因组坐标数据)中提取相应的序列数据,利用序列数据和基因组数据对甲基化位点进行识别。一组样本权重。由于每个训练集的正样本由5个单基分辨率组成,不同的单基分辨率对同一个站点有不同的标签。因此,根据不同的单碱基分辨率在同一位点的表现,为每个样本分配不同的权重(阳性样本为2,3,4,5,阴性样本为1)。阴性样本的生成。考虑到染色体上非甲基化位点的数量远大于甲基化位点的数量,实验选择阴性样本(阳性:阴性=1:10)。为了保证模型具有更好的预测性能和更强的泛化能力,利用GMM收集所有的负样本进行聚类,并聚为5类负样本。5个类别中采样数量相同且采样程度相同的阳性样本。
特征提取。基于序列的特征、理化性质和基因衍生特征,包括NCP、CKSNAP、DNC、Mismatch、PC-PseDNC、PC-PseTNC、SC-PseDNC、SC-PseTNC等14种基因特征提取方法。
特征选择和特征拼接。除NCP外的所有特征都使用MRMD进行选择,拼接在一起生成最终的特征向量。相对于单一特征的训练,特征的选择和拼接在原则上可以显著提高模型的性能。本文采用了三种特征提取方法进行特征提取。
使用XGBoost的训练模型。基于样本的权重信息和高级分类能力,XGBoost被认为是一种合适的分类算法。在此过程中,采用5倍交叉验证的方法进行模型的培养和构建。利用独立数据集1和2进一步证明了模型的分类能力和泛化能力。
对可识别甲基化位点的甲基化位点识别模型的性能评估。
交叉验证是机器学习中常用的模型验证方法,能够准确调整模型的超参数,有效防止模型过复杂度导致的过拟合。交叉验证被用来评价模型的预测性能,特别是新数据的预测性能,可以在一定程度上减少过拟合。交叉验证可以从有限的数据中提取尽可能多的有效信息。所有模型训练均采用5倍交叉验证。表2为HSM6AP在独立数据集1上的性能,如表2所示,在全转录本中,交叉验证的准确率超过96%,而在成熟mRNA中,所有交叉验证的准确率都超过89%。全转录本和成熟mRNA的性能意味着HSM6AP不仅具有强大的预测功能;而且泛化能力强,能有效防止过拟合。
表2
表3为本发明实施例HSM6AP在独立数据集2上的性能,表4为本发明实施例HSM6AP在独立数据集3上的性能。如表3和表4所示,在独立测试集1中,A549、CD8T、HEK293_abacm、HeLa、MOLM13各项指标均表现良好,SN、SP、F_score、ACC、AUC均超过了0.9。HEK_293sysy的效果不符合预期,AUC为0.937。全转录本的平均AUC为0.976,而成熟mRNA的平均AUC为0.899。独立数据集2的正样本量为40742,负样本量为3575,AUC作为综合评价标准更加合理。在独立数据集2中,全转录本的平均AUC为0.981,且几乎所有值相对较高。相反,成熟的mRNA的平均AUC是0.914。在独立数据集3中,全转录本的平均AUC为0.967,且几乎所有值相对较高。相反,成熟的mRNA的平均AUC是0.890。
表3
表4
本实施例甲基化位点识别模型与目前先进的甲基化位点识别模型的性能对比。
将基因特征和序列特征融合,并与支持向量机(SVM)相结合,构建了WHISLE模型。随着大数据时代的到来,深度学习被广泛应用于数据挖掘领域。DeepM6ASeq可以利用序列信息预测甲基化位点,这是甲基化位点识别领域的新加入。作为甲基化位点预测领域的先驱,SRAMP通过对三种碱基分类器进行投票来识别甲基化位点。
为了进一步证明HSM6AP的优越性,将WHISLE、DeepM6ASeq和SRAMP应用于对比实验。表5为HSM6AP与目前先进的甲基化位点识别方法的对比。这些方法的结果如表5所示。对于独立数据集1,HSM6AP的全转录模式AUC比WHISLE、DeepM6ASeq和SRAMP比WHISLE高0.028、0.277和0.301,成熟mRNA的AUC比WHISLE、DeepM6ASeq和SRAMP高0.019、0.266和0.117,如表5。对于独立数据集2,HSM6AP的AUC比WHISLE、DeepM6ASeq和SRAMP高0.019、0.266和0.117。HSM6AP对完整转录本和成熟mRNA的AUC值为0.981,比WHISLE高0.001,比DeepM6ASeq高0.307,比SRAMP高0.285。HSM6AP在成熟mRNA中的表现也更好,其AUC分别为0.01、0.281和0.132,优于WHISLE、DeepM6ASeq和SRAMP。实验结果表明,HSM6AP在预测人类甲基化位点方面优于目前最先进的方法。
表5
实施例2:
如图2所示,一种甲基化位点识别装置,包括:
获取模块,用于导入甲基化位点识别序列数据文件,并从所述甲基化位点识别序列数据文件中获取待处理的原始甲基化位点数据集;
处理模块,用于根据特征提取算法从所述原始甲基化位点数据集中提取数据特征,得到特征矩阵;
根据MRMD算法对所述特征矩阵进行特征选择,选择得到多个特征和与其关联的实例类别,根据所述多个特征和与其关联的实例类别得到特征子集;
根据Xgboost算法并基于所述特征子集中的多个目标特征分别生成对应的基分类器;
集成模块,用于将多个所述基分类器进行集成,得到可识别甲基化位点的甲基化位点识别模型。
具体地,所述处理模块具体用于:
所述特征提取算法包括基于序列特征的特征提取算法和基于物化性质的特征提取算法;
根据特征提取算法从所述原始甲基化位点数据集中提取数据特征,得到特征矩阵的过程包括:
基于序列特征的特征提取算法从所述原始甲基化位点数据集中提取Kmer,k间隔核苷酸对组成CKSNAP、核苷酸组成DNC和Mismatch组成,得到第一类数据特征;
基于物化性质的特征提取算法从所述原始甲基化位点数据集中提取并行相关伪三核苷酸组成PCPseTNC、系列相关伪二核苷酸组成SCPseDNC和序列相关伪三核苷酸组成SCPseTNC,得到第二类数据特征;
根据基因位点数据从所述原始甲基化位点数据集中提取基因衍生特征;
根据所述第一类数据特征、所述第二类数据特征和所述基因衍生特征得到特征矩阵。
实施例3:
一种甲基化位点识别装置,包括存储器、处理器以及存储在所述存储器中并可在所述处理器上运行的计算机程序,当所述处理器执行所述计算机程序时,实现如上所述的甲基化位点识别方法。
需要说明的是,在本文中,诸如第一和第二等之类的关系术语仅仅用来将一个实体或者操作与另一个实体或操作区分开来,而不一定要求或者暗示这些实体或操作之间存在任何这种实际的关系或者顺序。而且,术语“包括”、“包含”或者其任何其他变体意在涵盖非排他性的包含,从而使得包括一系列要素的过程、方法、物品或者设备不仅包括那些要素,而且还包括没有明确列出的其他要素,或者是还包括为这种过程、方法、物品或者设备所固有的要素。
以上所述仅为本发明的较佳实施例,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 一种低甲基化水平的差异甲基化位点识别方法
- 一种差异甲基化位点识别方法