双苯基苯氧基金属-配体络合物的烃基改性甲基铝氧烷助催化剂
文献发布时间:2024-04-18 19:52:40
相关申请的交叉引用
本申请要求于2020年7月17日提交的美国临时专利申请第63/053,354号的优先权,该美国临时专利申请的全部公开内容特此通过引用并入。
技术领域
本公开的实施方案总体上涉及用于包含具有三个原子醚接头的双苯基苯氧基金属-配体络合物的催化剂体系的改性烃基甲基铝氧烷活化剂。
背景技术
自Ziegler和Natta发现多相烯烃聚合以来,全球聚烯烃产量在2015年达到大约1.5亿吨每年,并且由于市场需求的增加而在上升。这一成功部分基于助催化剂技术的一系列重要突破。发现的助催化剂包括铝氧烷、硼烷以及具有三苯基碳鎓或铵阳离子的硼酸盐。这些助催化剂使均相单位点烯烃聚合催化剂活化,并且在工业上已经使用这些助催化剂来生产聚烯烃。
作为α-烯烃聚合反应中催化剂组合物的一部分,活化剂可具有有利于产生-烯烃聚合物和包括α-烯烃聚合物的最终聚合物组合物的特征。增加α-烯烃聚合物产量的活化剂特性包括但不限于:快速的主催化剂活化、高催化剂效率、高承温能力、一致的聚合物组成和选择性失活。
基于硼酸盐的助催化剂特别地显著有助于对烯烃聚合机制的基本理解,并通过有意地调节催化剂结构和方法,增强了对聚烯烃微观结构进行精确控制的能力。这导致了对机制研究的激发的兴趣,并导致开发了对聚烯烃微观结构和性能具有精确控制的新颖的均相烯烃聚合催化剂体系。然而,一旦活化剂或助催化剂的阳离子使主催化剂活化,活化剂的离子就可保留在聚合物组合物中。结果,硼酸根阴离子可能影响聚合物组成。特别地,硼酸根阴离子的大小、硼酸根阴离子的电荷、硼酸根阴离子与周围介质的相互作用以及硼酸根阴离子与可用抗衡离子的离解能将影响离子扩散通过周围介质(如溶剂、凝胶或聚合物材料)的能力。
改性的甲基铝氧烷(MMAO)可被描述为铝氧烷结构和三烃基铝物质的混合物。三烃基铝物质如三甲基铝用作清除剂以除去聚合过程中可能导致烯烃聚合催化剂失活的杂质。然而,据信三烃基铝物质在一些聚合体系中可能是有活性的。当三甲基铝在60℃下与二茂铪催化剂一起存在于丙烯均聚中时,注意到催化剂抑制(Busico,V.等人《大分子》2009,42,1789-1791)。然而,这些观察结果暗示了MAO活化与硼酸盐活化之间的差异,并且甚至在直接比较中仅可能捕获一些三甲基铝与没有三甲基铝之间的差异。另外,还不清楚这些观察结果是否扩展到其它催化剂体系、乙烯聚合或在更高温度下进行的聚合。无论如何,对可溶性MAO的偏好要求使用MMAO并且因此存在三烃基铝物质。
改性甲基铝氧烷(MMAO)在一些PE方法中用作活化剂,代替基于硼酸盐的活化剂。然而,已发现MMAO对一些催化剂(例如双联苯基苯氧基金属-配体络合物)的性能具有负面影响并且负面影响聚乙烯树脂的生产:对聚合方法的负面影响包括降低催化剂活性、加宽所产生的聚合物的组成分布以及负面影响粒料处理。
发明内容
持续需要产生一种催化剂体系,同时保持催化剂效率、反应性和产生具有良好物理特性的聚合物的能力。还需要生产均匀的聚合物组合物。
本公开的实施方案包括使烯烃单体聚合的方法。在一个或多个实施方案中,该方法包括使乙烯和任选的一种或多种烯烃单体在催化剂体系的存在下反应。催化剂体系包括改性烃基甲基铝氧烷和主催化剂。基于铝的总摩尔数,具有小于50摩尔的AlR
在式(I)中,M是钛、锆或铪。(X)
在式(I)中,R
在式(I)中,R
在式(II)、(III)和(IV)中,R
在式(I)中,Y是CH
在式(I)、(II)、(III)和(IV)中,式(I)中的每个R
附图说明
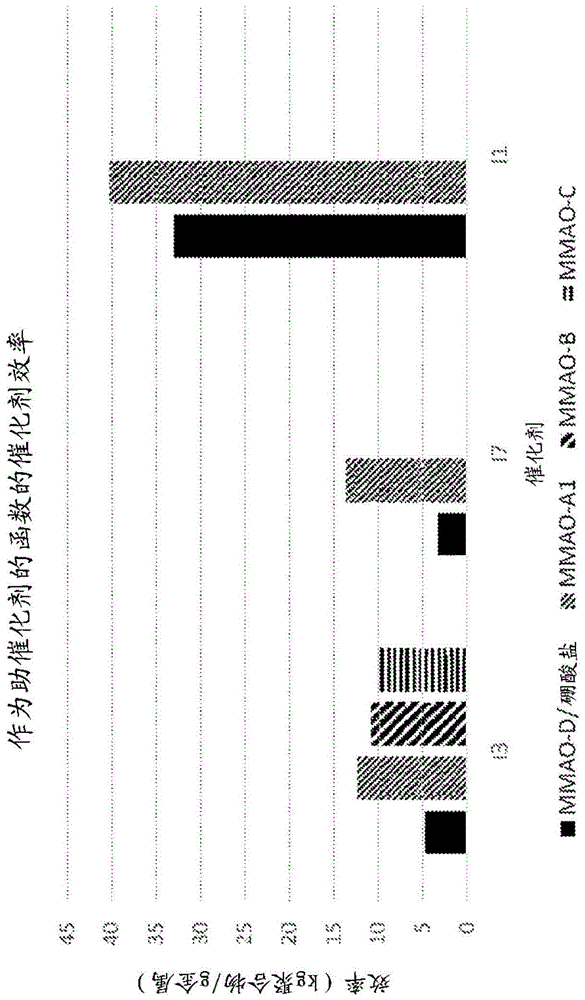
图1是金属-配体络合物I1、I3和I7的催化剂效率作为MMAO助催化剂类型的函数的图。
具体实施方式
现在将描述催化剂体系的具体实施方案。应当理解,本公开的催化剂体系可以不同的形式体现,并且不应被解释为限于本公开所阐述的具体实施方案。相反,提供实施方案使得本公开将是彻底且完整的,并且实施方案将向本领域技术人员充分传达主题的范围。
下文列出了常见的缩写:
Me:甲基;Et:乙基;Ph:苯基;Bn:苄基;i-Pr:异丙基;t-Bu:叔丁基;t-Oct:叔辛基(2,4,4-三甲基戊烷-2-基);Tf:三氟甲烷磺酸盐;THF:四氢呋喃;Et
术语“独立地选择”在本文中用于指示R基团,如R
术语“前催化剂”是指当与活化剂组合时具有烯烃聚合催化活性的过渡金属化合物。术语“活化剂”是指以将主催化剂转化为催化活性催化剂的方式与主催化剂化学反应的化合物。如本文所使用的,术语“助催化剂”和“活化剂”是可互换的术语。
当用于描述某些含碳原子的化学基团时,具有形式“(C
术语“取代”意指与对应的未取代的化合物或官能团中的碳原子键合的至少一个氢原子(-H)被取代基(例如R
术语“(C
术语(C
术语“(C
术语“卤素原子”或“卤素”意指氟原子(F)、氯原子(Cl)、溴原子(Br)或碘原子(I)的自由基。术语“卤化物”意指卤素原子的阴离子形式:氟离子(F
术语“饱和”意指缺少碳-碳双键、碳-碳三键以及(在含有杂原子的基团中)碳-氮双键、碳-磷双键和碳-硅双键。在饱和化学基团被一个或多个取代基R
本公开的实施方案包括使烯烃单体聚合的方法。在一个或多个实施方案中,该方法包括使乙烯和任选的一种或多种烯烃单体在催化剂体系的存在下反应。
在各种实施方案中,催化剂体系不含硼酸盐活化剂。
在一些实施方案中,烯烃单体是(C
在一个或多个实施方案中,催化剂体系包含烃基改性的甲基铝氧烷和主催化剂。基于铝的总摩尔数,烃基改性的甲基铝氧烷具有小于50摩尔%的AlR
术语“烃基改性的甲基铝氧烷”是指包含一定量的三烃基铝的甲基铝氧烷(MMAO)结构。烃基改性的甲基铝氧烷包括烃基改性的甲基铝氧烷基质和三烃基铝的组合。烃基改性的甲基铝氧烷中铝的总摩尔量由来自烃基改性的甲基铝氧烷基质的铝摩尔数和来自三烃基铝的铝摩尔数的铝贡献组成。基于烃基改性的甲基铝氧烷中铝的总摩尔数,烃基改性的甲基铝氧烷包括大于2.5摩尔%的三烃基铝。这些额外的烃基取代基可影响随后的铝氧烷结构并导致铝氧烷簇的分布和尺寸的差异(Bryliakov,K.P等人《大分子化学与物理(Macromol.Chem.Phys.2006,207,327-335)。额外的烃基取代基还可赋予铝氧烷在烃溶剂中的增加的溶解度,该烃溶剂例如但不限于己烷、庚烷、甲基环己烷和ISOPAR E
在本公开的实施方案中,催化剂体系包含一种或多种根据式(I)的金属-配体络合物。
在式(I)中,M为具有+2、+3或+4的形式氧化态的钛、锆或铪。(X)
在式(I)中,R
在式(I)中,R
在式(II)、(III)和(IV)中,R
在式(I)中,Y是CH
不受理论的束缚,据信这些优选的包含三原子桥(-CH
在式(I)、(II)、(III)和(IV)中,式(I)中的每个R
在实施方案中,基于烃基改性的甲基铝氧烷中铝的总摩尔数,聚合方法中的改性烃基甲基铝氧烷具有小于20摩尔%且大于5摩尔%的三烃基铝。在一些实施方案中,基于烃基改性的甲基铝氧烷中铝的总摩尔数,改性烃基甲基铝氧烷具有小于15摩尔%的三烃基铝。在一个或多个实施方案中,基于烃基改性的甲基铝氧烷中铝的总摩尔数,改性烃基甲基铝氧烷具有小于10摩尔%的三烃基铝。在各种实施方案中,改性烃基甲基铝氧烷是改性的甲基铝氧烷。
在一些实施方案中,三烃基铝具有式AlR
式(I)的金属-配体络合物中的基团R
在一些实施方案中,R
在一些实施方案中,当R
在实施方案中,当R
在一些实施方案中,当R
(C
在式(I)中,R
在一个或多个实施方案中,R
在各种实施方案中,R
在一些实施方案中,R
在各种实施方案中,R
在一个或多个实施方案中,R
在一些实施方案中,R
在各种实施方案中,在式(I)的金属-配体络合物中,R
在一些实施方案中,式(I)的金属-配体络合物的化学基团(例如,X和R
在式(I)、(II)、(III)和(IV)中,每个R
在一些实施方案中,在根据式(I)的金属-配体络合物中,R
在根据式(I)的金属-配体络合物中,X通过共价键或离子键与M键合。在一些实施方案中,X可以是净形式氧化态为-1的单阴离子配体。每个单阴离子配体可以独立地为氢化物、(C
在一些实施方案中,X为卤素、未经取代的(C
在另外的实施方案中,X选自:甲基;乙基;1-丙基;2-丙基;1-丁基;2,2-二甲基丙基;三甲基甲硅烷基甲基;苯基;苄基;或氯。X是甲基;乙基;1-丙基;2-丙基;1-丁基;2,2-二甲基丙基;三甲基甲硅烷基甲基;苯基;苄基;和氯。在一个实施方案中,n为2,并且至少两个X独立地为单阴离子单齿配体。在具体的实施方案中,n为2,并且这两个X基团接合以形成二齿配体。在另外的实施方案中,二齿配体为2,2-二甲基-2-二甲基硅烷-l,3-二基或1,3-丁二烯。
在一个或多个实施方案中,每个X独立地为–(CH
在一个或多个实施方案中,X为–(CH
在一些实施方案中,X为-CH
助催化剂组分
通过本领域已知的用于使烯烃聚合反应的基于金属的催化剂活化的任何技术,可以使包括式(I)的金属-配体络合物的催化剂体系具有催化活性。举例来说,可通过使络合物与活化助催化剂接触或使络合物与活化助催化剂组合使根据式(I)的金属-配体络合物的主催化剂显现催化活性。另外,根据式(I)的金属-配体络合物包含中性的主催化剂形式和可能由于丧失单体离子配体(如苄基或苯基)而带正电荷的催化形式两者。本文中适合的活化助催化剂包括低聚铝氧烷或改性烷基铝氧烷。
聚烯烃
在烯烃(主要为乙烯和丙烯)聚合中使用前面段落中描述的催化体系,以形成基于乙烯的聚合物或基于丙烯的聚合物。在一些实施方案中,聚合方案中仅存在单一类型的烯烃或α-烯烃,从而形成均聚物。然而,可将另外的α-烯烃掺入到聚合程序中。另外的α-烯烃共聚单体通常具有不超过20个碳原子。例如,α-烯烃共聚单体可具有3个至10个碳原子或3个至8个碳原子。示例性α-烯烃共聚单体包括但不限于丙烯、1-丁烯、1-戊烯、1-己烯、1-庚烯、1-辛烯、1-壬烯、1-癸烯和4-甲基-1-戊烯。例如,一种或多种α-烯烃共聚单体可以选自由以下项组成的组:丙烯、1-丁烯、1-己烯和1-辛烯;或另选地,选自由以下项组成的组:1-己烯和1-辛烯。
基于乙烯的聚合物,例如乙烯和任选的一种或多种共聚单体(诸如α-烯烃)的均聚物和/或互聚物(包含共聚物),可以包含至少50摩尔%(mol%)的衍生自乙烯的单体单元。“至少50摩尔%”所涵盖的所有单个值和子范围在本文中作为单独的实施方案公开;例如,基于乙烯的聚合物,即乙烯与任选的一种或多种诸如α-烯烃的共聚单体的均聚物和/或互聚物(包括共聚物),可以包含至少60摩尔%(mol%)的衍生自乙烯的单体单元;至少70摩尔%的衍生自乙烯的单体单元;至少80摩尔%的衍生自乙烯的单体单元;或50摩尔%至100摩尔%的衍生自乙烯的单体单元;或80摩尔%至100摩尔%的衍生自乙烯的单体单元。
在一些实施方式中,基于乙烯的聚合物可包括至少90摩尔百分比的衍生自乙烯的单元。来自至少90摩尔百分比的所有个别值和子范围包含在本文中并且在本文中作为单独的实施例公开。例如,基于乙烯的聚合物可包含至少93摩尔%的衍生自乙烯的单元;至少96摩尔%的单元;至少97摩尔%的衍生自乙烯的单元;或另选地,90摩尔%至100摩尔%的衍生自乙烯的单元;90摩尔%至99.5摩尔%的衍生自乙烯的单元;或97摩尔%至99.5摩尔%的衍生自乙烯的单元。
在基于乙烯的聚合物的一些实施方案中,另外的α-烯烃的量小于50mol%;其它实施方案包括至少1摩尔百分比(mol%)至25mol%;并且在另外的实施方案中,另外的α-烯烃的量包括至少5mol%至103mol%。在一些实施方案中,另外的α-烯烃是1-辛烯。
可以采用任何常规聚合工艺来产生乙烯类聚合物。这种常规聚合方法包括但不限于例如使用一种或多种常规反应器的溶液聚合法、浆相聚合法和其组合,所述一种或多种常规反应器如环式反应器、等温反应器、搅拌槽反应器、并联或串联的间歇反应器或其任何组合。
在一个实施方案中,基于乙烯的聚合物可在双反应器体系例如双环管反应器体系中经由溶液聚合来产生,其中乙烯和任选的一种或多种α-烯烃在如本文所述的催化剂体系和任选的一种或多种助催化剂存在下聚合。在另一个实施方案中,基于乙烯的聚合物可以在双反应器系统,例如双环管反应器系统中通过溶液聚合产生,其中乙烯和任选地一种或多种α-烯烃在本公开中的并且如本文所描述的催化剂体系以及任选的一种或多种其它催化剂存在下聚合。如本文所描述的催化剂体系可以任选地与一种或多种其它催化剂组合地用于第一反应器或第二反应器中。在一个实施方案中,基于乙烯的聚合物可以在双反应器系统,例如双环管反应器系统中经由溶液聚合产生,其中乙烯和任选的一种或多种α-烯烃在如本文所描述的催化剂体系存在下在这两个反应器中聚合。
在另一个实施方案中,基于乙烯的聚合物可以在单反应器体系(例如,单环式反应器体系)中通过溶液聚合来产生,其中乙烯和任选的一种或多种α-烯烃在存在如本公开内所述的催化剂体系以及任选的如前述段落中所述的一种或多种助催化剂的情况下聚合。
基于乙烯的聚合物可以进一步包括一种或多种添加剂。这种添加剂包括但不限于抗静电剂、颜色增强剂、染料、润滑剂、颜料、主抗氧化剂、次级抗氧化剂、加工助剂、UV稳定剂和它们的组合。基于乙烯的聚合物可以含有任何量的添加剂。基于乙烯的聚合物可包含按基于乙烯的聚合物和一种或多种添加剂的重量计约0至约10%的这类添加剂的组合重量。基于乙烯的聚合物可以进一步包括填充剂,所述填充剂可以包含但不限于有机或无机填充剂。按基于乙烯的聚合物和所有添加剂或填充剂的组合重量计,基于乙烯的聚合物可以含有约0至约20重量百分比的填充剂,例如,碳酸钙、滑石粉或Mg(OH)
在一些实施方式中,用于生产基于乙烯的聚合物的聚合方法可包括在根据本公开的催化剂体系存在下使乙烯和至少一种额外的α-烯烃聚合。根据ASTM D792(全文以引用方式并入本文),由掺入式(I)的金属-配体络合物的此类催化剂体系产生的聚合物的密度可以是例如0.850g/cm
在另一种实施方式中,由根据本公开的催化剂体系产生的聚合物具有的熔体流动比(I
在一些实施方式中,由根据本公开的催化剂体系产生的聚合物具有的分子量分布(MWD)为1至25,其中MWD定义为M
凝胶渗透色谱法(GPC)
色谱系统由配备有内部IR5红外检测器(IR5)的PolymerChar GPC-IR(西班牙,巴伦西亚)高温GPC色谱仪组成。自动取样器烘箱室设定为160摄氏度,并且柱室设定为150摄氏度。所使用的柱是4根安捷伦(Agilent)“MixedA”30cm20微米线性混合床柱。所用的色谱溶剂为1,2,4三氯苯并且含有200ppm的丁基羟基甲苯(BHT)。溶剂源是氮气喷射的。所使用的注射体积为200微升,并且流动速率为1.0毫升/分钟。
GPC柱组的校准用分子量范围为580至8,400,000的至少20个窄分子量分布聚苯乙烯标准物进行,并且排列在6个“鸡尾酒”混合物中,在各个分子量之间具有至少十倍的间隔。标准品购自Agilent Technologies。对于分子量等于或大于1,000,000,在50毫升溶剂中制备0.025克聚苯乙烯标准物,对于分子量小于1,000,000,在50毫升溶剂中制备0.05克聚苯乙烯标准物。将聚苯乙烯标准物在80摄氏度下溶解并轻轻搅拌30分钟。使用等式1将聚苯乙烯标准物峰值分子量转化为聚乙烯分子量(如Williams和Ward,《聚合物科学杂志(J.Polym.Sci.)》,Polym.Let.)》,第6卷,第621页(1968)中所述):
M
其中M为分子量,A具有0.4315的值,并且B等于1.0。
多顶式第5阶用于拟合对应聚乙烯等效校准点。对A进行小的调整(从大约0.40至0.44)以校正柱分辨率和谱带变宽效应,使得在52,000Mw下获得NIST标准NBS 1475。
高温热梯度相互作用色谱(HT-TGIC或TGIC)
使用市售结晶洗脱分级仪器(CEF)(西班牙的珀里莫查公司(Polymer Char,Spain))进行高温热梯度相互作用色谱(HT-TGIC或TGIC)测量(Cong等人,《大分子(Macromolecules)》,2011,44(8),3062-3072)。CEF仪器配备有IR-5检测器。石墨已经被用作HT TGIC柱中的固定相(Freddy,A.Van Damme等人,美国专利8,476,076;Winniford等人,US 8,318,896.)。使用单个石墨柱(250×4.6mm)进行分离。使用干填充技术,然后是湿填充技术将石墨填充到色谱柱中(Cong等人,EP 2714226B1以及引用的参考文献)。实验参数为:顶烘箱/输送管/针温度为150℃,溶解温度为150℃,溶解搅拌设定为2,泵稳定时间为15秒,清洁柱的泵流动速率为0.500mL/m,柱负载的泵流动速率为0.300mL/分钟,稳定温度为150℃,稳定时间(预,在载入柱之前)为2.0分钟,稳定时间(后,在载入柱之后)为1.0分钟,SF(可溶物级分)时间为5.0分钟,从150℃至30℃的冷却速率为3.00℃/分钟,在冷却过程期间的流动速率为0.04mL/分钟从30℃至160℃的加热速率为2.00℃/分钟,在160℃下的等温时间为10分钟,洗脱流动速率为0.500mL/分钟以及注射回路尺寸为200微升。
根据石墨柱的长度调节冷却过程中的流动速率,使得在冷却循环结束时所有的聚合物级分必须保留在柱上。
样品通过PolymerChar自动进样器以在ODCB(定义如下)中4.0mg/mL的浓度在150℃下维持120分钟来制备。在使用前,在真空烘箱中,在160℃下干燥硅胶40(粒度0.2-0.5mm,目录号10181-3,EMD)约两小时。2对于配备有具有N2吹扫能力的自动取样器的CEF仪器,将硅胶40填充到两个“300mm×7.5mm”GPC尺寸的不锈钢柱中,并将硅胶40柱安装在CEF仪器的泵的入口处以纯化ODCB。并且不将BHT添加到流动相中。用硅胶40干燥的ODCB现在称为“ODCB”。TGIC数据在PolymerChar(西班牙)“GPC One”软件平台上处理。用约4mg至6mg的二十烷、14.0mg的全同立构均聚物聚丙烯iPP(多分散性为3.6至4.0,分子量Mw报告为聚乙烯当量150,000至190,000,并且多分散性(Mw/Mn)为3.6至4.0)的混合物进行温度校准,其中iPP DSC熔融温度测量为158-159℃(下文描述的DSC方法)。在装有7.0mL ODCB的10mL小瓶中加入14.0mg的聚乙烯均聚物HDPE(共聚单体含量为零,重均分子量(Mw)以聚乙烯当量计为115,000至125,000,并且多分散性为2.5至2.8)。在160℃下溶解时间为2小时。
HT-TGIC对聚合物样品的数据处理
在与聚合物样品相同的实验条件下运行溶剂空白(纯溶剂注射)。对聚合物样品的数据处理包括:对每个检测器通道的溶剂空白的扣除、如校准工艺中所述的温度外推、用由校准工艺测定的延迟体积进行的温度补偿以及如由校准的加热速率计算的将洗脱温度轴调节到30℃和160℃范围。
色谱图(IR-5检测器的测量通道)与PolymerChar“GPC One”软件集成。当峰值在高洗脱温度下降低到平坦基线(扣除空白色谱图中的大致零值)和在可溶级分(SF)的高温侧上的检测器信号的最小或平坦区域时,由可见差异绘制直线基线。
TGIC谱的宽度指数(B-指数)
TGIC色谱图与共聚单体含量及其分布有关。它可以与催化剂活性位点的数目有关。色谱相关的实验因素可以在一定程度上影响TGIC谱(Stregel等人,“Modern size-exclusion liquid chromatography,Wiley,第2版,第3章)。TGIC宽度指数(B-指数)可用于对具有不同组成和分布的样品的TGIC色谱的宽度进行定量比较。可计算最大谱高度的任何部分的B指数。例如,“N”B指数可以通过在谱最大高度的第1/N处测量谱宽度并且利用以下等式来获得:
其中Tp是在谱中观察到最大高度的温度,其中N是整数2、3、4、5、6或7。在TGIC色谱图具有带有类似峰高度的多个峰的情况下,将最高洗脱温度下的峰定义为谱温度(Tp)。
TGIC谱的U指数(U指数)
TGIC用于测量聚合物的组成分布。为了评估组成分布的均匀性,根据以下等式将所得色谱图拟合至高斯分布(Guassian distribution):
该拟合通过使用上述函数使用最小二乘法来实现。对数据和函数之间的残差f(x
调整拟合函数,以提供用于求和的最小值。除了最小二乘法之外,拟合方程进一步与加权函数组合,以阻止对峰形的过度估计。
其中对于(y
如前面部分所述,由于洗脱温度,低密度聚合物通常比高密度聚合物具有更宽的分子量分布(MWD)。TGIC谱可受聚合物MWD影响(Abdulaal等人,Macromolecular Chem Phy,2017,218,1600332)。因此,当使用TGIC分析MWD曲线的宽度时,曲线的宽度不是聚合物化学组成的准确指示。
由于形成的聚合物的高分子量和掺入聚合物中的共聚单体的量,本公开中所描述的催化剂体系的实施方案产生独特的聚合物性质。
本公开的一个或多个特征根据如下实施例进行说明:
实施例
用于连续过程反应器聚合的程序:原材料(乙烯、1-辛烯)和工艺溶剂(窄沸点范围高纯度异链烷烃溶剂,以商标ISOPAR E商购自埃克森美孚公司(ExxonMobilCorporation))用分子筛纯化,随后引入到反应环境中。氢气在加压气缸中以高纯度级别供应并且不进行进一步的纯化。将反应器单体进料(乙烯)流加压到大于反应压力。将溶剂和共聚单体进料加压到大于反应压力。用纯化的溶剂将各催化剂组分(金属-配体络合物和助催化剂)手动分批稀释至规定的组分浓度,并加压到大于反应压力。所有反应进料流均用质量流量计测量并用计算机自动化阀门控制系统独立控制。
连续溶液聚合在连续搅拌釜反应器(CSTR)中进行。进料到反应器的组合的溶剂、单体、共聚单体和氢气的温度控制在5℃至50℃之间,并且通常为15℃-25℃。将所有组分与溶剂进料一起进料到聚合反应器中。将催化剂进料到反应器中以达到指定的乙烯转化率。助催化剂组分基于计算的指定摩尔比或ppm量来单独进料。来自聚合反应器的流出物(包括溶剂、单体、共聚单体、氢、催化剂组分和聚合物)离开反应器并与水接触。另外,此时可添加各种添加剂,如抗氧化剂。然后,料流经过静态混合器以均匀分散混合物。
在添加添加剂之后,流出物(包括溶剂、单体、共聚单体、氢、催化剂组分和熔融聚合物)穿过热交换器以提高料流温度,从而准备将聚合物与其它较低沸点组分分离。料流然后穿过反应器压力控制阀,跨过该阀,压力大大降低。从此处起,该流出物进入由脱挥发器和真空挤出机组成的两级分离体系,其中溶剂和未反应的氢、单体、共聚单体和水从聚合物中去除。在挤出机的出口处,所形成的熔融聚合物的线料通过冷水浴,在该冷水浴中固化。然后,所述丝束通过丝束切碎机进料,在所述切碎机中,聚合物在风干之后切割成粒料。
用于间歇式反应器聚合的程序。将原料(乙烯、1-辛烯)和工艺溶剂(ISOPAR E)在引入反应环境之前用分子筛纯化。向搅拌的高压釜反应器中装入ISOPAR E和1-辛烯。然后将反应器加热至温度并装入乙烯以达到压力。任选地,还添加氢气。在干燥箱中在惰性气氛下通过将金属-配体络合物和任选的一种或多种添加剂与另外的溶剂混合来制备催化剂体系。然后将催化剂体系注射到反应器中。通过在聚合反应期间进料乙烯,并且根据需要冷却反应器,使反应器压力和温度保持恒定。10分钟后,关闭乙烯进料并且将溶液转移到经氮气吹扫的树脂锅中。使聚合物在真空烘箱中彻底干燥,并且在各聚合运行之间用热ISOPAR E彻底冲洗反应器。
测试方法
除非本文另有指示,否则以下分析方法用于描述本公开的各个方面:
熔融指数
聚合物样品的熔体指数I
密度
根据ASTM D4703制备用于密度测量的样品。根据ASTM D792,方法B在按压样品的一小时内进行测量。
烃基改性的甲基铝氧烷的分析
实施例
在氮气氛手套箱中,将具有式AlR
烃基改性的烷基铝氧烷中AlR
使用先前描述的方法分析AlR
金属络合物通过涉及过渡金属源和中性多官能配体源的标准金属化和配体交换程序方便地制备。另外,络合物还可以通过酰胺消除和烃基化方法从对应的过渡金属四酰胺和烃基化药剂(如三甲基铝)开始制备。所采用的技术与美国专利号6,320,005、6,103,657、WO 02/38628、WO 03/40195、US-A-2004/0220050中公开的那些技术相同或类似。
用于合成金属-配体络合物I1至18和C1至C3的合成程序可以在以下程序中找到,并且在先前公开的情况下,在以下公开中找到:US20040010103A1、WO2007136494A2、WO2012027448A1、WO2016003878A1、WO2016014749A1、WO2017058981A1、WO2018022975A1。
制备I1(WO2018022975 A1中公开的配体)
6',6”'-(((二异丙基硅烷二基)双(亚甲基))双(氧基))双(3-(3,6-二叔丁基-9H-咔唑-9-基)-3'-氟-5-(2,4,4-三甲基戊-2-基)-[1,1'-联苯]-2-醇)二甲基锆(I1)的合成:将MeMgBr于乙醚中的溶液(3.00M,5.33mL,16.0mmol)添加到ZrCl
1
I5的合成
向100mL烘箱干燥的玻璃瓶中装入ZrCl
1
I6的合成路线
2-溴-4-氟-6-甲基-苯酚的合成:向1升玻璃瓶中装入乙腈(400mL)、4-氟-6-甲基-苯酚(50g,396.4mmol)和对甲苯磺酸(一水合物)(75.6g,396mmol),确保所有物质都在溶液中。将溶液用冰冷却至0℃持续25分钟(形成沉淀物)。将冷却的溶液用N-溴代琥珀酰亚胺(70.55g,396.4mmol)缓慢处理(经过大约5分钟的过程),并在搅拌过夜的同时使其达到室温。通过
双((2-溴-4-氟-6-甲基苯氧基)甲基)二异丙基锗烷的合成:在手套箱中,在配备有磁力搅拌棒的250mL烧瓶中,将95% NaH(1.76g)(小心产生H
1
I6配体的合成
向配备有搅拌棒的500mL玻璃瓶中装入2,7-二叔丁基-9-(2-((四氢-2H-吡喃-2-基)氧基)-3-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)-5-(2,4,4-三甲基戊-2-基)苯基)-9H-咔唑(在WO2014105411 A1中公开)(29.0g,41.9mmol)、双((2-溴-4-氟-6-甲基苯氧基)甲基)二异丙基锗烷(6.00g,8.65mmol,含有10%2-溴-4-氟-2-甲基-苯酚)和THF(80mL)。将溶液加热至55℃,并且在搅拌的同时用氯[(三叔丁基膦)-2-(2-氨基联苯)]钯(II)(tBu
1
I6的合成
向100mL烘箱干燥的玻璃瓶中装入ZrCl
1
I7的合成方案
双((2-溴-4-叔丁基苯氧基)甲基)二异丙基硅烷的制备
在手套箱中,在250mL单颈圆底烧瓶中将二异丙基氯硅烷(3.703g,20mmol,1.0当量)溶解于无水THF(120mL)中。将烧瓶用隔膜封盖,密封,从手套箱中取出,并且在干冰-丙酮浴中冷却至-78℃。添加溴氯甲烷(3.9mL,60mmol,3.0当量)。使用注射器泵在3h内将丁基锂(18.4mL,46mmol,2.3当量)的己烷溶液添加到烧瓶的冷却壁中。使混合物升温至室温过夜(16h),并且添加饱和NH
在手套箱中,向40mL小瓶中装入双(氯甲基)二异丙基硅烷(2.14g,10mmol,1.0当量)、4-叔丁基-2-溴苯酚(6.21g,27mmol,2.7当量)、K-
1
6
在手套箱中,向配备有搅拌棒的40mL小瓶中装入双((2-溴-4-叔丁基苯氧基)甲基)二异丙基硅烷(1.20g,2.0mmol,1.0当量)、2-(3',5'-二叔丁基-5-甲基-2-((四氢-2H-吡喃-2-基)氧基)-[1,1'-联苯基]-3-基)-4,4,5,5-四甲基-1,3,2-二氧杂硼烷(2.54g,5.0mmol,2.5当量)、tBu
1
I7的制备
在手套箱中,向烘箱干燥的具有搅拌棒的40mL小瓶中装入ZrCl
1
金属-配体络合物I1至I8具有根据式(I)的结构并且如下:
/>
金属-配体配合物C1至C3是比较例并且如下:
实施例
金属-配体络合物(MLC)I1至I8在连续聚合方法中使用MMAO-A1、MMAO-B、MMAO-C、MMAO-D1、MMAO-D2、MMAO-E或MMAO-F作为活化剂进行测试,并与比较金属配体络合物C1至C3进行比较,并且数据汇总在表2-9中。
表1:烷基铝氧烷组合物
*MMAO-A1和A2用正辛基取代基改性,使得甲基∶正辛基比为约6∶1。MMAO-B用正辛基取代基改性,使得甲基∶正辛基比为约19∶1。
表2:连续过程乙烯/1-辛烯共聚反应
在160℃的反应器温度下聚合,连续进料流量为3.4kg/h乙烯、3.3kg/h1-辛烯、21kg/h ISOPAR E,
表3:在连续操作下产生的聚合物数据
条目编号参考表2。
表4:在连续操作下产生的聚合物数据
条目编号参考表2。
表2至4中记录的数据表明本发明催化剂与MMAO活化剂组合产生具有窄MWD的聚合物,如U指数所示,与MMAO活化剂无关。
当U指数接近100时,拟合的面积和样品的面积是相似的,因此表明是单中心催化剂。如先前所述,据信本发明催化剂体系的取代模式防止了第二活性位点的形成并因此导致组成分布更窄。另外,组成分布越窄,预期的B指数将越小。表3和4中给出的本发明实施例均显示比具有未取代桥的比较例更小的B指数。
为了证明MMAO-A1、MMAO-B和MMAO-C的优点,连续过程数据示出于表5至8中。调节H
表5:连续过程乙烯/1-辛烯共聚反应
在160℃下聚合,连续进料流量为3.4kg/h乙烯、3.3kg/h 1-辛烯、21kg/h ISOPARE,14%固体,81%乙烯转化率。效率(Eff.)测量为10
表6:连续过程乙烯/1-辛烯共聚反应
在160℃下聚合,连续进料流量为3.4kg/h乙烯、3.3kg/h 1-辛烯、21kg/h ISOPARE,14%固体,81%乙烯转化率。效率(Eff.)测量为10
表7:连续过程乙烯/1-辛烯共聚反应
在175℃下聚合,连续进料流量为3.3kg/h乙烯、1.6kg/h 1-辛烯、22kg/h ISOPARE,14%固体,87% C2转化率。效率(Eff.)测量为10
图1是催化剂效率作为助催化剂类型的函数的图。当与MMAO-A1、MMAO-B和MMAO-C组合使用时,金属-配体配合物I1、I3和I7的效率大于与比较助催化剂MMAO-D/硼酸盐组合使用时的效率。
表8:连续过程乙烯/1-辛烯共聚反应
在175℃下聚合,连续流量为140lbs/h乙烯、30.7-35.0lbs/h 1-辛烯、900lbs/hISOPAR E,14%固体,93.8%乙烯转化率。效率(Eff.)测量为10
设备标准
除非另有说明,否则所有溶剂和试剂均从商业来源获得并按原样使用。通过活性氧化铝,在某些情况下,通过Q-5反应物纯化无水甲苯、己烷、四氢呋喃和二乙醚。用于在氮气填充的手套箱中进行的实验的溶剂通过在活化的
- 双苯基苯氧基金属-配体络合物的烃基改性的甲基铝氧烷助催化剂
- 在金属上具有烷氧基或酰胺基配体的用于改进溶解度的双-苯基-苯氧基聚烯烃催化剂