一种含有二硫键的N-乙酰- L-半胱氨酸衍生物及其制备方法与应用
文献发布时间:2023-06-19 19:30:30
技术领域
本发明属于有机合成和生物医药领域,特别涉及含有二硫键的N-乙酰-L-半胱氨酸(NAC)衍生物及其制备方法与应用。
背景技术
公开该背景技术部分的信息仅仅旨在增加对本发明的总体背景的理解,而不必然被视为承认或以任何形式暗示该信息构成已经成为本领域一般技术人员所公知的现有技术。
N-乙酰-L-半胱氨酸(NAC),作为被世界卫生组织批准和认可的基础性药物,临床上常用于粘液溶解剂和治疗对乙酰氨基酚过量。其抗氧化和抗炎能力是用于治疗与氧化应激和炎症相关疾病的生化基础。NAC有直接抗氧化作用和间接抗氧化作用。直接抗氧化作用表现在游离巯基和活性氧(ROS)作用并将其清除。间接抗氧化作用是源于其增加细胞内谷胱甘肽(GSH)浓度的能力,而谷胱甘肽是导致细胞氧化还原失衡的最关键的生物硫醇。NAC作为一种抗炎化合物,其可以通过抑制核因子(NF-κB)的活性来降低肿瘤坏死因子和白细胞介素的水平。
对与氧化应激和炎症应激相关的肺部疾病来说,NAC不仅可以通过抗氧化和调节细胞因子的产生,达到改善气道粘液分泌、调节免疫系统等作用,还能减少肺上皮细胞凋亡,促进凋亡细胞清除并减少胶原沉积,有利于保护肺组织细胞,促进炎症吸收。
尽管NAC具有相关的治疗潜力,但在多项实验研究中,其在针对不同病理状况的临床试验中的有效性仍然有限。此外,NAC具有游离巯基,本身稳定性不佳,容易被氧化,导致其半衰期较短,进一步影响其药理作用。其次,NAC的非疏水性造成细胞和组织渗透有限,导致给药剂量大。研究表明,NAC在发挥炎症治疗和免疫调节方面的作用有限。
论文《L-半胱氨酸系列衍生物的合成及其在医药上的应用》公开了多种含有单硫键的L-半胱氨酸衍生物及其在像镇咳、祛痰、消炎、退烧、止痛、抑制细菌、抗肿瘤等医药上的应用。但该文未涉及到抗呼吸系统疾病防治所必需的抗氧化药物,且由于其单硫键的L-半胱氨酸衍生物大大限制了其抗氧化和抗炎活性。
专利CN101232877A虽然公开了N-乙酰半胱氨酸酰胺(NAC酰胺)治疗与氧化应激有关的疾病和病症的应用,但所涉及的化合物仅局限于NAC酰胺类化合物,且该类化合物应用于呼吸系统疾病防治的效果较为有限。
对于像慢性阻塞性肺病(慢阻肺,COPD)、肺纤维化、急性肺损伤/急性呼吸窘迫综合征(ALI/ARDS)等氧化应激和炎症应激相关疑难病症的防治,目前尚未找到具有较好的抗氧化和抗炎活性的N-乙酰-L-半胱氨酸(NAC)衍生物。
发明内容
为了解决上述问题,本发明提供一种含有二硫键的N-乙酰-L-半胱氨酸(NAC)衍生物及其制备方法与应用。本发明为进一步提高NAC的活性而对其进行结构修饰,设计合成了一系列N-乙酰-L-半胱氨酸衍生物。同时,本发明合成路线简洁高效,操作简单安全,具有良好的实际应用价值。
为了实现上述目的,本发明采用如下技术方案:
本发明的第一个方面,提供了一种含有二硫键的N-乙酰-L-半胱氨酸衍生物,其选自如下式Ⅰ所示化合物,
式Ⅰ
其中,R选自:噻吩-2-基甲基、呋喃-2-基甲基、吡啶-4-基甲基、苄基、4-甲基苄基、4-乙基苄基、4-氟苄基、4-氯苄基、4-溴苄基、4-叔丁基苄基、2-甲基苄基、2-氯苄基、2-溴苄基、3-氟苄基、3-(三氟甲基)苄基、2,4,6-三甲基苄基、2,4-二氯苄基、萘-2-基甲基、苯乙基、1-苯基乙基、4-甲氧基苯基、环己基、异丙基、正丁基。
本发明通过将NAC进行适当的衍生化,得到一种含有二硫键结构的衍生物,在提高其稳定性的基础上,使其在体内能够释放活性巯基物,达到延长半衰期的目标。其次,在增强细胞和组织渗透性,减少给药剂量的基础上,使其具有更好的抗氧化、抗炎和免疫调节的效果,进一步提高其药理活性。
上述化合物还包括其药学上可接受的盐或酯或溶剂化物、互变异构体、内消旋体、外消旋体、立体异构体、代谢产物或前药;
根据本领域一般技术人员的理解,上述药学上可接受的盐包括上述化合物的碱金属盐形式(具体实例如钙盐或钠盐或钾盐),或上述化合物与无机盐如盐酸、硫酸、硝酸或氢溴酸形成的盐,以及与有机酸,例如甲磺酸、甲苯磺酸或三氟乙酸形成的盐。术语“药学可接受的”或者与其可互换使用的“可药用的”,例如在描述“药学可接受的盐”时,表示该盐其不但是受试者生理学上可接受,而且还可指在药学上有使用价值的合成物质,例如在为进行手性拆分时所形成的作为中间体的盐,虽然这种中间体的盐并不能直接给予受试者,但该盐可在为获得本发明终产物中起作用。
进一步地,通式Ⅰ所示NAC衍生物包括:
A1:N-乙酰-S-((噻吩-2-基甲基)硫代)-L-半胱氨酸;
A2:N-乙酰-S-((呋喃-2-基甲基)硫代)-L-半胱氨酸;
A3:N-乙酰-S-((吡啶-4-基甲基)硫代)-L-半胱氨酸;
A4:N-乙酰-S-(苄硫基)-L-半胱氨酸;
A5:N-乙酰-S-((4-甲基苄基)硫代)-L-半胱氨酸;
A6:N-乙酰-S-((4-乙基苄基)硫代)-L-半胱氨酸;
A7:N-乙酰-S-((4-氟苄基)硫代)-L-半胱氨酸;
A8:N-乙酰-S-((4-氯苄基)硫代)-L-半胱氨酸;
A9:N-乙酰-S-((4-溴苄基)硫代)-L-半胱氨酸;
A10:N-乙酰-S-((4-(叔丁基)苄基)硫代)-L-半胱氨酸;
A11:N-乙酰-S-((2-甲基苄基)硫代)-L-半胱氨酸;
A12:N-乙酰-S-((2-氯苄基)硫代)-L-半胱氨酸;
A13:N-乙酰-S-((2-溴苄基)硫代)-L-半胱氨酸;
A14:N-乙酰-S-((3-氟苄基)硫代)-L-半胱氨酸;
A15:N-乙酰-S-((3-(三氟甲基)苄基)硫代)-L-半胱氨酸;
A16:N-乙酰-S-((2,4,6-三甲基苄基)硫代)-L-半胱氨酸;
A17:N-乙酰-S-((2,4-二氯苄基)硫代)-L-半胱氨酸;
A18:N-乙酰-S-((萘-2-基甲基)硫代)-L-半胱氨酸;
A19:N-乙酰-S-(苯乙基硫基)-L-半胱氨酸;
A20:N-乙酰-S-((1-苯基乙基)硫代)-L-半胱氨酸;
A21:N-乙酰-S-((4-甲氧基苯基)硫代)-L-半胱氨酸;
A22:N-乙酰-S-(环己基硫基)-L-半胱氨酸;
A23:N-乙酰-S-(异丙基硫代)-L-半胱氨酸;
A24:N-乙酰-S-(丁基硫醇)-L-半胱氨酸;
本发明的第二个方面,提供了一种上述含有二硫键的N-乙酰-L-半胱氨酸衍生物的合成方法,包括:
采用路线一或路线二合成含有二硫键的N-乙酰-L-半胱氨酸衍生物;
进一步地,上述制备方法中,式Ⅰ所示化合物的合成路线一如下:
合成路线一
进一步地,上述式Ⅰ所示化合物的合成路线,具体包括:
(1)、将通式化合物Ⅱ溶于盐酸甲醇溶液中,在低温条件下将前述溶液加入甲氧羰基磺酰氯的甲醇溶液中继续低温搅拌反应,去除溶剂和反应原料后,得到固体经纯化后即Ⅲ;
(2)、将含有通式Ⅳ对应的化合物或其盐溶于甲醇中,搅拌条件下加入含有通式Ⅲ对应化合物的甲醇溶液,搅拌过夜后加入三乙胺和甲醇的混合溶液,继续搅拌,纯化后即得通式Ⅴ对应化合物。
(3)、将含有通式Ⅴ对应的化合物溶于乙酸中,搅拌条件下加入乙酸酐,反应温度为40℃,搅拌过夜后,去除溶剂,得到的油状粗品经纯化后即化合物Ⅰ
进一步地,步骤(1)中,低温条件具体为0℃左右,可采用冰水浴方式实现,通式化合物Ⅱ与甲氧羰基磺酰氯的摩尔比为1:1-3,在本发明的一个具体实施方式中,通式化合物Ⅱ与甲氧羰基磺酰氯的摩尔比为1.36:2.04;去除溶剂和反应原料后具体可以为采用旋转蒸发除去溶剂和过量的甲氧羰基磺酰氯,控制水浴温度<30℃;
进一步地,固体纯化的步骤包括:用乙醚反复洗涤并过滤。
进一步地,步骤(2)中,通式Ⅳ对于化合物或其盐与通式Ⅲ对应的化合物的摩尔比为1:0.1-1,优选为1:0.5;
进一步地,三乙胺和甲醇的摩尔体积比为1:1-3(mmol/mL),优选为1:2。
进一步地,纯化的具体步骤包括:过滤沉淀,然后用甲醇和乙醚分别洗涤后干燥。
进一步地,上述步骤(2)反应均可在室温条件下进行。
进一步地,步骤(3)中,通式Ⅴ对应的化合物与乙酸酐的摩尔比为1:1-8,优选为1:4;通式Ⅴ对应的化合物与溶剂乙酸的摩尔体积比为1:1-4(mmol/mL),优选为1:2;
进一步地,去除溶剂具体可以采用旋转蒸发,控制水浴温度在50-55℃;
进一步地,纯化步骤包括:正相柱层析,100-200目硅胶填充,流动相为二氯甲烷:甲醇(20-50:1),以体积比计;和/或反相中压制备色谱,流动相为甲醇:水(0.4-1:1),以体积比计。
进一步地,制备方法包括:
步骤(1):将通式中化合物(Ⅱ,1.36 mmol)溶于1.7 mL 盐酸甲醇溶液中(盐酸为0.19 mol/L),在冰水浴条件下,20 min内将化合物(Ⅱ,1.36 mmol)的甲醇溶液缓慢滴加至甲氧羰基磺酰氯(2.04 mmol)的甲醇(2 mL)溶液中,冰水浴条件下继续搅拌反应1 h。旋转蒸发除去溶剂和过量的甲氧羰基磺酰氯(水浴温度<30℃)。得到的固体用乙醚反复洗涤,过滤,得到无色无味白色结晶固体中间体(Ⅲ),干燥后于-20℃条件下密封保存。
步骤(2):将含有通式(Ⅳ)对应化合物或其盐(0.5mmol)溶于0.4 mL甲醇中,在室温并搅拌条件下,缓慢滴加含有通式(Ⅲ)对应化合物的甲醇溶液(0.25 mmol,溶于0.25 mL甲醇中),45 min滴完,继续于室温搅拌过夜后,滴入三乙胺(0.25 mmol)和0.5 mL甲醇的混合溶液,继续于室温搅拌30 min,过滤白色沉淀,甲醇和乙醚分别洗涤沉淀后干燥,得通式(Ⅴ)对应化合物。
步骤(3):将含有通式(Ⅴ)对应化合物(2 mmol)溶于4mL冰醋酸中,在室温并搅拌条件下,加入醋酸酐(8 mmol),体系升温至40℃搅拌过夜。旋转蒸发除去溶剂(水浴温度50-55℃),得到黄色油状粗品,经正相柱层析,100-200目硅胶填充,流动相为二氯甲烷:甲醇(20-50:1),以体积比计;和/或反相中压制备色谱,流动相为甲醇:水(0.4-1:1),以体积比计;得通式(Ⅰ)对应化合物。
进一步地,式Ⅰ所示化合物的合成路线二如下:
合成路线二
进一步地,上述式Ⅰ所示化合物的合成路线二,具体包括:
(4)、将通式化合物Ⅵ溶于干燥的二氯甲烷中,在氮气保护条件下于低温搅拌5min。将m-CPBA(间氯过氧苯甲酸)溶于干燥的二氯甲烷中,缓慢(超过30min)滴入上述溶液,低温下继续搅拌2h,反应液用碳酸氢钠溶液洗四次,有机相干燥,去除溶剂,得到固体经纯化后即Ⅶ;
(5)、将含有通式Ⅶ对应的化合物乙腈中,搅拌条件下加入含有通式Ⅷ对应化合物和三乙胺,搅拌过夜后去除溶剂,水稀释,乙醚萃取,有机相干燥,去除溶剂,纯化后即得通式Ⅰ对应化合物。
进一步地,步骤(4)中,所述低温条件具体为0℃左右,可采用冰水浴方式实现,所述通式化合物Ⅵ与m-CPBA(间氯过氧苯甲酸)的摩尔比为1:1-1.5,在本发明的一个具体实施方式中,所述通式化合物Ⅵ与m-CPBA的摩尔比为1:1.05;碳酸氢钠溶液可用5%的水溶液,有机相干燥可用无水硫酸钠或硫酸镁,去除溶剂可采用旋转蒸发除去溶剂,控制水浴温度<40℃;
进一步地,固体纯化的步骤包括:正相柱层析,100-200目硅胶填充,流动相为石油醚:乙酸乙酯(20-50:1),以体积比计。
进一步地,步骤(5)中,通式Ⅶ对应化合物与通式Ⅷ对应的化合物的摩尔比为1:2-8,优选为1:4;通式Ⅶ对应化合物与三乙胺的摩尔比为1:2-10,优选为1:5;通式Ⅶ对应化合物与溶剂乙腈的摩尔体积比为1:50(mmol/mL);
进一步地,去除溶剂具体可以采用旋转蒸发,控制水浴温度在50-55℃;
进一步地,纯化步骤包括:正相柱层析,100-200目硅胶填充,流动相为二氯甲烷:甲醇(20-50:1),以体积比计;和/或反相中压制备色谱,流动相为甲醇:水(0.4-1:1),以体积比计。
进一步地,制备方法包括:
步骤(4):将通式中化合物(Ⅵ,1.51 mmol)溶于6 mL 干燥的二氯甲烷中,在氮气保护和冰水浴条件下,搅拌5min。将化合物m-CPBA(1.59mmol)溶于5mL干燥的二氯甲烷中,缓慢滴加至上述溶液中,冰水浴条件下继续搅拌反应2 h。反应溶液用5%的碳酸氢钠溶液洗四次,有机相用水洗一次,无水硫酸钠干燥,旋转蒸发除去溶剂 (水浴温度<40℃)。得到的固体经正相柱层析,100-200目硅胶填充,流动相为石油醚:乙酸乙酯(20-50:1),以体积比计,得通式(Ⅶ)对应化合物。
进一步地,步骤(5):将含有通式(Ⅶ)对应化合物 (0.29mmol)溶于14.5 mL乙腈中,加入通式(Ⅷ,1.16mmol)对应化合物三乙胺(1.44mmol),在氮气保护并室温条件下搅拌过夜,旋转蒸发除去溶剂 (水浴温度50-55℃)。所得固体加入水(15mL)稀释,乙醚萃取三次,合并有机相,无水硫酸钠干燥,旋转蒸发除去溶剂,所得固体经正相柱层析,100-200目硅胶填充,流动相为二氯甲烷:甲醇(20-50:1),以体积比计;和/或反相中压制备色谱,流动相为甲醇:水(0.4-1:1),以体积比计,得通式(Ⅰ)对应化合物。
进一步地,本发明提供了一种药物组合物,上述药物组合物包括第一方面的化合物。更具体的,其作为活性成分发挥作用,另外,除第一方面的化合物之外,还可能包括其他具有抗氧化、抗炎作用的成分,在此不再做进行具体限定。
以及,一种药物制剂,其包含上述第一方面中所述化合物,和至少一种药学上可接受的辅料和/或载体。
本发明所述辅料是指药物组合物或药物制剂中除有效成分之外的成分,其对受试者无毒。本领域常用的辅料比如缓冲剂、稳定剂、防腐剂或赋型剂,常用的赋形剂例如粘合剂、填充剂、润湿剂、崩解剂等。
作为示例,本发明的所述制剂中可选用的赋形剂包括但不限于:所述赋形剂选自磷酸钙、硬脂酸镁、滑石粉、糊精、淀粉、凝胶纤维素、甲基纤维素、羧甲基纤维素钠盐和聚乙烯吡咯烷酮。
本发明所述药物载体可以是药学上可接受的溶剂、悬浮剂、囊泡、纳米材料等,用于将本发明上述第一方面所述的化合物递送至动物或人体内。载体可以是液体或固体,并按照计划的给药方式进行选择。蛋白和脂质体也是药物载体。
本领域技术人员可采用公知的技术将本发明的化合物配制成药物组合物或制剂。比如将本发明上述第一方面中披露的任意化合物(至少一种化合物)与药用辅料混合,然后如果需要,使所得混合物形成所需的形状。除本发明提到的除外,也可根据已知药物制剂进行药物制剂的制备。以及,除本发明提到的以外,合适的药用辅料是本领域已知的,例如参见2005年版的药用辅料手册(原著第四版),作者(英)R.C.罗(Raymond C. Rowe) (美)P.J.舍斯基(Paul J. Sheskey)。
本发明的第三个方面,提供了上述的含有二硫键的N-乙酰-L-半胱氨酸衍生物在制备抗呼吸系统疾病药物产品中的应用,所述的呼吸系统疾病包括:慢性阻塞性肺病、肺纤维化、急性肺损伤/急性呼吸窘迫综合征。
进一步地,所述产品具体为药品,所述药品具有抗氧化、抗炎等药理作用,用于多种适应症,包括溶解粘痰、解救对乙酰氨基酚药物中毒、慢性阻塞性肺病(COPD)、肺纤维化、急性肺损伤/急性呼吸窘迫综合征(ALI/ARDS)、高氧引起得肺损伤、人类免疫缺陷病毒(HIV)感染、良性肿瘤或恶性肿瘤。
本发明的第四个方面,提供一种抗氧化、抗炎的方法,其包括向受试者施用治疗有效量的本发明第一方面所述的化合物或本发明第三方面所述的药物组合物或药物制剂。
本发明所述受试者是指已经是治疗、观察或实验的对象的动物,优选指哺乳动物,最优选指人。
本发明所述治疗有效量是指包括本发明化合物在内的活性化合物或药物制剂的量,该量可引起研究者、兽医、医生或其他医疗人员所追求的组织系统、动物或人的生物学或医学响应,所述响应包括减轻或部分减轻所述治疗疾病、综合征、病症或障碍的症状。
本领域的研究者、兽医、医生或其他医疗人员可根据临床试验或者本领域其他公知的手段获知可使用的治疗有效量的范围。
本发明上述含有二硫键的N-乙酰-L-半胱氨酸(NAC)衍生物具有较好的抗氧化和抗炎活性。抗氧化活性结果显示,该类衍生物如A2、A3、A9、A10、A11、A16、A17、A18、A20、A21、A22使Nrf2蛋白表达升高趋势比较明显,Nrf2蛋白表达升高量超过NAC,具有显著性差异。抗炎活性结果显示,该类衍生物如A3、A6、A7、A8、A9、A10、A11、A12、A14、A15、A16、A17、A18、A19、A21等能够显著抑制RAW264.7细胞分泌NO,减轻LPS刺激产生的炎症反应,其抗炎活性优于阳性对照药福多司坦,具有较高的抗炎活性。
本发明上述含有二硫键的N-乙酰-L-半胱氨酸(NAC)衍生物可以用于像慢性阻塞性肺病(慢阻肺,COPD)、肺纤维化、急性肺损伤/急性呼吸窘迫综合征(ALI/ARDS)等氧化应激和炎症应激相关疾病的防治。因此,本发明上述的N-乙酰-L-半胱氨酸(NAC)衍生物具有开发为抗氧化、抗炎药物的潜力。
本发明的有益效果
(1)本发明提供了一系列含有二硫键的N-乙酰-L-半胱氨酸衍生物,为N-乙酰-L-半胱氨酸衍生物类药物的开发提供了潜在的活性化合物。上述技术方案通过试验证明,制备的化合物具有较高的抗氧化、抗炎活性,因此可用于制备效果优于NAC的药物。另外,上述技术方案提供的化合物,其制备工艺简便高效,适合工业放大生产,因此具有良好的应用前景。
(2)本发明中的N-乙酰-L-半胱氨酸(NAC)衍生物可使Nrf2蛋白表达升高趋势比较明显,也能够显著抑制RAW264.7细胞分泌NO,减轻LPS刺激产生的炎症反应,对于像慢性阻塞性肺病(慢阻肺,COPD)、肺纤维化、急性肺损伤/急性呼吸窘迫综合征(ALI/ARDS)等氧化应激和炎症应激相关疾病的防治具有较好的效果,具有开发为相关抗氧化、抗炎药物的潜力。
(3)本发明通过将NAC进行适当的衍生化,得到一种含有二硫键结构的衍生物,在提高其稳定性的基础上,使其在体内能够释放活性巯基物,达到延长半衰期的目标。其次,在增强细胞和组织渗透性,减少给药剂量的基础上,使其具有更好的抗氧化、抗炎和免疫调节的效果,进一步提高其药理活性。
(4)本发明制备方法简单、实用性强,易于推广。
附图说明
构成本发明的一部分的说明书附图用来提供对本发明的进一步理解,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。
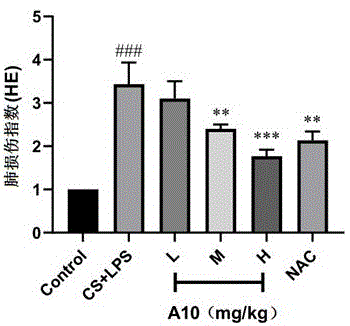
图1为NAC衍生物A10对COPD小鼠的肺组织损伤指数的影响;
图2为NAC衍生物A10对COPD小鼠肺灌洗液(BALF)中总细胞数(a)和中性粒细胞数(b)的影响;
图3为NAC衍生物A10对COPD小鼠肺组织中SOD1(a)和GPX2 (b)mRNA表达的影响;
图4为NAC衍生物A10对纤维化小鼠肺纤维化面积的影响;
图5为NAC衍生物A10对纤维化小鼠肺损伤指数的影响;
图6为NAC衍生物A10对纤维化小鼠肺水肿干湿重比的影响;
图7为NAC衍生物A10对纤维化小鼠肺灌洗液(BALF)中细胞总数(a)和中性粒细胞数(b)的影响;
图8为NAC衍生物A10对肺纤维化小鼠肺匀浆中炎症因子IL-1β(a)和转化生长因子TGF-β(b)的影响;
图9为NAC衍生物A10对ALI小鼠肺损伤指数的影响;
图10为NAC衍生物A10对ALI小鼠肺水肿干湿重比的影响;
图11为NAC衍生物A10对ALI小鼠肺灌洗液(BALF)中细胞总数(a)和中性粒细胞数(b)的影响;
图12为NAC衍生物A10对小鼠肺匀浆中炎症因子IL-1β(a)和转化生长因子TNF-α(b)的影响;
图13为NAC衍生物A10对小鼠肺匀浆中抗氧化因子GSH(a)和SOD(b)的影响。
具体实施方式
应该指出,以下详细说明都是示例性的,旨在对本发明提供进一步的说明。除非另有指明,本发明使用的所有技术和科学术语具有与本发明所属技术领域的普通技术人员通常理解的相同含义。
下面结合具体的实施例,对本发明做进一步的详细说明,应该指出,所述具体实施例是对本发明的解释而不是限定。
实施例1:
N-乙酰-S-((噻吩-2-基甲基)硫代)-L-半胱氨酸(A1)的制备
将0.215 g(1.36 mmol)L-无水半胱氨酸溶于1.7 mL 盐酸甲醇溶液中(盐酸约为0.19 mol/L),在冰水浴条件下,20 min内将L-无水半胱氨酸溶液缓慢滴加至甲氧羰基磺酰氯(2.04 mmol)溶于2 mL甲醇的溶液中,于冰水浴条件下继续搅拌反应1 h。旋转蒸发除去溶剂和过量的甲氧羰基磺酰氯(水浴温度<30℃)。得到的固体用乙醚反复洗涤后,过滤,得到白色结晶固体中间体Ⅲ。将原料2-噻吩甲基硫醇(65 mg,0.5 mmol)溶于0.4 mL甲醇中,在室温并搅拌条件下,缓慢滴加中间体M1的甲醇溶液(0.25 mmol,62.5 mg溶于0.25 mL甲醇中),45 min滴完,继续于室温搅拌过夜后,滴入三乙胺(35 μL,0.25 mmol)和0.5 mL甲醇的混合溶液,继续于室温搅拌30min,过滤白色沉淀,用甲醇和乙醚分别洗涤沉淀后干燥,得白色粉末中间体Ⅴ。将中间体Ⅴ(2 mmol)溶于4 mL冰醋酸中,在室温并搅拌条件下,加入醋酸酐(8 mmol),体系升温至40℃搅拌过夜。旋转蒸发除去溶剂(水浴温度50-55℃),得到黄色油状粗品,经正相柱层析,100-200目硅胶填充,流动相为二氯甲烷:甲醇(20-50:1),以体积比计;和/或反相中压制备色谱,流动相为甲醇:水(0.4-1:1),以体积比计,得白色粉末化合物A1。
实施例2:
N-乙酰-S-((呋喃-2-基甲基)硫代)-L-半胱氨酸(A2)的制备
制备方法参考实施例1。得白色粉末,
实施例3:
N-乙酰-S-((吡啶-4-基甲基)硫代)-L-半胱氨酸(A3)的制备
制备方法参考实施例1。得白色粉末,
实施例4:
N-乙酰-S-(苄硫基)-L-半胱氨酸(A4)的制备
制备方法参考实施例1。得白色粉末,
实施例5:
N-乙酰-S-((4-甲基苄基)硫代)-L-半胱氨酸(A5)的制备
制备方法参考实施例1。得白色粉末,
实施例6:
N-乙酰-S-((4-乙基苄基)硫代)-L-半胱氨酸(A6)的制备
制备方法参考实施例1。得白色粉末,
实施例7:
N-乙酰-S-((4-氟苄基)硫代)-L-半胱氨酸(A7)的制备
制备方法参考实施例1。得白色粉末,
实施例8:
N-乙酰-S-((4-氯苄基)硫代)-L-半胱氨酸(A8)的制备
制备方法参考实施例1。得白色粉末,
实施例9:
N-乙酰-S-((4-溴苄基)硫代)-L-半胱氨酸(A9)的制备
将0.607 g(1.51 mmol) 1,2-双(4-溴苄基)二硫溶于6 mL干燥的二氯甲烷中,在氮气保护和冰水浴条件下,搅拌5min。将化合物m-CPBA(85%,321mg,1.59mmol)溶于5mL干燥的二氯甲烷中,缓慢滴加至上述溶液中,冰水浴条件下继续搅拌反应2 h。反应溶液用5%的碳酸氢钠溶液洗四次,有机相用水洗一次,无水硫酸钠干燥,旋转蒸发除去溶剂 (水浴温度<40℃)。得到的固体经正相柱层析,100-200目硅胶填充,流动相为石油醚:乙酸乙酯(20-50:1),以体积比计,得S-(4-溴苄基)(4-溴苯基)甲磺酸酯。将原料S-(4-溴苄基)(4-溴苯基)甲磺酸酯0.121g(0.29mmol)溶于14.5 mL乙腈中,加入N-乙酰半胱氨酸(Ⅷ, 1.16mmol)和三乙胺0.145g(1.44mmol),在氮气保护并室温条件下搅拌过夜,旋转蒸发除去溶剂 (水浴温度50-55℃)。所得固体加入水(15mL)稀释,乙醚萃取三次,合并有机相,无水硫酸钠干燥,旋转蒸发除去溶剂,所得固体经正相柱层析,100-200目硅胶填充,流动相为二氯甲烷:甲醇(20-50:1),以体积比计;和/或反相中压制备色谱,流动相为甲醇:水(0.4-1:1),以体积比计,得白色粉末化合物A9。
实施例10:
N-乙酰-S-((4-(叔丁基)苄基)硫代)-L-半胱氨酸(A10)的制备
制备方法参考实施例9。得白色粉末,
实施例11:
N-乙酰-S-((2-甲基苄基)硫代)-L-半胱氨酸(A11)的制备
制备方法参考实施例1。得白色粉末,
实施例12:
N-乙酰-S-((2-氯苄基)硫代)-L-半胱氨酸(A12)的制备
制备方法参考实施例1。得白色粉末,
实施例13:
N-乙酰-S-((2-溴苄基)硫代)-L-半胱氨酸(A13)的制备
制备方法参考实施例1。得白色粉末,
实施例14:
N-乙酰-S-((3-氟苄基)硫代)-L-半胱氨酸(A14)的制备
制备方法参考实施例1。得白色粉末,
实施例15:
N-乙酰-S-((3-(三氟甲基)苄基)硫代)-L-半胱氨酸(A15)的制备
制备方法参考实施例1。得白色粉末,
实施例16:
N-乙酰-S-((2,4,6-三甲基苄基)硫代)-L-半胱氨酸(A16)的制备
制备方法参考实施例9。得白色粉末,
实施例17:
N-乙酰-S-((2,4-二氯苄基)硫代)-L-半胱氨酸(A17)的制备
制备方法参考实施例1。得白色粉末,
实施例18:
N-乙酰-S-((萘-2-基甲基)硫代)-L-半胱氨酸(A18)的制备
制备方法参考实施例1。得白色粉末,
实施例19:
N-乙酰-S-(苯乙基硫基)-L-半胱氨酸(A19)的制备
制备方法参考实施例1。得白色粉末,
实施例20:
N-乙酰-S-((1-苯基乙基)硫代)-L-半胱氨酸(A20)的制备
制备方法参考实施例1。得白色粉末,
实施例21:
N-乙酰-S-((4-甲氧基苯基)硫代)-L-半胱氨酸(A21)的制备
制备方法参考实施例9。得白色粉末,
实施例22:
N-乙酰-S-(环己基硫基)-L-半胱氨酸(A22)的制备
/>
制备方法参考实施例9。得白色粉末,
实施例23:
N-乙酰-S-(异丙基硫代)-L-半胱氨酸(A23)的制备
制备方法参考实施例1。得白色粉末,1H NMR (400 MHz, MeOD) δ4.69 (dd, J =9.0, 4.3 Hz, 1H), 3.21 (dd, J = 13.7, 4.4 Hz, 1H), 3.09 – 2.88(m, 2H), 2.00(s, 3H), 1.29 (d, J = 6.7 Hz, 6H). HRMS: calculated for C
实施例24:
N-乙酰-S-(丁基硫醇)-L-半胱氨酸(A24)的制备
制备方法参考实施例1。得白色粉末,
实施例25:
上述实施例化合物抗氧化活性测定。
采用Western Blot技术考察NAC系列衍生物对人支气管上皮细胞16HBE中Nrf2蛋白表达影响的能力,以NAC为阳性药,通过对比筛选出具有潜力的先导化合物。
16HBE细胞为贴壁细胞,培养环境为:10 cm的无菌培养皿中含有8 mL DMEM完全培养基,培养温度为37°C、CO
采用BCA法测定蛋白含量。按照待测孔数配置BCA工作液(BCA:Cu为50:1),混匀充分至透亮淡蓝色。取出少量牛血清白蛋白(BSA)5 mg/mL标准品用PBS稀释至1 mg/mL并在96孔板中配制BSA标准溶液。另起一排加入20 μL稀释后的样品,再加入200 μL BCA工作液混合反应,放于37°C条件下反应30 min,结束后在562 nm处测定OD值。通过对 BSA标准液的OD值进行拟合,进而求出测定样品中蛋白质含量。最后Western Blotting(蛋白免疫印迹技术)检测Nrf2蛋白表达情况。如表1所示,衍生物A2、A3、A9、A10、A11、A16、A17、A18、A20、A21、A22在内的这几个化合物的促进Nrf2蛋白表达量超过阳性对照NAC,具有良好的抗氧化活性。
表1:N-乙酰-L-半胱氨酸(NAC)衍生物影响Nrf2蛋白表达能力
实验重复三次,数据为给药组相对于空白组的倍数,表示为mean±SD(均数加减标准差)。
实施例26:
实施例1至实施例24的化合物抗炎活性测定。
实验采用LPS诱导构建体外细胞炎症模型,对NAC系列衍生物的体外抗炎作用进行评价。首先,选择细胞培养上清NO水平为检测指标,用不同浓度的LPS(0-5 μg/mL)与RAW264.7细胞共培养,通过NO检测试剂盒检测其水平变化情况,选择适宜的LPS刺激浓度和时间。结果表明,当LPS浓度为4 μg/mL,与细胞共培养12 h和24 h条件下,NO相对水平显著提高。因此,抗炎活性筛选实验中的炎症模型选用LPS浓度为4 μg/mL诱导RAW264.7细胞12h建立。造模后,给予2.5、5 μM NAC系列衍生物,阳性药选择临床用于治疗支气管炎等疾病的福多司坦,检测上清NO浓度,作为抗炎活性筛选结果。如表2所示,该系列化合物如A3、A6、A7、A8、A9、A10、A11、A12、A14、A15、A16、A17、A18、A19、A21等抗炎活性优于阳性对照福多司坦,具有良好的抗炎活性。
表2:N-乙酰-L-半胱氨酸(NAC)衍生物影响LPS刺激RAW264.7细胞上清液NO水平
*实验重复三次,数据为各组相对于Ctrl组(对照组)的倍数,Fudostein(福多司坦)。
实施例27:
具有较好抗氧化和抗炎活性化合物A10抗慢阻肺活性评价。
选择主动吸烟且气管滴注LPS建立慢阻肺(COPD)小鼠模型,选择C57BL/6小鼠作为研究对象,以N-乙酰-L-半胱氨酸(NAC)为阳性对照药;为了增加A10的溶解度,将A10制备成甲基-β-环糊精包合物,小鼠腹腔注射后来评价抗慢阻肺功效。
将C57BL/6实验小鼠(共42只)按每组7只小鼠随机分成6组:(1)空白对照组(Control):在气管滴注和药物治疗时,给予小鼠相同量的PBS;(2)COPD模型组(CS+LPS):COPD组和治疗组的小鼠在第1天和14天,接受气管滴注LPS(10 μg-50 μL),然后除第1天和14天,小鼠需每天暴露在8支烟的烟雾中,每天2h,持续30天;(3)治疗组:低中高剂量分别为20 mg/kg (L)、40 mg/kg (M)、80mg/kg(H)的A10包合物,从第2天开始腹腔注射给药,一天一次;(4)NAC组:80 mg/kg剂量,与A10给药时间相同。
A10治疗后的COPD模型小鼠肺组织病理学变化、气道与肺泡中炎性细胞数量变化、小鼠肺部抗氧化相关因子的测定结果分别列于图1、图2和图3。如图1至图3所示,注射给药高剂量(80 mg/kg,H)的A10对COPD的肺损伤指数(图1)、肺灌洗液中总细胞数(图2中a)、中性粒细胞数(图2中b)、肺组织中SOD1和GPX2 mRNA(图3)等治疗效果明显优于同等剂量的NAC,而且中等剂量(40 mg/kg,M)A10抗氧化活性(如SOD1和GPX2 mRNA)也明显超过了NAC,显示了所优选的NAC衍生物(如A10)能够显著地提高抗慢阻肺的疗效。
实施例28:
具有较好抗氧化和抗炎活性化合物A10抗肺纤维化性评价。
使用高压喷雾针向C57BL/6小鼠气管内滴注硫酸博来霉素(BLM)建立小鼠肺纤维化模型,灌胃给药A10和阳性对照NAC,通过记录体重变化,考察A10各组抗肺纤维化情况、肺组织损伤、肺水肿、级联炎症等治疗效果。将小鼠随机分为空白对照组(Ctrl),A10对照组(80 mg/kg, A10),BLM模型组(BLM),BLM+A10低剂量组(20 mg/kg,L),BLM+A10中剂量组(40mg/kg,M),BLM+A10高剂量组(80 mg/kg,H),BLM+NAC阳性药组(500 mg/kg, NAC),共7组。麻醉小鼠后,通过高压喷雾针将硫酸BLM(5 mg/kg)溶液由小鼠气管内滴注入小鼠肺部,注入后保持小鼠呈坐立状态并左右轻摇小鼠使BLM和生理盐水在肺部均匀分布,建立小鼠肺纤维化模型。24 h后分别灌胃给予生理盐水、A10(L、M、H)和NAC,不间断给药天后,无痛苦处死所有动物,收集支气管肺泡灌洗液(Bronchoalveolar lavagefluid,BALF)和肺组织作为样本用于后续实验研究。
A10对肺纤维化面积、肺组织损伤、肺水肿、气道与肺泡中炎性细胞数量变化、肺匀浆中炎症等抗肺纤维化治疗效果分别列于图4、图5、图6、图7和图8。如图4-图8所示,口服给药高剂量(80 mg/kg,H)A10抗肺纤维化、肺损伤、肺水肿、炎性应激等治疗效果明显优于500mg/kg的NAC;显示了所优选的NAC衍生物(如A10)能够显著地提高了药物口服生物利用度,提高了抗肺纤维化的疗效。
实施例29:
具有较好抗氧化和抗炎活性化合物A10抗急性肺损伤评价。
使用高压喷雾针向C57BL/6小鼠气管内滴注脂多糖(LPS)建立小鼠急性肺损伤(ALI)模型,灌胃给药A10和阳性对照地塞米松磷酸钠(DEX),通过记录体重变化,考察A10各组抗肺组织损伤、肺水肿、氧化应激、炎症应激等治疗效果。将小鼠随机分为空白对照组(C),LPS模型组(M),LPS+A10低剂量组20 mg/kg(20),LPS+A10中剂量组40 mg/kg(40),LPS+A10高剂量组80 mg/kg(80),LPS+DEX阳性药组(5 mg/kg,P),共7组。麻醉小鼠后,通过高压喷雾针将硫酸LPS(5 mg/kg)溶液由小鼠气管内滴注入小鼠肺部,注入后保持小鼠呈坐立状态并左右轻摇小鼠使LPS和生理盐水在肺部均匀分布,建立小鼠肺纤维化模型。1小时后分别灌胃给予生理盐水、A10(20、40、80)和DEX,不间断给药24小时后,无痛苦处死所有动物,收集支气管肺泡灌洗液(Bronchoalveolar lavagefluid,BALF)和肺组织作为样本用于后续实验研究。
A10对肺组织损伤、肺水肿、气道与肺泡中炎性细胞数量变化、肺匀浆中炎症因子和抗氧化因子等抗急性肺损伤治疗效果分别列于图9、图10、图11、图12和图13。如图9-图13所示,注射给药高剂量(80 mg/kg,80)A10抗肺损伤、肺水肿、炎性应激和氧化应激等治疗效果明显优于阳性对照药地塞米松;显示了所优选的NAC衍生物(如A10)具有抗急性肺损伤的显著疗效。
以上所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 一种N-乙酰-L-半胱氨酸的制备方法
- 一种双-氟喹诺酮噁二唑脲类N-乙酰基环丙沙星衍生物及其制备方法和应用
- 一种双-氟喹诺酮噁二唑脲类N-乙酰诺氟沙星衍生物及其制备方法和应用
- 一种N-苯乙酰基-L-脯氨酸的制备方法
- 用于发酵制备L-半胱氨酸、L-胱氨酸、N-乙酰丝氨酸或四氢噻唑衍生物的微生物和方法
- 用于发酵制备L-半胱氨酸、L-胱氨酸、N-乙酰丝氨酸或四氢噻唑衍生物的微生物和方法