方法和组合物
文献发布时间:2023-06-19 12:07:15
发明领域
本发明涉及将2,3-二氨基丙酸(DAP)遗传引入多肽中;包含DAP的非天然氨基酸;用于用包含DAP的非天然氨基酸装载tRNA的tRNA合成酶;以及使用所得多肽例如在酶促反应中捕获底物和/或中间体的方法。
许多酶通过与酶活性位点中的丝氨酸或半胱氨酸侧链结合的共价中间体进行反应
稳定捕获这些中间体的策略使天然底物的鉴定以及其他难捕捉的中间体和功能状态的表征成为可能。在已知的方法中,可使用带有亲电替换羰基的底物类似物来捕获酰基酶中间体的类似物
酰基酶中间体——在酶的半胱氨酸或丝氨酸残基的巯基或羟基侧链与底物的羰基之间形成的——在多种生物转化中普遍存在,包括由非核糖体肽合成酶(NRPS)和蛋白酶介导的那些生物转化。这些重要的硫酯和酯中间体不稳定,通常具有几分钟到几小时的半衰期,这使其表征具有挑战性。这是本领域中的问题。
现有技术中已经公开了包含2,3-二氨基丙酸(DAP)的多肽。具体地,现有技术中已经通过固相合成/缀合技术生产了一种或多种带有DAP的多肽(参见Virdee,S.,Macmillan,D.,&Waksman,G.(2010)Chemistry&Biology vol 17第274-284页“Semisynthetic Src SH2domains demonstrate altered phosphopeptide specificity induced byincorporation of unnatural lysine derivatives.”)。然而,通过常规固相合成/缀合技术生产包含DAP的多肽存在许多问题。例如,使用现有技术将DAP引入具有最大生物学意义的结构域或基序(例如酶的活性位点)是极其困难或不可能的。这可能是因为这些位点无法进行化学偶联,和/或因为通过固相合成制得的蛋白质需要进行化学重折叠才能获得正确的确认。除了费力外,这还非常不可靠,并且是不可预测的,并且常常是不成功的。这些是本领域中的问题。
本发明提供的关键技术是一种新的生产包含2,3-二氨基丙酸(DAP)的多肽的方式。这对于将DAP引入到现有技术中难以或不可能修饰的位置(例如引入到酶活性位点中)特别有用。
这些方法基于新的非天然氨基酸,其用于通过天然存在的翻译机构(例如细胞自身的核糖体)引入到多肽中。所述方法还涉及能够用新的非天然氨基酸装载正交tRNA的新的tRNA合成酶。一旦将新的非天然氨基酸引入到多肽中,就可以很容易地对其进行脱保护,以将DAP留在多肽骨架中。
因此,本发明能够实现将DAP引入到一定范围的蛋白质中,和/或引入到蛋白质内的一定范围的位置中,这是当前使用现有技术无法实现的。
因此,在一方面,本发明提供了式(I)或式(II)的非天然氨基酸:
或其盐、溶剂化物、互变异构体、异构体或混合物;
其中:
R
R
q是1、2或3;
每个R
X是X
X
X
X
X
R
Y是选自以下的保护基:
R
R
每个R’独立地选自C
R
R
R
X
Y
Y
M
Z是Si或Ge;
R
R
Y
一方面,本发明提供了包含如上所述的非天然氨基酸的多肽,其中所述非天然氨基酸通过肽键连接至所述多肽。
一方面,本发明涉及一种制备包含DAP的多肽的方法,该方法包括如上所述对多肽进行脱保护。合适地,所述脱保护包括在365nm、35mWcm
一方面,本发明涉及包含突变Y271C、N311Q、Y349F和V366C的PylRStRNA合成酶。合适地,所述PylRS tRNA合成酶是包含所述突变的巴氏甲烷八叠球菌(Methanosarcinabarkerii)PylRS(MbPylRS)tRNA合成酶。
在一个方面,本发明涉及一种生产包含2,3-二氨基丙酸(DAP)的多肽的方法,所述方法包括将如上所述的非天然氨基酸遗传引入到多肽中,以及任选地将所述非天然氨基酸脱保护为2,3-二氨基丙酸(DAP)。
合适地,生产多肽包括:
(i)提供编码所述多肽的核酸,所述核酸包含编码根据权利要求1至10中任一项所述的非天然氨基酸的正交密码子;
(ii)在能够识别所述正交密码子的正交tRNA合成酶/tRNA对的存在下翻译所述核酸,并将所述非天然氨基酸引入到多肽链。
合适地,所述正交密码子包括琥珀密码子(TAG),所述tRNA包括MbtRNA
合适地,所述非天然氨基酸包含:
一方面,本发明涉及如上所述的多肽或如上所述的方法,其中所述多肽是酶,并且其中在对应于所述酶的活性位点内的氨基酸残基的位置引入所述非天然氨基酸。
一方面,本发明涉及如上所述的多肽,其中所述多肽是酶,并且其中根据酶的国际命名法和分类,所述酶是EC 3.4肽酶、EC 3.4.22.44肽酶或EC 2.3.2.23 E2泛素结合酶。
一方面,本发明涉及如上所述的多肽,其包含一至二十个2,3-二氨基丙酸(DAP)基团。合适地,所述多肽包含单一2,3-二氨基丙酸(DAP)基团。
一方面,本发明涉及如上所述的多肽或如上所述的方法,其中在对应于野生型多肽中的半胱氨酸、丝氨酸或苏氨酸残基的位置,任选地在对应于野生型多肽中的半胱氨酸或丝氨酸残基的位置,引入所述非天然氨基酸。
一方面,本发明涉及如上所述的非天然氨基酸在生产包含2,3-二氨基丙酸(DAP)的多肽中的用途。合适地,多肽的生产包括进行如上所述的方法。
一方面,本发明涉及一种捕获酶的底物的方法,该方法包括:
a)提供在其活性位点包含至少一个2,3-二氨基丙酸(DAP)基团的酶,
b)使所述酶与所述酶的候选底物接触,和
c)孵育以允许所述DAP基团与所述候选底物反应。
合适地,所述底物是代谢产物。
合适地,所述酶是肽酶或泛素结合酶或水解酶或形成碳硫键的酶。
在这里,我们公开了遗传编码的2,3-二氨基丙酸如何实现对酶促反应的结构见解。特别地,我们通过缬氨霉素生物合成中的酰基硫酯酶中间体举例说明了这种方法。
本发明实现了遗传引导将2,3-二氨基丙酸(DAP)有效引入到在(例如)大肠杆菌(E.coli)中产生的重组蛋白中的策略。我们教导了如何用DAP替换催化残基,例如半胱氨酸或丝氨酸残基。这实现了对通过稳定酰胺键连接的酰基酶复合物的有效捕获。
例如,通过阐明生物合成途径来展示本发明,通过该生物合成途径,缬氨霉素合成酶的硫酯酶结构域(Vlm TE)促进线性四缩肽顺序三聚化为十二缩肽以及随后的十二缩肽环化为缬氨霉素。通过捕获Vlm TE的催化循环中的第一个和最后一个酰基-TE中间体作为DAP缀合物,使用本发明提供了对NRPS的TE结构域中的构象变化如何控制从线性底物的低聚到环化的转换的结构见解。诸如通过本发明实现的这种策略的策略可用于促进多种酰基酶中间体的表征的应用。此外,本发明可用于实现功能未知的酶的天然底物的捕获和鉴定的应用。
一方面,本发明涉及如上所述的同源重组多肽。合适地,所述多肽通过如上所述的方法制备。
一方面,本发明涉及根据本文所述方法生产的多肽。这种多肽不仅是这些新方法的产物,还具有包含如上所述的非天然氨基酸或包含DAP的技术特征。
突变有它在本领域中的标准含义,并且可以是指提到的残基、基序或结构域的取代或截短或删除。可以在多肽水平上实现突变,例如通过合成含有突变序列的多肽,或可以在核苷酸水平上实现突变,例如通过制备编码突变序列的核酸,可以随后翻译该核酸以产生突变多肽。在无氨基酸被指定为对给定突变位点的替换氨基酸的情况下,合适地,使用随机化的所述位点。作为默认(default)突变,可以使用丙氨酸(A)。合适地,在一个或多个特定位点使用的突变如本文所陈述。
片段的长度适合为至少10个氨基酸,适合为至少25个氨基酸,适合为至少50个氨基酸,适合为至少100个氨基酸,适合为至少200个氨基酸,适合为至少250个氨基酸,适合为至少300个氨基酸,适合为至少313个氨基酸,或适合为目标多肽的大多数。
可以在体内或体外实施本发明的方法。
在一个实施方案中,合适地,本发明的方法不应用于人体或动物体。合适地,本发明的方法是体外方法。合适地,所述方法不需要人体或动物体的存在。合适地,所述方法不是人体或动物体的诊断或手术或治疗方法。
术语‘包含’应理解成具有它在本领域中的标准含义,即,包括规定的特征或特征组,但该术语不排除也存在的任何其他规定特征或特征组。
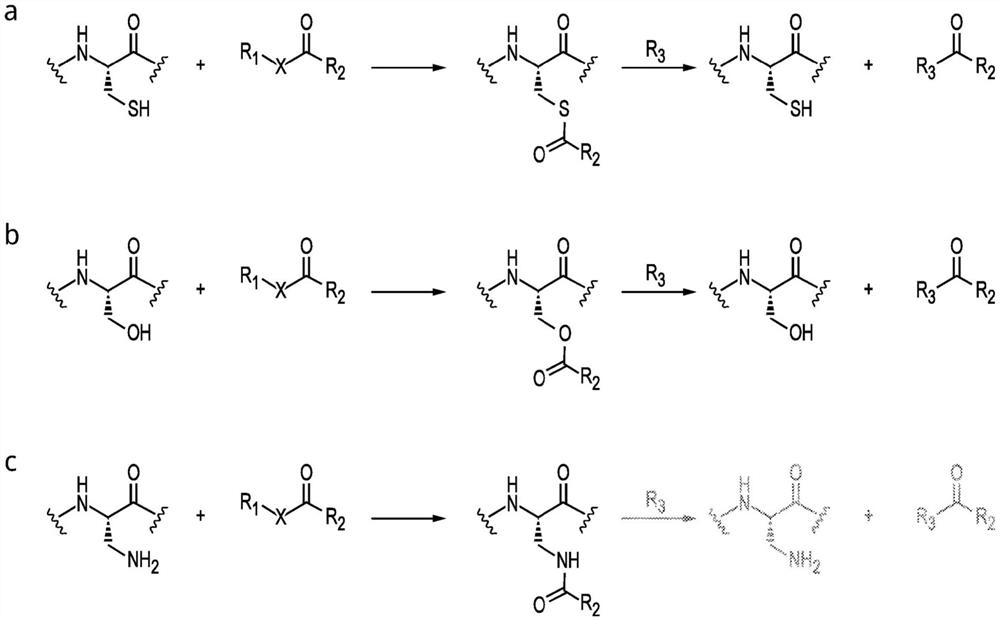
我们假设,用其中巯基或羟基被氨基替换的氨基酸(2,3-二氨基丙酸(DAP,1))选择性替换催化半胱氨酸或丝氨酸残基将允许捕获通过酰胺键连接的酰基酶中间体(图1C)。赖氨酸侧链中的胺的共轭酸的pKa为10.5,而DAP的β氨基的共轭酸的pKa为9.4
产生次级代谢产物的巨型酶(megaenzyme)非核糖体肽合成酶(NRPS)和聚酮合成酶(PKS)在其合成周期中生成高度复杂的酰基酶中间体。这些分子机器使用硫代模板化的生物合成途径将小的酰基分子组装到大量生物活性天然产物,包括临床抗癌剂、抗生素、抗真菌剂和免疫抑制剂(补充图1)。试图揭示其详细分子功能的现有技术尝试已受到了以高分辨率表征其多种酰基酶中间体的挑战的阻碍。这种挑战以来自NRPS途径的硫酯酶(TE)结构域为例,NRPS途径使线性化肽基或二肽基底物低聚并环化。这些TE结构域参与了抗生素短杆菌肽S
酰基-TE中间体的高分辨率结构将提供有关TE如何控制底物命运并代表着重大进展的机制见解。已经获得了少数高分辨率的酰基-TE结构,最值得注意的是形成聚酮苦霉素的TE和非天然底物类似物
本发明提供的获得稳定的酰基-TE中间体的能力具有显著的益处,使本领域技术人员能够表征非核糖体肽生物合成以及聚酮和脂肪酸生物合成中TE结构域选择性的机制。
如下面更详细描述的,发明人已经提出了引入可在温和条件下翻译后转化为DAP的氨基酸的氨基酰基-tRNA合成酶/tRNA
我们证明了本发明用于阐明将四缩肽转化为缬氨霉素的生物合成途径。通过用DAP替换Vlm TE中的催化丝氨酸,我们证明了如何获得稳定的脱氧-四缩肽基-N-TE
合适地,多肽包含单个DAP基团和/或如上所述的非天然氨基酸残基。这具有保持对可能针对如上所述的DAP基团/非天然氨基酸的任何进一步化学修饰的特异性和/或当DAP存在于酶的活性位点时的捕获特异性的优点。例如,当目标多肽中只有单一的如上所述的DAP基团/非天然氨基酸时,则有利地避免可能的部分修饰/部分脱保护的问题或同一多肽中交替的DAP基团之间反应微环境发生变化的问题(这可能导致在该多肽中在不同位置的不同DAP基团之间具有不等同的反应性)。
合适地,该多肽包含两个DAP基团;合适地,该多肽包含三个DAP基团;合适地,该多肽包含四个DAP基团;合适地,该多肽包含五个DAP基团;合适地,该多肽包含十个或甚至更多的DAP基团,例如15-20个DAP基团。最合适地,该多肽包含一个至五个DAP基团。更合适地,该多肽包含一个DAP基团。
原则上,可以通过相同或不同的正交密码子/正交tRNA对引入如上所述的多个非天然氨基酸(相同非天然氨基酸的多个拷贝,或两个或更多个不同非天然氨基酸中的每个的一个或多个拷贝)。合适地,通过插入/翻译多个琥珀密码子(与如本文所述的合适的正交tRNA合成酶一起)引入多个非天然氨基酸。
本文以常规方式使用通式描述了新型化学实体(NCE)。描述了式(I)或式(II)的非天然氨基酸:
或其盐、溶剂化物、互变异构体、异构体或混合物;
其中:
R
R
q是1、2或3;
每个R
X是X
X
X
X
X
R
Y是保护基,其选自:
R
R
每个R’独立地选自C
R
R
R
X
Y
Y
M
Z是Si或Ge;
R
R
Y
术语“或其盐、溶剂化物、互变异构体、异构体或其混合物”是指还包括所示结构的盐、溶剂化物、互变异构体、异构体形式。其混合物是指可以存在这些形式的混合物,例如,本发明的化合物可以包含互变异构形式的盐。
“药学上可接受的”物质是指在合理的医学判断范围内适合与受试者的组织接触而没有不适当的毒性、刺激性、过敏反应等的那些物质,其具有合理的收益风险比,并且对它们的预期用途有效。
“药物组合物”是指一种或多种药物物质和一种或多种赋形剂的组合。
如本文所用,“溶剂化物”是指由溶质(例如式(I)-(II)或本文中的任何其他化合物或其盐)和溶剂形成的可变化学计量的复合物。结晶化合物可以形成药学上可接受的溶剂化物,其中在结晶过程中溶剂分子引入到晶格中。引入的溶剂分子可以是水分子或非水分子,例如但不限于乙醇、异丙醇、二甲亚砜、乙酸、乙醇胺和乙酸乙酯分子。
“独立地选自”用于以下陈述的上下文中,例如,“每个R
在本说明书中,C
在本说明书中,“氨基酸残基”是指氨基酸的胺或羧酸端已被肽键替换的氨基酸。
C
“卤代”或“卤素”:是指-F、-Cl、-Br或-I。
“C
“C
单环杂芳基的实例包括但不限于衍生自以下的那些单环杂芳基:
N
O
S
N
N
N
N
N
N
N
包含稠环的杂芳基的实例包括但不限于衍生自以下的那些杂芳基:
O
N
S
N
N
O
N
O
S
N
N
N
N
“C
与双环环烷基一样,双环杂环基可以包括分开的环、螺环、稠环和桥环。杂环基可以在任何环原子处连接至母体基团或连接至底物,并且可以包括一个或多个非氢取代基,除非这种连接或取代会违反化合价要求或导致化学上不稳定的化合物。
单环杂环基的实例包括但不限于衍生自以下的那些单环杂环基:
N
O
S
O
O
N
N
N
N
O
N
取代的单环杂环基的实例包括衍生自环状形式的糖的那些,环状形式的糖例如呋喃糖,例如阿拉伯呋喃糖、来苏呋喃糖(lyxofuranose)、呋喃核糖和呋喃木糖;以及吡喃糖,例如aliopyranose、阿卓吡喃糖、吡喃葡萄糖、吡喃甘露糖、吡喃古洛糖、吡喃艾杜糖、吡喃半乳糖和吡喃塔洛糖。
如本文所用,术语“肽”是指包含多个氨基酸残基的线性分子,所述多个氨基酸残基通过肽键彼此结合。
保护基是通过官能团的化学修饰引入到分子中以暂时掩盖官能团的特征性化学过程以防止其干扰另一反应的基团。可通过光解反应去除的保护基(即通过光解反应能够被去除/可去除/可被脱保护)是可通过诸如光的辐射能去除的基团。保护基描述在Wuts,P.G.M.和Greene,T.W.,Protective Groups in Organic Synthesis,第4版,Wiley-lnterscience,2007和P.Kocienski,Protective Groups,第3版(2005)中。
“糖”取代基表示单糖或多糖,其中来自糖的羟基的H已被将糖取代基连接至式(I)或式(II)的化合物的其余部分的键所替换。合适地,糖是单糖。合适地,糖是葡萄糖、甘露糖或半乳糖。
合适地,保护基是光不稳定的,因此可在大于300nm的波长λ
一方面,合适地,式(I)或式(II)的非天然氨基酸为:
或其盐、溶剂化物、互变异构体、异构体或混合物。因此,在这个方面,式(I)或式(II)的非天然氨基酸为D-形式。
更合适地,式(I)或式(II)的非天然氨基酸为:
或其盐、溶剂化物、互变异构体、异构体或混合物。因此,在这个更合适的方面,式(I)或式(II)的非天然氨基酸为L-形式。
合适地,式(I)或式(II)的非天然氨基酸为:
或其盐、溶剂化物、互变异构体、异构体或混合物。合适地,上述非天然氨基酸式(I)或式(II)为D-形式。更合适地,上述非天然氨基酸式(I)或式(II)为L-形式。
更合适地,式(I)或式(II)的非天然氨基酸为:
或其盐、溶剂化物、互变异构体、异构体或混合物。
合适地,非天然氨基酸是式(I)的非天然氨基酸或其盐、溶剂化物、互变异构体、异构体或混合物。
合适地,式(I)的非天然氨基酸具有下式:
或其盐、溶剂化物、互变异构体、异构体或混合物。
合适地,式(I)的非天然氨基酸具有下式:
或其盐、溶剂化物、互变异构体、异构体或混合物。
合适地,式(I)的非天然氨基酸具有下式:
或其盐、溶剂化物、互变异构体、异构体或混合物。
合适地,X是X
合适地,X
合适地,X
合适地,X
合适地,X
R
合适地,R
合适地,q是1或2。更合适地,q是1。
合适地,每个R
合适地,每个R
合适地,每个R
合适地,R
更合适地,R
当R
在一些方面,更合适地,Y在R
在一些方面,更合适地,Y对于在R
合适地,式(I)或式(II)的化合物或其盐、溶剂化物、互变异构体、异构体或混合物包含基团Y。
合适地,Y是:
更合适地,非天然氨基酸包含基团Y,该基团为:
更合适地,非天然氨基酸包含基团Y,该基团为:
合适地,R
更合适地,R
合适地,R
在一些方面,更合适地,R
更合适地,R
合适地,每个R’独立地选自H、CH
合适地,R
合适地,R
合适地,R
合适地,在一方面,M
在一方面,M
在一方面,Z是Si。
在另一方面,Z是Ge。
合适地,R
在一方面,合适地,R
更合适地,Y是:
在一些方面,Y是
在一个实施方案中,式(I)或式(II)的化合物是
或其盐、溶剂化物、互变异构体、异构体或混合物。
一个高度合适的实施方案表示为:
或其盐、溶剂化物、互变异构体、异构体或混合物。
在本文中,本实施方案有时被称为‘6’或‘化合物6’或‘DAP 5’;这些名称中的每一个均是指与上面所示相同的化学结构。
在一方面,更合适地,非天然氨基酸为:
或其盐、溶剂化物、互变异构体、异构体或混合物。
更合适地,非天然氨基酸是式(III)或式(IV)的化合物或其盐、溶剂化物、互变异构体、异构体或混合物。在一方面,非天然氨基酸是式(III)的化合物或其盐、溶剂化物、互变异构体、异构体或混合物。在另一方面,非天然氨基酸是式(IV)的化合物或其盐、溶剂化物、互变异构体、异构体或混合物。
在一方面,图3的化合物2、3、4或5中的一个或多个可以用于体外翻译系统,其使用如上所述的合成酶,或例如Nguyen,D.P.et al.(2014)[J.Am.Chem.Soc.,2014,136(6),pp2240–2243]中描述的合成酶,即合成酶“PCC1RS”,其是具有突变N311M、C313Q、V366G、W382N、R85H的MbPylRS。这种PCC1RS合成酶确实引入了非常相似的非天然氨基酸(光笼型半胱氨酸),发明人主张它将接受DAP的受保护形式,它们的区别仅在于1个原子(或两个氢原子)。另一种技术将包括用所谓的“灵活的酶(flexizyme)”加载tRNA,所述“灵活的酶”实际上可用任何氨基酸加载任何tRNA(例如,参见Morimoto et al.,2011,Acc.Chem Res.44)。然后可以将它们用于体外系统。不希望被理论所束缚,发明人相信图3的类似物化合物2、3、4或5在细胞中不起作用,因为它们不能进入细胞。
除非另有说明,否则以上包括的是这些取代基的众所周知的离子、盐或溶剂化物形式。例如,提及羧酸(-COOH)还包括其阴离子(羧酸根)形式(-COO
某些化合物可能以一种或多种特定的几何、光学、对映异构、非对映异构、差向异构、阿托异构(atropic)、立体异构、互变异构、构象或端基异构形式,包括但不限于顺式和反式形式;E-和Z-形式;c-、t-和r-形式;内-和外-形式;R-、S-和中间-形式;D-和L-形式;d-和l-形式;(+)和(-)形式;酮、烯醇和烯醇化物形式;顺式和反式;向斜和背斜形式;α-和β-形式;轴向和赤道形式;船式、椅式、扭船式、信封式和半椅式;及其组合,在下文中统称为“异构体”(或“异构体形式”)。
注意,除下文关于互变异构形式的讨论外,本文所用的术语“异构体”明确排除结构(或构成)异构体(即,原子之间的连接不同而不仅仅是原子在空间中的位置不同的异构体)。例如,提及甲氧基-OCH
提及一类结构可以很好地包括落入该类中的结构异构形式(例如C
上述排除不适用于互变异构形式,例如,酰胺/亚氨基醇-NH-C(=O)-/-N=C(-OH)-;或酮、烯醇和烯醇化物形式,例如以下互变异构对:酮/烯醇、亚胺/烯胺、酰胺/亚氨基醇、脒/脒、亚硝基/肟、硫酮/烯硫醇、N-亚硝基/羟基偶氮和硝基/酸硝基。
注意,术语“异构体”中明确包括具有一个或多个同位素取代的化合物。例如,H可以是任何同位素形式,包括
除非另有说明,提及特定化合物包括所有此类异构体形式,包括(全部或部分)外消旋体及其其他混合物。其他指定键的一个实例是DAP中的CH基团,其如下所示具有指定的立体化学:
此类异构体形式的制备(例如不对称合成)和分离(例如分步结晶和色谱手段)方法是本领域已知的,或者通过以已知方式调整本文教导的方法或已知方法容易地获得。
除非另有说明,否则提及特定化合物还包括其离子、盐、溶剂化物和保护形式,例如,如下所述。
在一些实施方案中,式(I)或式(II)的化合物或其盐、溶剂化物、互变异构体、异构体或混合物包含式(I)或式(II)的化合物的药学上可接受的盐。
式(I)或式(II)的化合物(其包括以上具体命名的化合物)可以形成药学上可接受的复合物、盐、溶剂化物和水合物。这些盐包括无毒酸加成盐(包括二酸)和碱式盐。
如果化合物是阳离子的或具有可以是阳离子的官能团(例如-NH
例如,如果化合物是阴离子的,或具有可以是阴离子的官能团(例如-COOH可以是–COO
药学上可接受的盐可以使用各种方法来制备。例如,可以使式(I)或式(II)的化合物与适当的酸或碱反应以得到所需的盐。也可以使式(I)或式(II)的化合物的前体与酸或碱反应以除去酸或碱不稳定的保护基或打开该前体的内酯或内酰胺基团。另外,通过用适当的酸或碱处理或通过与离子交换树脂接触,可以将式(I)或式(II)的化合物的盐转化为另一种盐。反应后,如果盐从溶液中沉淀出来,则可以通过过滤分离盐,或者通过蒸发来回收盐。盐的离子化程度可以从完全离子化到几乎未离子化。
制备、纯化和/或处理活性化合物的相应溶剂化物可能是方便或期望的。术语“溶剂化物”描述了包含化合物和一种或多种药学上可接受的溶剂分子(例如,EtOH)的分子复合物。术语“水合物”是其中溶剂是水的溶剂化物。药学上可接受的溶剂化物包括其中溶剂可以被同位素取代的那些(例如D
目前公认的有机化合物的溶剂化物和水合物的分类系统是区分分离的位点、通道和金属离子配位溶剂化物和水合物的一种系统。参见例如K.R.Morris(H.G.Brittain ed.)Polymorphism in Pharmaceutical Solids(1995)。分离的位点溶剂化物和水合物是其中通过介入有机化合物的分子使溶剂(例如水)分子从彼此直接接触分开的溶剂化物和水合物。在通道溶剂化物中,溶剂分子位于晶格通道中,在此其与其他溶剂分子相邻。在金属离子配位溶剂化物中,溶剂分子键合至金属离子。
当溶剂或水紧密结合时,配合物将具有定义明确的化学计量,而与湿度无关。但是,当溶剂或水结合较弱时(例如在通道溶剂化物和吸湿性化合物中),水或溶剂的含量将取决于湿度和干燥条件。在这种情况下,通常会观察到非化学计量。
对于通过遗传引入生产根据本发明的多肽,所述遗传引入优选使用正交或扩展的遗传密码,其中已分配一个或多个特定正交密码子来编码目标非天然氨基酸,从而使得可通过使用正交tRNA合成酶/tRNA对来遗传引入它。正交tRNA合成酶/tRNA对原则上可以是任何能够用目标非天然氨基酸装载tRNA、并且能够响应于正交密码子将该目标非天然氨基酸引入到多肽链中的此类对。
正交密码子可以是正交琥珀密码子、赭石密码子、卵白石(opal)密码子或四联体密码子。密码子仅仅必须对应于将用于携带目标非天然氨基酸的正交tRNA。最合适地,正交密码子是琥珀密码子。
应注意,本文示出的许多特定实例已使用琥珀密码子和对应的tRNA/tRNA合成酶。如上文指出的,这些可以是变化的。或者,为了在不费力使用或选择能够用目标非天然氨基酸工作的替代tRNA/tRNA合成酶对的情况下使用其他密码子,仅仅可以用所选密码子所期望的反密码子区域调换tRNA的反密码子区域。反密码子区域不涉及tRNA的装载或引入功能,也不由tRNA合成酶识别,因此此类调换完全在熟练操作人员的范围内。因此,在一些实施方案中,用于本发明中的tRNA反密码子区域例如MbtRNA
因此,如果需要的话,可以使用替代正交tRNA合成酶/tRNA对,只要保留了所需的装载活性。
巴氏甲烷八叠球菌PylT基因编码MbtRNA
tRNAcua
MbPylT(菌株MS,来自Genbank登记号AY064401)
gggaacctgatcatgtagatcgaatggactctaaatccgttcagccgggttagattcccggggtttccgcca
可以使用的这种tRNA有两种变体。一个以ggg(如上所述)开始,另一个以gga(如下所示)开始:
tRNAcua“gga”变体
MbPylT(菌株MS,来自Genbank登记号AY064401)
ggaaacctgatcatgtagatcgaatggactctaaatccgttcagccgggttagattcccggggtttccgcca
这两个变体之间没有实质性差异。
巴氏甲烷八叠球菌PylS基因编码MbPylRS tRNA合成酶蛋白。
tRNA合成酶
如果需要,本领域技术人员可以通过使MbPylRS tRNA合成酶蛋白突变来调整它,以便针对所使用的特定非天然氨基酸进行优化。对突变(如果有的话)的需要取决于所使用的特定非天然氨基酸。可能需要使MbPylRS tRNA合成酶突变的一个实例是当所使用的特定非天然氨基酸未被MbPylRS tRNA合成酶蛋白处理。
在本发明中,发明人将大量的智力投入到新型合成酶DAPRS的产生中。尤其参见实施例3。合适地,本发明的合成酶包含271位的C、311位的Q、349位的F和366位的C(即相对于MbPylRS,Y271C、N311Q、Y349F和V366C)或同等位置(如果使用来自其他物种的不同起始合成酶序列/主链合成酶序列)。
新型合成酶DAPRS的示例性序列:
DAPRS(突变的残基被加上下划线):
优选地,正交合成酶/tRNA对是巴氏甲烷八叠球菌MS吡咯赖氨酸tRNA合成酶(MbPylRS)及其同族琥珀抑制基因tRNA(MbtRNA
本发明的tRNA合成酶可以变化。尽管在实例中可能已使用特定tRNA合成酶序列,但本发明不意图仅限制于那些实例。
原则上,在本发明中可使用任何提供相同tRNA装载(氨酰化)功能的tRNA合成酶。
例如,tRNA合成酶可以来自任何合适的物种,例如来自古菌域,例如来自巴氏甲烷八叠球菌(Methanosarcina barkeri)MS;巴氏甲烷八叠球菌str.Fusaro;马氏甲烷八叠球菌(Methanosarcina mazei)Go1;嗜乙酸甲烷八叠球菌(Methanosarcina acetivorans)C2A;嗜热甲烷八叠球菌(Methanosarcina thermophila);或布氏拟甲烷球菌(Methanococcoides burtonii)。或者,tRNA合成酶可以来自细菌,例如来自哥本哈根脱亚硫酸菌(Desulfitobacterium hafniense)DCB-2;哥本哈根脱亚硫酸菌Y51;哥本哈根脱亚硫酸菌PCP1;醋酸氧化脱硫肠状菌(Desulfotomaculum acetoxidans)DSM 771。
来自这些生物的示例性序列是可公开得到的序列。提供以下实例作为吡咯赖氨酸tRNA合成酶的示例性序列:
>巴氏甲烷八叠球菌iMS/1-419/
巴氏甲烷八叠球菌MS
版本Q6WRH6.1 GI:74501411
>巴氏甲烷八叠球菌iF/1-419/
巴氏甲烷八叠球菌str.Fusaro
版本YP_304395.1GI:73668380
>马氏甲烷八叠球菌/1-454
马氏甲烷八叠球菌Go1
版本NP_633469.1GI:21227547
>嗜乙酸甲烷八叠球菌/1-443
嗜乙酸甲烷八叠球菌C2A
版本NP_615128.2 GI:161484944
>嗜热甲烷八叠球菌/1-478
嗜热甲烷八叠球菌,版本DQ017250.1GI:67773308
>布氏拟甲烷球菌/1-416
布氏拟甲烷球菌DSM 6242,版本YP_566710.1 GI:91774018
>哥本哈根脱亚硫酸菌_DCB-2/1-279
哥本哈根脱亚硫酸菌DCB-2
版本YP_002461289.1 GI:219670854
>哥本哈根脱亚硫酸菌_Y51/1-312
哥本哈根脱亚硫酸菌Y51
版本YP_521192.1 GI:89897705
>哥本哈根脱亚硫酸菌PCP1/1-288
哥本哈根脱亚硫酸菌
版本AY692340.1 GI:53771772
>醋酸氧化脱硫肠状菌/1-277
醋酸氧化脱硫肠状菌DSM 771
版本YP_003189614.1 GI:258513392
当已通过突变tRNA合成酶来提供特定tRNA装载(氨酰化)功能时,那么仅仅使用另一野生型tRNA合成酶序列(例如选自上述的序列)可能是不适当的。在这种情境下,保留相同的tRNA装载(氨酰化)功能将是重要的。这是通过将示例性tRNA合成酶中的一个或多个突变移入替代的(alternate)tRNA合成酶骨架(如选自上述的一个)来完成的。
以这种方式,应可能将选定的突变移至对应tRNA合成酶序列,例如来自除示例性巴氏甲烷八叠球菌i和/或马氏甲烷八叠球菌序列以外的其他生物的对应pylS序列。
可以通过与已知tRNA合成酶(例如示例性巴氏甲烷八叠球菌i和/或马氏甲烷八叠球菌序列)比对来选择靶tRNA合成酶蛋白质/骨架。
现在参考pylS(吡咯赖氨酸tRNA合成酶)序列来说明此主题,但所述原理同样应用于特定目标tRNA合成酶。
例如,可以准备所有PylS序列的比对。这些可具有低总%序列同一性。因此,如通过使序列与已知tRNA合成酶比对(而不是仅仅使用低序列同一性得分)来研究序列是重要的,以确保使用的序列确实是tRNA合成酶。
因此,合适地,当考虑到序列同一性时,合适地,在上述tRNA合成酶实例的序列间考虑。合适地,%同一性可如根据上述序列的比对而定义。
聚焦于催化区域可能是有用的。此目的在于提供tRNA催化区域,根据其可限定高%同一性,以捕获/鉴定适合于接受为了产生相同的tRNA装载(氨酰化)功能而移植的突变的骨架支架,例如新的或非天然氨基酸识别。
因此,合适地,当考虑到序列同一性时,合适地,在催化区域间考虑。合适地,%同一性可根据催化区域而定义。
可通过编码tRNA合成酶骨架的核苷酸序列的定点诱变来‘转移’或‘移植’突变至替代的tRNA合成酶骨架上。此技术是本领域众所周知的。基本上,骨架pylS序列被选定(例如使用上述活性位点比对),并且将选定的突变转移至(即,在其中制得)对应/同源位置。
当使用数字地址提及特定氨基酸残基时,除非以其他方式显而易见,否则使用MbPylRS(巴氏甲烷八叠球菌吡咯酰-tRNA合成酶)氨基酸序列作为参考序列(即,如由可公开得到的野生型巴氏甲烷八叠球菌PylS基因登录号Q46E77编码的)来进行编号:
如本领域熟知的,这用于定位目标残基。这并非总是严格的计数运用——必须注意背景或比对。例如,如果目标蛋白质的长度轻微不同,则在该序列中对应于(例如)L266的正确残基的位置可能需要比对序列并且挑选等价或对应残基,而不是仅仅采用目标序列的第266位残基。这很好地在有经验读者的能力范围内。
本文使用的突变符号是本领域中的标准。例如,L266M意指用M替换在野生型序列位置266处对应于L的氨基酸。
参考本文描述的示例性DAPRS合成酶,与M.b.参考序列(参见上文)相比,突变将被标记为Y271C、N311Q、Y349F和V366C,或者如果移植到在起始序列中那些位置具有不同氨基酸的骨架上,则可以理解为X271C、X311Q、X349F和X366C。
现在参考示例性巴氏甲烷八叠球菌i和马氏甲烷八叠球菌序列说明替代的tRNA骨架之间的突变移植,但相同的原理同样应用于至其他骨架上或来自其他骨架的移植。
例如,Mb AcKRS是用于AcK引入的工程化合成酶
亲本蛋白质/骨架:巴氏甲烷八叠球菌PylS
突变:L266V、L270I、Y271F、L274A、C317F
Mb PCKRS:用于PCK引入的工程化合成酶
亲本蛋白质/骨架:巴氏甲烷八叠球菌PylS
突变:M241F、A267S、Y271C、L274M
可通过将这些突变移植到马氏甲烷八叠球菌PylS中来得到具有相同底物特异性的合成酶。因此,可以通过将来自Mb骨架的突变移植到Mm tRNA骨架上来产生以下合成酶:
Mm AcKRS将突变L301V、L305I、Y306F、L309A、C348F引入到马氏甲烷八叠球菌PylS中,以及
Mm PCKRS将突变M276F、A302S、Y306C、L309M引入到马氏甲烷八叠球菌PylS中。
在下方给出这些示例性移植突变的合成酶的全长序列。
>Mb_PylS/1-419
>Mb_AcKRS/1-419
>Mb_PCKRS/1-419
>Mm_PylS/1-454
>Mm_AcKRS/1-454
>Mm_PCKRS/1-454
相同的原理同样应用于其他突变和/或其他骨架。
有利地,应测试以这种方式产生的移植多肽来确保已保留所需的功能/底物特异性。
在一个实施方案中,tRNA可以来自一个物种,例如巴氏甲烷八叠球菌(Methanosarcina barkeri),并且tRNA合成酶可以来自另一个物种,例如马氏甲烷八叠球菌(Methanosarcina mazei)。在另一个实施方案中,tRNA可以来自第一物种,例如马氏甲烷八叠球菌,并且tRNA合成酶可以来自第二物种,例如巴氏甲烷八叠球菌。当正交对包含来自不同物种的tRNA和tRNA合成酶时,总是有正交对能一起有效工作的限制条件,即,tRNA合成酶将有效氨基酰化目标氨基酸的tRNA。
最合适地,正交对包含来自相同物种的tRNA和tRNA合成酶。
tRNA合成酶的性质和有助于其活性的特定突变将在下面分别讨论。
可以生产嵌合的tRNA合成酶,前提是tRNA合成酶分子的装载/乙酰化部分基于或衍生自Pyl tRNA合成酶。换言之,tRNA分子的反密码子部分可以根据操作人员的选择而变化,例如变化为引导tRNA识别替代密码子,例如有义密码子、四联体密码子、琥珀密码子或另一“终止”密码子。然而,应保存tRNA分子的有功能的酰化/装载部分,以便保留如上所述的非天然氨基酸可用的装载活性。
巴氏甲烷八叠球菌和马氏甲烷八叠球菌tRNA中的任一种都是合适的。在任何情况下,这些tRNA相差仅一个核苷酸。这种一个核苷酸的差异对它们的活性没有影响。因此,任何一个tRNA在本发明中可同样适用。
使用的tRNA可以变化,如经突变。在所有情况下,任何此类Pyl tRNA的变体或突变体应总是保持与用于用如上所述的一种或多种非天然氨基酸装载tRNA的tRNA合成酶有成效地相互作用的能力。
巴氏甲烷八叠球菌和马氏甲烷八叠球菌物种吡咯赖氨酸tRNA合成酶是合适的,只要它们包含如本文所述的突变,其有助于用所述的非天然氨基酸装载tRNA。
显而易见的是,将本文所述的非天然氨基酸引入到多肽中的直接产物将是包含通过肽键加入到多肽骨架中的所述非天然氨基酸的残基的多肽。这意味着从严格意义上讲,该多肽将不包含准确的所提及的非天然氨基酸,而是将包含已经历缩合反应的其残基,因为所提及的非天然氨基酸的氨基酸基团与在多肽链中与其相邻的氨基酸残基反应,导致基于肽键的引入所提及的非天然氨基酸的残基加释放的一分子H
在一方面,本发明涉及包含如上所述的非天然氨基酸的多肽。
在一方面,本发明涉及包含如上所述的非天然氨基酸的多肽,其中所述非天然氨基酸通过肽键连接至所述多肽。
在一方面,本发明涉及包含如上所述的非天然氨基酸的多肽,其中所述非天然氨基酸通过肽键加入到所述多肽。
在一方面,本发明涉及包含通过肽键或经由肽键引入的如上所述的非天然氨基酸的多肽。
在一方面,本发明涉及包含通过肽键或经由肽键引入的如上所述的非天然氨基酸的残基的多肽。
在本说明书中,“非天然氨基酸”是指不是天然编码的氨基酸,也不是在任何生物体的遗传密码中发现的氨基酸。因此,非天然氨基酸是含有胺和羧酸官能团和侧链的化合物,但其不是翻译机制用来组装蛋白质的任何蛋白原氨基酸。
类似地,本文公开的示例性非天然氨基酸可以被称为“包含DAP的非天然氨基酸”或“包含DAP的氨基酸”或类似物。显然,从IUPAC命名惯例的最严格意义上讲,DAP具有两个NH
可将编码用于上述方法的目标多肽的多核苷酸引入到重组可复制载体中。载体可以用于在相容宿主细胞中复制核酸。因此,在一个另外实施方案中,本发明提供一种制备本发明的多核苷酸的方法,通过将本发明的多核苷酸引入到可复制载体中;将载体引入到相容宿主细胞中;并且使宿主细胞在引起载体复制的条件下生长来实现该方法。可以从宿主细胞回收载体。合适的宿主细胞包括细菌,如大肠杆菌(E.coli)。
优选地,将载体中的本发明的多核苷酸有效地连接至控制序列,该控制序列能够通过宿主细胞供应编码序列的表达,即,载体是表达载体。术语“有效地连接”意指所述组分处在容许它们以其预期方式起作用的关系中。将“有效地连接"至编码序列的调节序列以下述方式连接:使得在与控制序列相容的条件下实现编码序列的表达。
可以将本发明的载体转化或转染入如描述用来提供本发明的蛋白质表达的合适的宿主细胞。此方法可以包括在提供编码蛋白的编码序列的载体的表达的条件下培养用如上述表达载体转化的宿主细胞,并且任选地回收表达的蛋白质。
载体可以是例如具备复制起点,任选地用于所述多核苷酸表达的启动子以及任选地启动子的调节子的质粒或病毒载体。载体以可含有一个或多个可选择标记基因,例如在细菌质粒的情况下含有氨苄青霉素抗性基因。载体可以用于(例如)转染或转化宿主细胞。
被有效地连接至编码本发明的蛋白质的序列的控制序列包括启动子/增强子和其他表达调节信号。这些控制序列可被选择以与宿主细胞(表达载体被设计于其中使用)相容。术语启动子是本领域众所周知的,并且包括在尺寸和复杂性上处于一定范围的从最小启动子至包括上游元件和增强子的启动子的核酸区域。
本发明的另一个方面是一种将一个或多个包含DAP的非天然氨基酸遗传地和位点特异性地引入到所选蛋白质中的方法,例如体外方法,合适地,在宿主细胞中。通过所述方法遗传引入的一个优点在于它排除了包含DAP的蛋白质一旦形成后被递送到细胞中的需要,因为在此实施方案中,它们可以直接在靶细胞中合成。该方法包括以下步骤:
i)在编码该蛋白质的核苷酸序列中的期望位点处引入正交密码子,或用正交密码子例如琥珀密码子替换特定密码子,
ii)在细胞中引入正交tRNA合成酶/tRNA对的表达系统,例如DAPRS tRNA合成酶/tRNA对,
使细胞在具有根据本发明的包含DAP氨基酸的培养基中生长。
步骤(i)在蛋白质的遗传序列中的期望位点处需要或用正交密码子例如琥珀密码子替换特定密码子。这可以通过仅仅引入具有编码该蛋白质的核苷酸序列的构建体(例如质粒)来实现,其中将期望引入/替换的含DAP的非天然氨基酸的位点改变,以包含正交密码子例如琥珀密码子。这较好在本领域技术人员的能力范围内,并且在此处下方给出此类实例。
步骤(ii)需要正交表达系统以在期望位置(例如琥珀密码子)处特异性引入包含DAP的非天然氨基酸。因此,需要特定正交tRNA合成酶,例如正交DAPRS-tRNA合成酶,以及特定的对应的正交tRNA对,二者一起能够用包含DAP的非天然氨基酸装载所述tRNA。本文提供了这些的实例。
蛋白质表达和纯化
包含本发明的多核苷酸的宿主细胞可以用于表达本发明的蛋白质。
合适的宿主细胞包括细菌,例如大肠杆菌,或某些真核细胞。
对于真核细胞,针对光笼型半胱氨酸,许多使用PylS/PylT系统的UAA已显示在真核细胞中起作用(最接近的实例是Nguyen,D.P.et al.(2014)[J.Am.Chem.Soc.,2014,136(6),pp 2240–2243];该文献通过引用并入本文中用于教导在真核细胞中的操作。
真核细胞可以是任何合适的真核细胞,例如昆虫细胞(例如Sf9昆虫细胞)、哺乳动物细胞(例如小鼠细胞或人细胞)。
合适地,真核细胞是哺乳动物细胞。合适地,哺乳动物细胞是HEK293细胞,例如HEK293T细胞。
在一个实施方案中,合适地,真核细胞、哺乳动物细胞、HEK293细胞或HEK293T细胞是体外的。在该实施方案中,合适地,该细胞不是体内细胞。
合适地,宿主细胞是细菌细胞。合适地,宿主细胞是大肠杆菌。合适地,宿主细胞是大肠杆菌细胞。合适地,大肠杆菌细胞是BL21 DE3大肠杆菌细胞。
在一方面,本发明涉及生产包含如上所述的2,3-二氨基丙酸(DAP)的多肽的方法,其中所述方法在活细胞内部进行。
合适地,所述方法包括将如上所述的非天然氨基酸遗传引入到多肽中,以及任选地使所述非天然氨基酸脱保护为2,3-二氨基丙酸(DAP)。
合适地,所述活细胞包含大肠杆菌细胞,例如BL21 DE3大肠杆菌细胞。
合适地,所述活细胞包含哺乳动物细胞,例如HEK293T细胞。
可以在允许本发明的蛋白质表达的合适条件下培养宿主细胞。本发明的蛋白质的表达可以是组成型的,以使得蛋白质持续产生;或者是诱导型的,其需要刺激物以引发表达。在诱导型表达的情况下,需要时,可通过例如将诱导剂物质(例如地塞米松或IPTG)添加至培养基,来引发蛋白质产生。
HEK293T(具有大的T抗原的人胚肾)细胞可广泛获得,例如来自LGC Standards,Queens Road,Teddington,Middlesex,TW11 0LY,UK的
BL21 DE3大肠杆菌细胞可广泛获得,例如来自New England Biolabs NewEngland Biolabs,240County Road,Ipswich,MA 01938-2732,USA的C2527I或C2527H。这些可根据供应商的说明进行培养,如本领域中众所周知的那样。
可通过本领域已知的多种技术从宿主细胞中提取本发明的蛋白质,包括酶促、化学和/或渗透性裂解以及物理破坏。
可通过本领域已知的标准技术,例如制备色谱、亲和纯化或任何其他合适的技术来纯化本发明的蛋白质。
合适地,在对应于野生型多肽中的半胱氨酸、丝氨酸或苏氨酸残基的位置,任选地在对应于野生型多肽中的半胱氨酸或丝氨酸残基的位置,最合适地在对应于野生型多肽中的半胱氨酸残基的位置,引入如上所述的DAP和/或非天然氨基酸。
更合适地,在对应于野生型多肽中的催化半胱氨酸、催化丝氨酸或催化苏氨酸残基的位置,任选地在对应于野生型多肽中的催化半胱氨酸或催化苏氨酸残基的位置,最合适地在对应于野生型多肽中的催化半胱氨酸残基的位置,引入如上所述的DAP和/或非天然氨基酸。
本发明发现了通过引入DAP来修饰酶的一个或多个活性位点的特殊应用。合适地,在对应于野生型酶的活性位点内的氨基酸的位置引入本文所述的DAP或非天然氨基酸。合适地,在对应于野生型酶的活性位点内的催化氨基酸的位置引入本文所述的DAP或非天然氨基酸。
合适地,多肽是酶。合适地,生成酯或硫酯中间体的酶。合适地,本发明的多肽是遵循该一般机理的酶;更合适地,该多肽可以是丝氨酸水解酶(其由人类基因组中1%的基因编码);合适地,该酶可以是蛋白酶、肽酶、酰胺酶、去泛素酶、脂肪酶、胆碱酯酶、硫酯酶、磷脂酶、聚糖水解酶
更合适地,本文所述的DAP或非天然氨基酸被引入到酶(例如蛋白酶)中。
参考酶系统的国际命名法和分类(由国际生物化学和分子生物学联盟命名委员会(NC-IUBMB)在与IUPAC-IUBMB生化命名委员会(JCBN)协商后准备和更新的)(例如,参见
本发明尤其提供了一种生产包含2,3-二氨基丙酸(DAP)的多肽的新方法。这具有广泛的工业应用,例如利用其反应特性共价捕获目标分子,例如正在研究的酶的底物。这也使得能够通过将DAP引入到酶活性位点心脏能够实现的类似方法来剖析代谢途径。应用还包括小分子药物的研究/捕获,以鉴定它们如何被酶修饰/代谢,和/或鉴定其蛋白质靶标。
蛋白质的结构表征通常代表许多药物开发项目的起点。由于DAP可用于任何通过与酶活性位点中的丝氨酸或半胱氨酸侧链结合的共价中间体进行的蛋白质催化反应,因此它具有广阔的应用潜力。DAP可用于在结构上表征这些共价中间体。除此之外,DAP还允许鉴定这些酶的新底物,这可能再次代表了新药开发策略的起点。
我们的技术允许将DAP特异性地引入到重组蛋白中。DAP可用于任何通过与酶活性位点中的丝氨酸或半胱氨酸侧链结合的共价中间体进行的蛋白质催化反应中。DAP可用于在结构上表征这些共价中间体,还可以允许鉴定这些酶的新底物。
我们已经开发了一种引入氨基酸(DAP5)的氨酰基-tRNA合成酶/tRNA
在一个实施方案中,本发明提供一种氨基酸(DAP5),其通过使用吡咯赖氨酸tRNA合成酶的突变体而引入蛋白质中,该突变体已经进化为用DAP5(DAPRS)加载其同族tRNA。这允许合成具有位点特异性引入的DAP5的重组蛋白,DAP5可在温和条件下脱保护为DAP。
合适地,光照以脱保护持续少于一分钟,合适地持续大约一分钟,合适地持续一分钟,合适地持续至少一分钟。合适地,光照以脱保护持续1毫秒至120秒,更合适地持续1毫秒至60秒。最合适地,光照以脱保护持续一分钟。
合适地,随后在光照后在高于冰点的任何温度下孵育蛋白质。孵育是脱保护的第二阶段。完成所需的时间取决于引入了DAP的蛋白质。在实施例中,样品在光照后孵育1-2小时。
在一方面,本发明涉及一种捕获酶的底物的方法。
合适的底物是那些通过酶对所述底物的作用而产生硫酯或酯中间体键的底物(注意,底物不含有酯或硫酯-合适地,底物包含化学结构,使得这些部分在催化过程中产生)。合适地,这些键是在酶对其底物的亲核攻击之后或作为其结果而产生的。在用根据本发明的DAP取代活性位点残基的情况下,产生的键是酰胺键(在下文中更详细地讨论)。
合适地,所述底物作为稳定的酰胺类似物被捕获。
合适地,所述底物可以是天然存在的底物的类似物。
合适地,所述酶是半胱氨酸蛋白酶或硫酯酶。
合适地,所述酶通过一种或多种酰基酶中间体、更合适地通过一种或多种半胱氨酸或丝氨酸结合的酰基酶中间体起作用。
在一个实施方案中,本发明可以用于观察通过添加小分子(药物)形成的中间体酶-底物复合物。这使得能够阐明药物如何在不同的复合物形成过程中破坏这种酶-底物的相互作用。因此,如果小分子是酶的底物,则这是本发明的有用的应用。只要DAP替换任何亲核(催化)残基(合适地,半胱氨酸和/或丝氨酸),它将捕获任何底物。
本发明发现了在经由编码的2,3-二氨基丙酸对生物合成酰基酶中间体的研究见解中的应用。
另外的特定和优选方面陈述于随附的独立和从属权利要求中。从属权利要求的特征可适当地与独立权利要求的特征结合,并且可能以不同于在权利要求中明确列出的其他组合方式来结合。
其中设备特征被描述为有效的,以提供功能的,将理解这包括提供该功能或经调试或配置来提供功能的设备特征。
现在将参考附图进一步描述本发明的实施方案,其中:
图1显示了在活性位点酶与半胱氨酸和丝氨酸亲核体通过酰基酶中间体进行的一般机理。
a,b,活性位点丝氨酸或半胱氨酸亲核体与羰基反应形成四面体中间体(未示出),该四面体中间体通过失去R
图2显示了缬氨霉素合成酶和提出的缬氨霉素生物合成。
缬氨霉素合成酶亚基Vlm1和Vlm2以顺序方式将D-α-羟基异戊酸(D-α-hiv)、D-缬氨酸(D-val)、L-乳酸(L-lac)和L-缬氨酸(L-val)缩合为形成四缩肽基(D-hiv-D-val-L-lac-L-val)中间体。D-α-hiv和L-lac是由专门的模块1和3(其包括酮还原酶(KR)结构域)选择和酮还原其前体酮酸而产生的。将四缩肽基中间体低聚为八缩肽基中间体,然后为十二缩肽基中间体,其通过末端硫酯酶(TE)结构域环化,从而产生缬氨霉素。A:腺苷酸化结构域,PCP:肽基载体蛋白结构域;C:缩合结构域。有关规范NRPS的合成循环,参见补充图1。
图3显示了在重组蛋白中遗传指导DAP引入。
a,DAP的结构和本文研究的保护形式。1:2,3-二氨基丙酸(DAP)。2:(S)-3-(((烯丙氧基)羰基)氨基)-2-氨基丙酸。3:(S)-2-氨基-3-((2-硝基苄基)氨基)丙酸4:(2S)-2-氨基-3-((1-(6-硝基苯并[d][1,3]二氧杂环戊烯-5-基)乙基)氨基)丙酸5:(2S)-2-氨基-3-(((1-(6-硝基苯并[d][1,3]二氧杂环戊烯-5-基)乙氧基)羰基)氨基)丙酸6:(2S)-2-氨基-3-(((2-((1-(6-硝基苯并[d][1,3]二氧杂环戊烯-5-基)乙基)硫代)乙氧基)羰基)氨基)丙酸。b-f,通过LC-MS测定法对提取物进行的测定化合物2-6的细胞内浓度。深蓝色迹线代表每种化合物的100μM标准溶液。浅蓝色迹线代表每种化合物的10μM标准溶液。红色迹线是由在不存在化合物的情况下生长的细胞产生的。褐色的迹线是由在不存在化合物的情况下生长的但是用化合物加标至10μM的细胞产生的。绿色迹线是由在存在1mM化合物的情况下生长的细胞产生的。g,DAPRS/tRNA
图4显示了用TEV(C151DAP)稳定捕获酰基酶中间体。
a,将所示的TEV蛋白酶变体与Ub-tev-His一起孵育。TEV(wt)的使用导致TEV裂解序列的裂解。TEV(C151A)的使用导致最小的裂解。DEV在TEV的活性位点中的存在导致考马斯凝胶中出现一条新带,代表了异肽连接的TEV(Ci5iDAP)-Ub复合物(左)。反应的αUb和αStrep的Western印迹证实了该复合物的身份(TEV构建体含有Strep标签)。b,异肽连接的TEV(C151DAP)–Ub复合物的串联质谱。串联质谱法明确地鉴定了所需位点的DAP修饰和残基上预期的tev-Gly-Gly修饰,这与DAP上的Ub捕获一致。
图5显示了Vlm TE从四缩肽基-SNAC制备的小分子产物描绘了低聚途径。
从四缩肽基-SNAC 7(1.7mM)和Vlm TE(6.5μM)的反应的HR-LC-ESI-MS提取的离子色谱图(EIC)。a,TE
图6显示了TE
a,TE
图7显示了PCP结构域与TE结构域的相互作用和推定途径的建模。
a,十二缩肽基-TE
图8显示了补充图1。
图9显示了补充图2。
图10显示了补充图3。
图11显示了补充图4。
图12显示了补充图5。
图13显示了补充图6。
图14显示了补充图7。
图15显示了补充图8。
图16显示了补充图9。
图17显示了补充图10。
图18显示了补充图11。
图19显示了补充图12。
图20显示了补充图13。
图21显示了补充图14。
图22显示了补充图15。
图23显示了照片。采用UBE2L3(C86DAP)进行泛素化测试。A:采用UBE2L3(wt)、UBE2L3(C86A)或UBE2L3(C86DAP)在存在或不存在Ub和β-巯基乙醇的情况下进行的泛素化反应的考马斯染色。采用UBE2L3(wt)、UBE2L3(C86A)或UBE2L3(C86DAP)在存在或不存在Ub和β-巯基乙醇的情况下进行的泛素化反应的αHA(B)和αUBE2L3(111-125)(C)western印迹。Ub~UBE2L3:硫酯连接的(UBE2L3[wt])或异肽连接的(UBE2L3[C86DAP])E2-Ub复合物。在UBE2L3(C86DAP)和Ub之间形成的复合物对β-巯基乙醇的存在不敏感,这与UBE2L3(wt)和Ub之间的复合物不同,其在β-巯基乙醇的存在下被还原。
图24显示了照片。
图25显示了照片。
实施例:在重组蛋白中遗传编码DAP衍生物
实施例1
发明人认为,DAP与组成性存在于细胞中的半胱氨酸和丝氨酸的结构相似性表明,发现选择性地引入DAP的氨酰基-tRNA合成酶可能具有挑战性。因此,我们设计并合成(图3a,补充图2)了DAP的四种受保护形式(化合物2-5)。我们预期,发现这些氨基酸的氨酰基-tRNA合成酶—tRNA
相对于我们先前分别使用PCC1RS/tRNA
为了发现引入氨基酸2-5的正交氨酰基-tRNA合成酶,我们查询了MbPylRS/tRNA
对化合物2-5的预测logP值进行检查后发现,它们具有很强的亲水性;促使我们检查它们是否进入细胞。使用基于LC-MS的氨基酸摄取测定法
实施例2:优选的包含DAP的非天然氨基酸
为了解决编码受保护的DAP形式的挑战,发明人设计并合成了氨基酸6(补充图2),我们预期可以将其翻译后脱保护以显示侧链胺。该氨基酸比氨基酸1-5具有更有利的预测logP值,并且我们发现向细胞培养基中添加1mM 6导致细胞内浓度约为2mM(图3f)。因此,与氨基酸2-5相反,氨基酸6可以在大肠杆菌中以毫摩尔浓度积累。
实施例3:创建DAP tRNA合成酶(“DAPRS”)
鉴于许多早期方法的失败,发明人设计并创建了一个全新的库DAPRSlib,其中基于PylRS活性位点中的6的模型仔细选择了待随机化的位置(补充图3和补充表1)。
作为这种智力练习的结果,决定将五个位置(Y271;N311;Y349;V366;W382)随机分配给所有二十个规范氨基酸,从而导致具有3.4x 10
我们获得了一个克隆,该克隆在6存在时赋予高水平的氯霉素抗性,而在6不存在时赋予最小的氯霉素抗性(图3g)。
相对于MbPylRS,所选的合成酶含有四个活性位点突变(Y271C、N311Q、Y349F和V366C)。
(应注意的是,该库含有5个随机化位置,但DAPRS中仅存在4个突变——在DAPRS中第5个位置是野生型(即W382)。
实施例4:包含DAP的非天然氨基酸引入到多肽中。
为了进一步表征6的遗传定向、位点特异性引入到蛋白质中,在DAPRS/tRNA
引入6的GFP(GFP(6))的产量与BocK引入的产量相当,证明了DAPRS/tRNA
电喷雾电离质谱法(ESI-MS)证实了DAPRS/tRNA
实施例5:使包含DAP的氨基酸脱保护
在这里,我们证明了包含DAP的氨基酸的脱保护,将DAP基团留在多肽骨架中,从而得到了包含DAP的多肽。
我们预期用365nm的光照射引入氨基酸6的蛋白质将显示含有巯基的中间体,该中间体可能经历进一步的反应,导致DAP(通过巯基的5exo-trig环化作用到氨基甲酸酯的羰基上和所得四面体中间体的塌陷来释放氨基,或通过形成环硫化物和二氧化碳来释放氨基)。实际上,光照GFP(6)(365nm,35mWcm
实施例6:稳定地捕获半胱氨酸蛋白酶酰基酶中间体
半胱氨酸蛋白酶(如烟草蚀纹病毒(TEV)蛋白酶)通常含有Cys-His-Asp催化三分子,并在用其同族底物处理后生成硫酯中间体
为了证明用DAP替换催化半胱氨酸使得能够捕获共价蛋白酶-底物中间体,我们将TEV
实施例7:Vlm TE的活性和合成途径
为了深入了解TE结构域的功能以及制备其以与DAP引入系统一起使用,我们克隆并表达了Vlm TE,并纯化了所得的Vlm TE(TE
在缬氨霉素合成中检测到的合成中间体揭示了Vlm TE
实施例8:可视化TE结构域介导的缬氨霉素合成中的关键中间体
我们接着获得并优化了用于使TE
Vlm TE与缩肽基-SNAC分子一起孵育不产生稳定的缀合物(补充图12a-c和补充表4),并且试图用缩肽基-SNAC分子浸泡TE
为了使Vlm TE的酰基酶复合物可视化,我们生产了其中活性位点丝氨酸2463被DAP替换(TE
为了提供对Vlm TE催化循环中第一个酰基-TE中间体的了解,我们捕获了一个四缩肽基-N-TE
我们假设TE
为了确定脱氧-四缩肽基-N-TE
接下来,我们专注于通过捕获十二缩肽基-N-TE
十二缩肽基-TE
将Vlm TE盖子在载脂蛋白/四缩肽结合的结构中的位置与盖子在十二缩肽结合的结构中的位置进行比较,证明并强调了其极大的迁移性。为了从一种盖子构象过渡到另一种盖子构象,螺旋Lα5-6保持其位置,Lα3-4旋转~45°并移位
这种独特的盖构象直接影响缩肽的可能位置。在盖子的载脂蛋白/四缩肽结合的构象中,螺旋Lα1的C末端位于Ser/DAP2463的
晶体结构的模型和结构因子存放在蛋白质数据库中,登录号为6ECB、6ECC、6ECD、6ECE和6ECF。
方法
一般合成程序。
所有试剂均购自Sigma-Aldrich,但以下除外:L-乳酸购自Fisher Scientific,EDC购自Oakwood Chemicals(Estill,SC),具有最高的可用纯度,并且无需进一步纯化即可使用。缬氨霉素购自Sigma-Aldrich和BioShop Canada。所有溶剂均购自FisherScientific。除非另有说明,所有反应均在氩气气氛下使用干燥溶剂进行。使用BrukerAVANCE II(
缩写:M=摩尔浓度;conc.=浓;mol=摩尔;mmol=毫摩尔;℃=摄氏度;eq.=当量;h=小时;min=分钟;r.t.=室温;cat.=催化;aq.=水溶液;Su=琥珀酰亚胺基;DIPEA=二异丙基乙胺;atm=大气,Boc=叔丁氧羰基;
氨基酸的合成
制备氨基酸的一般合成方案如下所示,所用试剂和条件如下:
试剂和条件:(i)2a(10.0mmol),HCl(4M的1,4-二氧六环溶液)(8.0eq.),Et
(S)-3-{[(烯丙氧基)羰基]氨基}-2-氨基丙酸(2)
将Boc-Dap(Alloc)-OH 2a(4.325g,15.0mmol,1.0eq.,购自Bachem Ltd.)装载到干燥的250mL单颈圆底烧瓶中并溶解于HCl(4M的1,4-二氧六环溶液,60.0mL,240.0mmol,16.0eq.)中。在室温下向该溶液中添加干燥的Et
(S)-2-[(叔丁氧羰基)氨基]-3-[(2-硝基苄基)氨基]丙酸叔丁酯(3a)
将Boc-L-Dap-O
(S)-2-氨基-3-[(2-硝基苄基)氨基]丙酸3
将(S)-2-[(叔丁氧羰基)氨基]-3-[(2-硝基苄基)氨基]丙酸叔丁酯3a(4.59g,11.607mmol,1.0eq.)装入100mL干燥的单颈圆底烧瓶中。加入HCl(25mL,4M的1,4-二氧六环溶液,100.0mmol,8.615eq.),接着加入干燥的Et
4',5'-亚甲二氧基-2'-硝基苯乙酮(4b)
按照McGall et al.
(R,S)-1-[4',5'-(亚甲二氧基)-2'-硝基苯基]乙醇(4c)
在2升单颈圆底烧瓶中将4',5'-亚甲二氧基-2'-硝基苯乙酮4b(43.714g,0.209mol,1.0eq.)悬浮于CH
(R,S)-1-溴-1-[4',5'-(亚甲二氧基)-2'-硝基苯基]乙烷(4d)
使用热风枪在>100℃下将1升3颈圆底烧瓶在真空中干燥15min,并用干燥的氩气吹扫,然后冷却至室温。向其中加入(R,S)-1-[4',5'-(亚甲二氧基)-2'-硝基苯基]乙醇4c(15.838g,75.0mmol,1.0eq.)。将4c溶解在干燥的CH
(2S)-2-[(叔丁氧羰基)氨基]-3-{[1-(6-硝基苯并[d][1,3]二氧环戊烯-5-基)乙基]氨基}丙酸叔丁酯(4e)
在干燥的1升三颈圆底烧瓶中将Boc-L-Dap-O
(2S)-2-氨基-3-{[1-(6-硝基苯并[d][1,3]二氧环戊烯-5-基)乙基]氨基}丙酸(4)
在用铝箔包裹以避光的干燥250mL圆底烧瓶中将(2S)-2-[(叔丁氧羰基)氨基]-3-{[1-(6-硝基苯并[d][1,3]二氧环戊烯-5-基)乙基]氨基}丙酸叔丁酯4e(4.303g,9.489mmol,1.0eq.)溶解于干燥的CH
2,5-二氧代吡咯烷-1-基(1-(6-硝基苯并[d][1,3]二氧环戊烯-5-基)乙基)碳酸酯(5a)
将(R,S)-1-[4',5'-(亚甲二氧基)-2'-硝基苯基]乙醇4c(42.234g,200.0mmol,1.0eq.)装入到干燥的2升3颈圆底烧瓶中,并溶解于干燥的CH
(2S)-2-[(叔丁氧羰基)氨基]-3-({[1-(6-硝基苯并[d][1,3]二氧环戊烯-5-基)乙氧基]羰基}-氨基)丙酸(5b)
在用铝箔包裹以避光的干燥1升单颈圆底烧瓶中将Boc-L-Dap-OH 1a(2.553g,12.5mmol,1.25eq.)悬浮于干燥的THF(180mL)和干燥的CH
(2S)-2-氨基-3-({[1-(6-硝基苯并[d][1,3]二氧环戊烯-5-基)乙氧基]羰基}-氨基)丙酸(5)
在用铝箔包裹的干燥1L单颈圆底烧瓶中将新鲜蒸馏的CF
2-{[1-(6-硝基苯并[d][1,3]二氧环戊烯-5-基)乙基]硫代}乙-1-醇(6a)
将新鲜制备的NaOH溶液(0.5M,8g在40mL去离子H
2,5-二氧代吡咯烷-1-基-(2-{[1-(6-硝基苯并[d][1,3]二氧环戊烯-5-基)乙基]硫代}乙基)碳酸酯(6b)
从醇6a开始原位合成中间体6b,其用于随后的合成DAP衍生物6c和6d的反应。使用热风枪在真空中干燥3颈500mL圆底烧瓶,并用氩气吹扫;在使用之前,重复此程序三次。在干燥的烧瓶中装入溶解于干燥的CH
(2S)-2-[(叔丁氧羰基)氨基]-3-{[(2-{[1-(6-硝基苯并-[d][1,3]二氧环戊烯-5-基)乙基]硫代}乙氧基)羰基]氨基}丙酸叔丁酯(6c)
将Boc-L-Dap-O
(2S)-2-[(叔丁氧羰基)氨基]-3-{[(2-{[1-(6-硝基苯并[d][1,3]-二氧环戊烯-5-基)乙基]硫代}乙氧基)羰基]氨基}丙酸(6d)
在氩气下,将Boc-L-Dap-OH(13.479g,66.0mmol,1.082eq.)以一份添加到6b(26.70g,如上所述制备的)的干燥的CH
(2S)-2-氨基-3-{[(2-{[1-(6-硝基苯并[d][1,3]二氧环戊烯-5-基)乙基]硫代}乙氧基)羰基]氨基}丙酸(6)
方法I(从6c制备):
将(2S)-2-[(叔丁氧羰基)氨基]-3-{[(2-{[1-(6-硝基苯并[d][1,3]-二氧环戊烯-5-基)乙基]硫代}-乙氧基)羰基]氨基}丙酸酯6c(6.728g,12.066mmol,1.0eq.)的干燥样品装载到干燥的250mL单颈圆底烧瓶中,并溶解于干燥的CH
保存:将DAP氨基酸6·TFA的干燥样品在冷、干燥和黑暗的环境中保存在气密的深色玻璃小瓶中,稳定超过3年,且无分解。
处理:DAP氨基酸6·TFA对光敏感,并且在暴露于潮湿的空气中略微吸湿,因此其在小瓶中的样品始终在黑暗且干燥的环境中进行处理。值得注意的是,从冰箱或冰柜中取出的含有6·TFA的小瓶在打开和处理之前总是要加热到室温。
方法II(从6d制备):
将(2S)-2-[(叔丁氧羰基)氨基]-3-{[(2-{[1-(6-硝基苯并[d][1,3]二氧环戊烯-5-基)乙基]硫代}乙氧基)羰基]氨基}丙酸6d(26.70g,53.2395mmol,1.0eq.)的干燥样品装载到干燥的1升单颈圆底烧瓶中,并溶解于干燥的CH
VlmTE底物合成
缩肽基-SNAC化合物7和8的合成方案。
a,脱氧四缩肽基-SNAC 8的合成。a)8c,EDC,DMAP,72%;b)TFA,DCM,99%;c)TBSCl,Imid.,DCM;d)LiOH,THF,78%,2步;e)(COCl)
b,四缩肽基-SNAC 7的合成。a)烯丙基Br,Cs
c脱氧四缩肽基-SNAC 8的结构和
d四缩肽基-SNAC 7的结构。
(S)-S-(2-乙酰氨基乙基)2-((叔丁氧羰基)氨基)-3-甲基丁烷硫酸酯(7a)
将Boc-L-缬氨酸(7.29g,33.56mmol,1.0当量)溶解在CH
(S)-S-(2-乙酰氨基乙基)2-氨基-3-甲基丁烷硫酸酯(7b)
在圆底烧瓶中,将7a(0.5g,1.57mmol,1.0当量)溶解在CH
(S)-2-((叔丁基二甲基甲硅烷基)氧基)丙酸(7c)
在圆底烧瓶中,将L-乳酸乙酯(5.08g,43.0mmol,1.0当量)溶解在CH
(S)-2-((叔丁基二甲基甲硅烷基)氧基)丙酰氯(7d)
在圆底烧瓶中,将7c(3.7g,18mmol,1.0当量)溶解在DMF(45mL)中,并使用冰浴将溶液冷却至0℃。加入草酰氯(13.6mL的2.0M DCM溶液,10.0当量)和催化量的DMF。反应从0℃至环境温度进行2h。浓缩反应混合物,并将粗油状物不经纯化即用于随后的反应中。
TBSO-L-Lac-L-Val-SNAC(7e)
在圆底烧瓶中,将7b(1.95g,9mmol,1.0当量)溶解在CH
HO-L-Lac-L-Val-SNAC(7f)。
在50mL聚丙烯Falcon管中,将化合物7e(250mg,0.617mmol,1.0当量)溶解在乙腈(20mL)中。加入吡啶(249μL,3.09mmol,5当量)和HF(48wt.%aq.533μL,30.9mmol,50当量)。将反应在环境温度下搅拌16h。将反应混合物用NH
(S)-2-羟基丙酸烯丙酯(7g)
在圆底烧瓶中,将1g L-乳酸(11.11mmol,1当量)和3.8g碳酸铯(11.67mmol,1.05当量)溶解在13mL DMF中。在环境温度下滴加烯丙基溴(3.75mL,5.37g,44.44mmol,4当量)。添加完成后,将反应在环境温度下搅拌48h。完成后,通过旋转蒸发除去过量的烯丙基溴,并将剩余溶液用水稀释,然后用Et
(R)-(S)-1-(烯丙氧基)-1-氧代丙-2-基2-((叔丁氧羰基)氨基)-3-甲基-丁酸酯(7h)。
在圆底烧瓶中,将1g 7g(7.69mmol,1当量)和1.67g Boc-D-Val(8.46mmol,1.1当量)溶解在39mL CH
Boc-D-Val-L-Lac-L-Val-SNAC(7i)
在圆底烧瓶中,在氮气气氛下,将250mg 7h(0.76mmol,1当量)溶解在4mL CH
HO-D-Hiv-D-Val-L-Lac-L-Val-SNAC(7)
向圆底烧瓶中加入118mg 7i(0.24mmol,1当量)的最少量THF溶液,并将其冷却至0℃。向其中加入1mL 4M HCl的二氧六环溶液(Sigma),并使反应温热至环境温度。通过TLC监测反应,完成时通过旋转蒸发除去所有溶剂。未经纯化的中间体7k立即用于随后的反应中。将中间体7k溶解在2mL CH
(R)-3-甲基-2-(3-甲基丁酰氨基)丁酸甲酯(8a)
在圆底烧瓶中,将D-缬氨酸甲酯盐酸盐(250mg,1.5mmol,1.0当量)溶解在CH
(R)-3-甲基-2-(3-甲基丁酰氨基)丁酸(8b)
在圆底烧瓶中,将8a(180mg,1.2mmol,1.0当量)溶解在MeOH(24mL)和THF(24mL)中,并使用冰浴将溶液冷却至0℃。滴加LiOH(1M,24mL),并历经4小时使溶液从0℃升至环境温度。将溶液浓缩至三分之一体积,并将所得水溶液用10%HCl酸化至pH 3。溶液用CH
8(A)和8c(B)
(R)-(S)-1-(((S)-1-((2-乙酰氨基乙基)硫代)-3-甲基-1-氧代丁-2-基)氨基)-1-氧代丙-2-基3-甲基-2-(3-甲基丁酰氨基)丁酸酯(8)
在圆底烧瓶中,将醇7f(25.2mg,0.087mmol,1.0当量)和羧酸8b(35mg,0.174mmol,2.0当量)溶解在DMF(1mL)中。使用干燥的冰/丙酮浴将溶液冷却至–20℃,并加入EDC(67mg,0.35mmol,4.0当量)和DMAP(21mg,0.174mmol,2.0当量)。使混合物温热至环境温度,并使反应进行16h。用NH
实施例1-8的总结
我们描述了一种在重组蛋白中遗传编码DAP的策略。我们表明遗传编码DAP代替催化半胱氨酸或丝氨酸能够捕获不稳定的硫酯或酯中间体作为其稳定的酰胺类似物。我们举例说明了该方法对半胱氨酸蛋白酶和硫酯酶的实用性,并提供了通过Vlm TE合成缬氨霉素中的中间体的独特见解。我们的结果显示,与十二肽基结合的Vlm TE相关的巨大的盖子重排。重要的是,DAP系统允许使用广泛使用的、具有反应能力的底物(例如,含有蛋白酶位点的天然蛋白质)、底物类似物(在这种情况下为SNAC)和商业上可获得的天然产物(此处为缬氨霉素和可能的其他环状产物
这些结构中不存在PCP结构域,它是TE结构域催化循环中的关键角色,但其结合位点可从信息丰富的终端抑制剂捕获的EntF的PCP-TE结构
因此,人们可以将已知结构组装成低聚和环化的一种假想途径(图7)。在观察到的载脂蛋白/四缩肽结合的构象中,Vlm TE的Lα1可以抑制任何连接的缩肽在周围卷曲以进行环化(图7)。PCP结合可以诱导TE构象,其类似于我们对十二缩肽结合的TE观察到的构象,其可以容纳与PCP结构域结合的
在十二缩肽-TE
即使在TE结构域与真正的底物共价结合的情况下,在其他研究中看到的以及在此处明显看到的盖子的迁移性极好,而且盖子与TE结构域的其余部分之间缺乏特异性的相互作用使得不太可能在合成循环的这些步骤中的任何一个步骤中存在单个完整定义的构象。已提出形成环化底物的预定义/模板构象以促进短杆菌酪肽合成酶中的环化
尽管我们专注于利用编码的DAP提供有关缬氨霉素合成中的硫酯酶酰基酶中间体的见解,但DAP系统为研究以半胱氨酸或丝氨酸结合的酰基酶中间体为特征的多种酶具有广大的前景
实施例9-UBE2L3-DAP的表达、纯化和活性测试
还测试了通过GST-tag亲和纯化,然后通过TEV蛋白酶裂解GST-tag和Strep-tag亲和纯化来纯化UBE2L3(C86DAP5)。这种策略导致了干净的产物。
通过LC-ESI-MS确定纯化的UBE2L3(C86DAP5)-Strep的质量:
在UV光照射前UBE2L3(C86DAP5)-Strep的LC-ESI-MS:
UBE2L3(C86DAP5)-Strep[1]:预期:19050.57Da,观测:19048.79Da;UBE2L3(C86INT);Strep[2]:预期:18857.53Da,观测:18853.15Da。
在两个不同的步骤中发生DAP5的脱保护。首先,在UV光的作用下除去光笼基团,从而形成半脱保护的中间体。第二分子内反应最终导致完全脱保护的DAP。纯化UBE2L3(C86DAP5)后,发现大多数蛋白质都含有半脱保护的中间体(UBE2L3[C86INT]),尽管其中一些以完整的光笼形式存在。UV光照射后,再次通过LC-ESI-MS评估蛋白质质量:
UV光照射后UBE2L3(C86DAP5)-Strep的LC-ESI-MS:UBE2L3(C86INT)-Strep[2]:预期:18857.53Da,观测:18853.15Da。
如预期的那样,在UV光照射后不再能检测到UBE2L3(C86DAP5)-Strep。实际上,仅检测到UBE2L3(C86INT)-Strep。随后将蛋白质在37℃下孵育3h,并通过LC-ESI-MS评估其质量。如预期的那样,与UBE2L3(C86INT)-Strep一起检测到UBE2L3(C86DAP)-Strep。
UV光照射并在37℃孵育3h后UBE2L3(C86DAP5)-Strep的LC-ESI-MS:UBE2L3(C86INT)-Strep[2]:预期:18857.53Da,观测:18853.15Da;
UBE2L3(C86DAP)-Strep[3]:预期:18753.59Da,观测:18753.13Da。
不幸的是,在37℃下较长的孵育时间(6h和16h)并未导致UBE2L3(C86INT)-Strep脱保护的改善。实际上,含有DAP的蛋白质的比例似乎没有变化,占UV光照射和较长孵育时间后UBE2L3(C86DAP5)-Strep的总蛋白质LC-ESI-MS的约30%。在37℃下将UBE2L3(C86INT)-Strep孵育6h或16h。没有观察到脱保护的改善,含DAP的蛋白质部分(约30%)在很大程度上保持不变。
为了测试已用UV线照射并在37℃下孵育过夜的UBE2L3(C86DAP5)是否可以用Ub装载,建立了在E2上样缓冲液中含有0.2μM E1和带有HA标签的Ub的反应。每个反应(阳性对照[wt]、阴性对照[C86A]和[C86DAP])在有和没有Ub的情况下进行(参见图23)。
如预期的那样,可以观察到UBE2L3(wt)的较高分子量的带,对应于硫酯连接的E2-Ub复合物。另外,还可以检测到UBE2L3[C86DAP]的较高分子量的带,对应于异肽连接的E2-Ub复合物。UBE2L3不完全转化为与Ub的复合物(图23),与UBE2L3中DAP5的不完全脱保护一致。
UBE2L3(C86DAP)和Ub之间新形成的异肽键对氧化还原不敏感,在β-巯基乙醇存在下不能还原。这与对氧化还原敏感的由UBE2L3(wt)和Ub形成的复合物形成对比(图23)。
为了进一步表征不同带的身份,进行了抗HA和抗UBE2L3印迹(图23B和C),清楚地表明,在HAUb和UBE2L3(C86DAP)都存在的情况下形成了较高分子量的带含有UBE2L3和Ub。
最后,为了表征新形成的键的化学性质,在胰蛋白酶消化后,将与UBE2L3(C86DAP)–Ub复合物相对应的带切除并通过串联质谱法进行分析(由ProteomicsFacility,University of Bristol进行),异肽连接的UBE2L3(C86DAP)–Ub复合物的串联质谱法。
串联质谱法明确鉴定了所需位点的DAP修饰和预期的对残基的Gly-Gly修饰,这与DAP上的Ub负载一致。
分析明确证实了UBE2L3(C86DAP)和Ub之间形成了稳定的酰胺键。
实施例10-在活细胞中的应用
在本实施例中,我们证实了在活细胞中的技术。
在本实施例中,我们显示了在大肠杆菌细胞(BL21)和哺乳动物细胞(HEK293T)中的发明。
我们参考图24和25中提供的TEV-GFP WB数据。
特别地,我们参考图24,该图显示了在活的大肠杆菌细胞中Dap介导的底物捕获。GFP(在C末端带有TEV切割位点的GFP)和C末端带有Strep标签的TEV蛋白酶的不同变体(WT/Ala/TAG(有或没有Dappc;(在本实施例中,化合物‘DAP5’称为‘Dappc’))在20℃下在大肠杆菌BL21细胞中共表达。表达后20h,将细胞直接用UV光(35mW/cm
此外,我们参考图25,该图显示了在哺乳动物HEK293T细胞中Dap介导的底物捕获。GFP(在C末端带有TEV切割位点的GFP)和C末端带有Strep标签的TEV蛋白酶的不同变体(WT/Ala/TAG(有或没有Dappc(DAP5)))被共转染到HEK293T细胞中。转染后48h,将细胞直接用UV光(8mW/cm
实施例10的其他方法:
在20℃诱导与含有GFP的质粒和含有TEV的质粒共转化的BL21(DE3)细胞来表达蛋白质。将0.1mM Dappc(DAP5)加入到培养基中以引入Dappc(DAP5)。20h后,将细胞转移至50mL锥形离心falcon管中,并在温和搅拌下用UV光(365nm,35mW/cm
将HEK293T细胞与含有GFP的质粒、含有TEV的质粒共转染。转染后30min添加1mMDappc(DAP5)用于琥珀抑制。转染后48h,用UV光(365nm,10mW/cm
补充方法
本研究中使用的引物列表。突变的残基以大写字母表示。
使用质粒pBK-pylS作为模板
用DAP衍生物选择活性aaRS
使用以下文库,如先前报道的
GFP(150TAG)His6表达与纯化
在含有pBK_DAPRS或pBK_PylRS载体的MegaX DH10B T1R细胞中,从pSF-sfGFP150TAG表达在第150位引入了6的超折叠绿色荧光蛋白(sfGFP)。用转化细胞接种补充了12.5μg/mL四环素、25μg/mL卡那霉素和1mM 6或N
His6-硫辛酰基-TEV-Strep表达和纯化
将BL21(DE3)细胞用pNHD-His6-硫辛酰基-TEV
Ub
将BL21(DE3)细胞用pNHD-Ub-tev-His6转化,并在含有25μg/mL四环素的LB琼脂板上于37℃下过夜生长。用从转化得到的一些菌落接种含有25μg/mL四环素的LB培养基。将培养物以1:100稀释到含有12.5μg/mL四环素的新鲜LB培养基中;一旦OD 600达到0.5,就使用1mM IPTG诱导培养物,并在37℃下进行蛋白质表达6h。通过离心收获细胞并重悬50mMtris-HCl pH 7.5、150mM NaCl、2mMβ-巯基乙醇、1片Roche抑制剂鸡尾酒片剂/50mL、0.5mg/mL溶菌酶(Sigma)、50μg/mL DNA酶(Sigma),并通过超声裂解。通过以39’000×g离心30min来澄清裂解物,并通过0.4μm PES膜进行过滤。使用镍亲和色谱法(HisTrap HP柱,GEHealthcare)并采用咪唑线性梯度(30mM至500mM)来纯化Ub。将蛋白质在4℃下针对10mMtris-HCl透析过夜,并使用在50mM乙酸铵pH 4.5中的NaCl梯度(0-1M mM)通过离子交换色谱法(HiTrapS 5mL柱,GE Healthcare)来进一步纯化Ub。合并纯的部分,然后针对20mMtris-HCl pH 7.4透析过夜。然后使用Amicon Ultra-15(3kDa MWCO)离心过滤器装置(Millipore)将样品浓缩至~15mg/mL。
TEV与Ub
将15μg His6-硫辛酰基-TEV-Strep在30℃下与60μg Ub
DAP衍生物的细胞内浓度的分析
如先前所述
Vlm TE构建体的克隆、表达和纯化
通过ATUM(以前为DNA 2.0)在pJExpress411载体中合成含有vlm2
TE结构域在用pJExpress411-vlm2-TE
对于蛋白质纯化,将TE
如对Te
晶体学
在气相扩散结晶试验中使用商业上可获得的筛选(Qiagen)和10mg mL
在与TE
对于十二缩肽基-TE
通过向结晶滴中加入10μL(TE
TE
使用iMosflm
使用iMosflm
缩肽基-TE复合物形成
将终浓度为0.2mg mL
完整蛋白质的LC-ESI-MS分析
对于图2c、扩展数据图3b、7c中所示的实验,蛋白质样品先经过液相色谱(LC)系统(Agilent 1200系列),然后在6130四极杆质谱仪上进行在线电喷雾电离质谱(ESI-MS)。使用Jupiter 5μ C4 300A柱,150mm x 2.00mm(Phenomenex),通过LC系统使用含0.1%(v/v)甲酸的水(溶剂A)和含0.1%(v/v)甲酸的乙腈梯度溶液(在6min内从10%到75%,以及在1.5min内从75%到95%)(溶剂B)运行蛋白质。通过监测200和280nm处的UV吸光度来检测蛋白质。使用OpenLAB CDS软件(Agilent Technologies),通过在正离子模式下从MS采集中进行去卷积来计算蛋白质质量。
对于图4a,b、扩展数据图6d-f,7d,e所示的实验,将缓冲液T中的蛋白质浓度调整为0.1mg mL
串联MS/MS分析
蛋白质用MES缓冲液在4-12%NuPAGE Bis-Tris凝胶(Invitrogen)上运行,并使用InstantBlue(Expedeon)进行短暂染色。切下带并保存在20mM Tris pH 7.4中。胰蛋白酶消化和串联MS/MS分析由Kate Heesom(Proteomics Facility,University of Bristol)进行。
Vlm TE反应产物的LC-ESI-MS分析
将0.2mg mL
实施例1-9的参考文献
1.Holliday,G.L.,Mitchell,J.B.O.&Thornton,J.M.Understanding theFunctional Roles of Amino Acid Residues in Enzyme Catalysis.Journal ofmolecular biology 390,560-577(2009).
2.Di Cera,E.Serine proteases.IUBMB Life 61,510-515(2009).
3.Hedstrom,L.Serine protease mechanism and specificity.Chem Rev 102,4501-4523(2002).
4.Long,J.Z.&Cravatt,B.F.The Metabolic Serine Hydrolases and TheirFunctions in Mammalian Physiology and Disease.Chem Rev 111,6022-6063(2011).
5.Verma,S.,Dixit,R.&Pandey,K.C.Cysteine Proteases:Modes of Activationand Future Prospects as Pharmacological Targets.Front Pharmacol7(2016).
6.Otto,H.H.&Schirmeister,T.Cysteine proteases and theirinhibitors.Chem Rev 97,133-171(1997).
7.Swatek,K.N.&Komander,D.Ubiquitin modifications.Cell Res 26,399-422(2016).
8.Yang,W.&Drueckhammer,D.G.Understanding the relative acyl-transferreactivity of oxoesters and thioesters:computational analysis of transitionstate delocalization effects.Journal of the American Chemical Society 123,11004-11009(2001).
9.Liu,B.,Schofield,C.J.&Wilmouth,R.C.Structural analyses onintermediates in serine protease catalysis.Journal of Biological Chemistry281,24024-24035(2006).
10.Ngo,P.D.,Mansoorabadi,S.O.&Frey,P.A.Serine Protease Catalysis:AComputational Study of Tetrahedral Intermediates and Inhibitory Adducts.JPhys Chem B 120,7353-7359(2016).
11.Cleary,J.A.,Doherty,W.,Evans,P.&Malthouse,J.P.G.Quantifyingtetrahedral adduct formation and stabilization in the cysteine and the serineproteases.Bba-Proteins Proteom 1854,1382-1391(2015).
12.Scaglione,J.B.et al.Biochemical and structural characterization ofthe tautomycetin thioesterase:analysis of a stereoselective polyketidehydrolase.Angew Chem Int Ed Engl 49,5726-5730(2010).
13.Cappadocia,L.&Lima,C.D.Ubiquitin-like Protein Conjugation:Structures,Chemistry,and Mechanism.Chem Rev 118,889-918(2018).
14.Plechanovova,A.,Jaffray,E.G.,Tatham,M.H.,Naismith,J.H.&Hay,R.T.Structure of a RING E3 ligase and ubiquitin-loaded E2 primed forcatalysis.Nature 489,115-U135(2012).
15.Hay,R.W.&Morris,P.J.Interaction of Dl-2,3-Diaminopropionic Acidand Its Methyl Ester with Metal Ions.1.Formation Constants.J Chem Soc A,3562-&(1971).
16.Lan,Y.et al.Incorporation of 2,3-Diaminopropionic Acid into LinearCationic Amphipathic Peptides Produces pH-Sensitive Vectors.Chembiochem 11,1266-1272(2010).
17.Radzicka,A.&Wolfenden,R.Rates of uncatalyzed peptide bondhydrolysis in neutral solution and the transition state affinities ofproteases.Journal of the American Chemical Society 118,6105-6109(1996).
18.Hoyer,K.M.,Mahlert,C.&Marahiel,M.A.The iterative gramicidin sthioesterase catalyzes peptide ligation and cyclization.Chemistry&biology 14,13-22(2007).
19.Alonzo,D.A.,Magarvey,N.A.&Schmeing,T.M.Characterization ofcereulide synthetase,a toxin-producing macromolecular machine.PloS one 10,e0128569(2015).
20.Magarvey,N.A.,Ehling-Schulz,M.&Walsh,C.T.Characterization of thecereulide NRPS alpha-hydroxy acid specifying modules:activation of alpha-ketoacids and chiral reduction on the assembly line.Journal of the AmericanChemical Society 128,10698-10699(2006).
21.Shaw-Reid,C.A.et al.Assembly line enzymology by multimodularnonribosomal peptide synthetases:the thioesterase domain of E.coli EntFcatalyzes both elongation and cyclolactonization.Chemistry&biology 6,385-400(1999).
22.May,J.J.,Wendrich,T.M.&Marahiel,M.A.The dhb operon of Bacillussubtilis encodes the biosynthetic template for the catecholic siderophore 2,3-dihydroxybenzoate-glycine-threonine trimeric ester bacillibactin.J BiolChem 276,7209-7217(2001).
23.Zhou,Y.et al.Iterative Mechanism of Macrodiolide Formation in theAnticancer Compound Conglobatin.Chemistry&biology 22,745-754(2015).
24.Robbel,L.,Hoyer,K.M.&Marahiel,M.A.TioS T-TE--a prototypicalthioesterase responsible for cyclodimerization of the quinoline-andquinoxaline-type class of chromodepsipeptides.FEBS J 276,1641-1653(2009).
25.Jaitzig,J.,Li,J.,Sussmuth,R.D.&Neubauer,P.Reconstitutedbiosynthesis of the nonribosomal macrolactone antibiotic valinomycin inEscherichia coli.ACS Synth Biol 3,432-438(2014).
26.Akey,D.L.et al.Structural basis for macrolactonization by thepikromycin thioesterase.Nat Chem Biol 2,537-542(2006).
27.Samel,S.A.,Wagner,B.,Marahiel,M.A.&Essen,L.O.The thioesterasedomain of the fengycin biosynthesis cluster:a structural base for themacrocyclization of a non-ribosomal lipopeptide.Journal of molecular biology359,876-889(2006).
28.Tseng,C.C.et al.Characterization of the surfactin synthetase C-terminal thioesterase domain as a cyclic depsipeptide synthase.Biochemistry41,13350-13359(2002).
29.Bruner,S.D.et al.Structural basis for the cyclization of thelipopeptide antibiotic surfactin by the thioesterase domain SrfTE.Structure10,301-310(2002).
30.Li,J.et al.Palladium-triggered deprotection chemistry for proteinactivation in living cells.Nat Chem 6,352-361(2014).
31.Baker,A.S.&Deiters,A.Optical Control of Protein Function throughUnnatural Amino Acid Mutagenesis and Other Optogenetic Approaches.AcsChemical Biology 9,1398-1407(2014).
32.Nguyen,D.P.et al.Genetic Encoding of Photocaged Cysteine AllowsPhotoactivation of TEV Protease in Live Mammalian Cells.Journal of theAmerican Chemical Society 136,2240-2243(2014).
33.Nguyen,D.P.,Elliott,T.,Holt,M.,Muir,T.W.&Chin,J.W.GeneticallyEncoded 1,2-Aminothiols Facilitate Rapid and Site-Specific Protein Labelingvia a Bio-orthogonal Cyanobenzothiazole Condensation.Journal of the AmericanChemical Society 133,11418-11421(2011).
34.Neumann,H.,Peak-Chew,S.Y.&Chin,J.W.Genetically encoding N-epsilon-acetyllysine in recombinant proteins.Nature Chemical Biology 4,232-234(2008).
35.Chin,J.W.Expanding and Reprogramming the Genetic Code of Cells andAnimals.Annu Rev Biochem 83,379-408(2014).
36.Liu,C.C.&Schultz,P.G.Adding New Chemistries to the GeneticCode.Annual Review of Biochemistry,Vol 79 79,413-444(2010).
37.Zhang,M.S.et al.Biosynthesis and genetic encoding ofphosphothreonine through parallel selection and deep sequencing.Nat Methods14,729-736(2017).
38.Virdee,S.,Ye,Y.,Nguyen,D.P.,Komander,D.&Chin,J.W.Engineereddiubiquitin synthesis reveals Lys29-isopeptide specificity of an OTUdeubiquitinase.Nature Chemical Biology 6,750-757(2010).
39.Phan,J.et al.Structural basis for the substrate specificity oftobacco etch virus protease.Journal of Biological Chemistry 277,50564-50572(2002).
40.Trauger,J.W.,Kohli,R.M.,Mootz,H.D.,Marahiel,M.A.&Walsh,C.T.Peptidecyclization catalysed by the thioesterase domain of tyrocidinesynthetase.Nature 407,215-218(2000).
41.Zhou,Y.,Prediger,P.,Dias,L.C.,Murphy,A.C.&Leadlay,P.F.Macrodiolideformation by the thioesterase of a modular polyketide synthase.Angew Chem IntEd Engl 54,5232-5235(2015).
42.Horsman,M.E.,Hari,T.P.A.&Boddy,C.N.Polyketide synthase and non-ribosomal peptide synthetase thioesterase selectivity:Logic gate or a victimof fate?Natrual Products Reports(2015).
43.Frueh,D.P.et al.Dynamic thiolation-thioesterase structure of anon-ribosomal peptide synthetase.Nature 454,903-906(2008).
44.Whicher,J.R.et al.Structure and function of the RedJ protein,athioesterase from the prodiginine biosynthetic pathway in Streptomycescoelicolor.J Biol Chem 286,22558-22569(2011).
45.Ekici,O.D.,Paetzel,M.&Dalbey,R.E.Unconventional serine proteases:variations on the catalytic Ser/His/Asp triad configuration.Protein Sci 17,2023-2037(2008).
46.Liu,Y.,Zheng,T.&Bruner,S.D.Structural basis forphosphopantetheinyl carrier domain interactions in the terminal module ofnonribosomal peptide synthetases.Chemistry&biology 18,1482-1488(2011).
47.Trauger,J.W.,Kohli,R.M.&Walsh,C.T.Cyclization of backbone-substituted peptides catalyzed by the thioesterase domain from the tyrocidinenonribosomal peptide synthetase.Biochemistry 40,7092-7098(2001).
48.Aggarwal,A.et al.Development of a Novel Lead that TargetsM.tuberculosis Polyketide Synthase 13.Cell 170,249-259e225(2017).
49.Cravatt,B.F.,Wright,A.T.&Kozarich,J.W.Activity-based proteinprofiling:from enzyme chemistry to proteomic chemistry.Annu Rev Biochem 77,383-414(2008).
50.McGall,G.H.et al.The efficiency of light-directed synthesis of DNAarrays on glass substrates.Journal of the American Chemical Society 119,5081-5090,doi:DOI 10.1021/ja964427a(1997).
51.Pendrak,I.,Wittrock,R.&Kingsbury,W.D.Synthesis and Anti-HsvActivity of Methylenedioxy Mappicine Ketone Analogs.J Org Chem 60,2912-2915,doi:DOI 10.1021/jo00114a050(1995).
补充方法的参考文献:
31Alonzo,D.A.,Magarvey,N.A.&Schmeing,T.M.Characterization ofcereulide synthetase,a toxin-producing macromolecular machine.PloS one 10,e0128569,doi:10.1371/journal.pone.0128569(2015).
32Shaw-Reid,C.A.et al.Assembly line enzymology by multimodularnonribosomal peptide synthetases:the thioesterase domain of E.coli EntFcatalyzes both elongation and cyclolactonization.Chemistry&biology 6,385-400,doi:10.1016/S1074-5521(99)80050-7(1999).
33May,J.J.,Wendrich,T.M.&Marahiel,M.A.The dhb operon of Bacillussubtilis encodes the biosynthetic template for the catecholic siderophore 2,3-dihydroxybenzoate-glycine-threonine trimeric ester bacillibactin.J BiolChem 276,7209-7217,doi:10.1074/jbc.M009140200(2001).
34Zhou,Y.et al.Iterative Mechanism of Macrodiolide Formation in theAnticancer Compound Conglobatin.Chemistry&biology 22,745-754,doi:10.1016/j.chembiol.2015.05.010(2015).
35Robbel,L.,Hoyer,K.M.&Marahiel,M.A.TioS T-TE--a prototypicalthioesterase responsible for cyclodimerization of the quinoline-andquinoxaline-type class of chromodepsipeptides.FEBS J 276,1641-1653,doi:10.1111/j.1742-4658.2009.06897.x(2009).
36 Liu,Y.,Zheng,T.&Bruner,S.D.Structural basis forphosphopantetheinyl carrier domain interactions in the terminal module ofnonribosomal peptide synthetases.Chemistry&biology 18,1482-1488,doi:10.1016/j.chembiol.2011.09.018(2011).
37 Trauger,J.W.,Kohli,R.M.&Walsh,C.T.Cyclization of backbone-substituted peptides catalyzed by the thioesterase domain from the tyrocidinenonribosomal peptide synthetase.Biochemistry 40,7092-7098(2001).
38 Aggarwal,A.et al.Development of a Novel Lead that TargetsM.tuberculosis Polyketide Synthase 13.Cell 170,249-259 e225,doi:10.1016/j.cell.2017.06.025(2017).
39 Neumann,H.,Peak-Chew,S.Y.&Chin,J.W.Genetically encoding N(epsilon)-acetyllysine in recombinant proteins.Nat Chem Biol 4,232-234,doi:10.1038/nchembio.73(2008).
40 Rogerson,D.T.et al.Efficient genetic encoding of phosphoserine andits nonhydrolyzable analog.Nat Chem Biol 11,496-503,doi:10.1038/nchembio.1823(2015).
41 Zhang,M.S.et al.Biosynthesis and genetic encoding ofphosphothreonine through parallel selection and deep sequencing.Nat Methods14,729-736,doi:10.1038/nmeth.4302(2017).
42 McGall,G.H.et al.The efficiency of light-directed synthesis of DNAarrays on glass substrates.Journal of the American Chemical Society 119,5081-5090,doi:DOI 10.1021/ja964427a(1997).
43 Nguyen,D.P.et al.Genetic encoding of photocaged cysteine allowsphotoactivation of TEV protease in live mammalian cells.Journal of theAmerican Chemical Society 136,2240-2243,doi:10.1021/ja412191m(2014).
44 Pendrak,I.,Wittrock,R.&Kingsbury,W.D.Synthesis and Anti-HsvActivity of Methylenedioxy Mappicine Ketone Analogs.J Org Chem 60,2912-2915,doi:DOI 10.1021/jo00114a050(1995).
45 Mayer,S.C.,Ramanjulu,J.,Vera,M.D.,Pfizenmayer,A.J.&Joullie,M.M.Synthesis of New Didemnin B Analogs for Investigations of Structure/Biological Activity Relationships.J Org Chem 59,5192-5205,doi:10.1021/jo00097a022(1994).
46 Faure,S.et al.Asymmetric intramolecular[2+2]photocycloadditions:alpha-and beta-hydroxy acids as chiral tether groups.J Org Chem 67,1061-1070,doi:10.1021/jo001631e(2002).
47 Battye,T.G.,Kontogiannis,L.,Johnson,O.,Powell,H.R.&Leslie,A.G.iMOSFLM:a new graphical interface for diffraction-image processing withMOSFLM.Acta crystallographica.Section D,Biological crystallography 67,271-281,doi:10.1107/S0907444910048675(2011).
48 Winter,G.et al.DIALS:implementation and evaluation of a newintegration package.Acta crystallographica.Section D,Structural biology 74,85-97,doi:10.1107/S2059798317017235(2018).
49 Evans,P.Scaling and assessment of data quality.Actacrystallographica.Section D,Biological crystallography 62,72-82,doi:10.1107/S0907444905036693(2006).
50 McCoy,A.J.et al.Phaser crystallographic software.Journal ofapplied crystallography 40,658-674,doi:10.1107/S0021889807021206(2007).
51 Bunkoczi,G.&Read,R.J.Improvement of molecular-replacement modelswith Sculptor.Acta crystallographica.Section D,Biological crystallography 67,303-312,doi:10.1107/S0907444910051218(2011).
52 Tanovic,A.,Samel,S.A.,Essen,L.O.&Marahiel,M.A.Crystal structure ofthe termination module of a nonribosomal peptide synthetase.Science 321,659-663,doi:10.1126/science.1159850(2008).
53 Adams,P.D.et al.PHENIX:a comprehensive Python-based system formacromolecular structure solution.Acta crystallographica.Section D,Biologicalcrystallography 66,213-221,doi:10.1107/S0907444909052925(2010).
54 Terwilliger,T.C.et al.Iterative model building,structurerefinement and density modification with the PHENIX AutoBuild wizard.Actacrystallographica.Section D,Biological crystallography 64,61-69,doi:10.1107/S090744490705024X(2008).
55 Emsley,P.,Lohkamp,B.,Scott,W.G.&Cowtan,K.Features and developmentof Coot.Acta crystallographica.Section D,Biological crystallography 66,486-501,doi:10.1107/S0907444910007493(2010).
56 Bond,C.S.TopDraw:a sketchpad for protein structure topologycartoons.Bioinformatics 19,311-312(2003).
57 Long,F.et al.AceDRG:a stereochemical description generator forligands.Acta crystallographica.Section D,Structural biology 73,112-122,doi:10.1107/S2059798317000067(2017).
58 Vagin,A.A.et al.REFMAC5 dictionary:organization of prior chemicalknowledge and guidelines for its use.Acta crystallographica.Section D,Biological crystallography 60,2184-2195,doi:10.1107/S0907444904023510(2004).
59 Murshudov,G.N.et al.REFMAC5 for the refinement of macromolecularcrystal structures.Acta crystallographica.Section D,Biologicalcrystallography 67,355-367,doi:10.1107/S0907444911001314(2011).
60 Korman,T.P.et al.Structure and function of an iterative polyketidesynthase thioesterase domain catalyzing Claisen cyclization in aflatoxinbiosynthesis.Proceedings of the National Academy of Sciences of the UnitedStates of America 107,6246-6251,doi:10.1073/pnas.0913531107(2010).
61 Gehret,J.J.et al.Terminal alkene formation by the thioesterase ofcuracin A biosynthesis:structure of a decarboxylating thioesterase.J BiolChem 286,14445-14454,doi:10.1074/jbc.M110.214635(2011).
62 Tsai,S.C.et al.Crystal structure of the macrocycle-formingthioesterase domain of the erythromycin polyketide synthase:versatility froma unique substrate channel.Proceedings of the National Academy of Sciences ofthe United States of America 98,14808-14813,doi:10.1073/pnas.011399198(2001).
63Tsai,S.C.,Lu,H.,Cane,D.E.,Khosla,C.&Stroud,R.M.Insights intochannel architecture and substrate specificity from crystal structures of twomacrocycle-forming thioesterases of modular polyketide synthases.Biochemistry41,12598-12606(2002).
64Argyropoulos,P.et al.Towards a characterization of the structuraldeterminants of specificity in the macrocyclizing thioesterase fordeoxyerythronolide B biosynthesis.Biochim Biophys Acta 1860,486-497,doi:10.1016/j.bbagen.2015.11.007(2016).
65Giraldes,J.W.et al.Structural and mechanistic insights intopolyketide macrolactonization from polyketide-based affinity labels.Nat ChemBiol 2,531-536,doi:10.1038/nchembio822(2006).
66Koglin,A.et al.Structural basis for the selectivity of the externalthioesterase of the surfactin synthetase.Nature 454,907-911,doi:10.1038/nature07161(2008).
67Drake,E.J.et al.Structures of two distinct conformations of holo-non-ribosomal peptide synthetases.Nature 529,235-238,doi:10.1038/nature16163(2016).
68Gavalda,S.et al.The polyketide synthase Pks13 catalyzes a novelmechanism of lipid transfer in mycobacteria.Chemistry&biology 21,1660-1669,doi:10.1016/j.chembiol.2014.10.011(2014).
69Guntaka,N.S.,Healy,A.R.,Crawford,J.M.,Herzon,S.B.&Bruner,S.D.Structure and Functional Analysis of ClbQ,an Unusual Intermediate-Releasing Thioesterase from the Colibactin Biosynthetic Pathway.ACS Chem Biol12,2598-2608,doi:10.1021/acschembio.7b00479(2017).
尽管本文已经参照附图详细公开了本发明的说明性实施方案,但是读者应该注意,本发明不限于那些精确的实施方案,并且本领域的技术人员可以在不脱离由所附权利要求及其等同方案所限定的本发明的范围的情况下在其中进行各种改变和修改。
- 化合物及其制造方法、组合物、光学部件形成用组合物、光刻用膜形成组合物、抗蚀剂组合物、抗蚀图案形成方法、辐射敏感组合物、非晶膜制造方法、光刻用下层膜形成材料、光刻用下层膜形成用组合物、光刻用下层膜制造方法、抗蚀图案形成方法、电路图案形成方法、纯化方法
- 集成导电聚合物粘合剂组合物、该粘合剂组合物的制备方法、包含该粘合剂组合物的储能装置、包含由该粘合剂组合物形成的感测部的传感器,及包含该粘合剂组合物作为活性成分的防腐涂料组合物