埃氏拟杆菌肝素酶I融合蛋白及其编码基因和制备方法
文献发布时间:2023-06-19 19:30:30
技术领域
本发明涉及肝素酶融合蛋白制备技术领域,具体涉及埃氏拟杆菌肝素酶I融合蛋白及其编码基因和制备方法。
背景技术
肝素酶(heparinase)是一类作用于肝素或者乙酰肝素分子的多糖裂解酶,分别为肝素酶I、II、III。肝素酶主要从一些利用肝素为碳源的细菌中分离得到,最初来源于肝素黄杆菌(Flavobacteriumheparinum),此外,在许多微生物中也发现了肝素酶的存在,但至今肝素黄杆菌仍然是商品肝素酶的唯一来源。
自上世纪九十年代开始,已有大量的肝素酶I的基因工程表达研究和实践,其中以大肠杆菌为宿主的异源表达研究和应用最多。但是,在大肠杆菌中的表达肝素酶I,容易聚集成不溶性包涵体,形成包涵体肝素酶I很难再复性。此外,许多融合表达策略已被用于改善肝素酶I的生产。但是,针对肝素酶I的融合蛋白的研究,都是以肝素黄杆菌来源的肝素酶I为研究对象,并且表达的肝素酶I的活性和产量都难以达到工业化水平。因此寻找和建立表达效率高、酶活性高的肝素酶重组表达技术、以及寻找新型的可以商业化的肝素酶I,具有重要的理论和现实意义。
发明内容
本发明意在提供埃氏拟杆菌肝素酶I融合蛋白,以解决使用生物工程技术获得的肝素酶的酶活性有限的技术问题。
为达到上述目的,本发明采用如下技术方案:
埃氏拟杆菌肝素酶I融合蛋白,包括肝素酶I和麦芽糖结合蛋白,肝素酶I和麦芽糖结合蛋白之间连接有连接肽,所述连接肽的核苷酸序列如SEQ ID NO.11或者SEQ ID NO.12所示。
本方案的原理及优点是:肝素酶I大多以肝素黄杆菌来源的肝素酶I为研究对象,本方案以埃氏拟杆菌来源的肝素酶I为研究对象,拓宽了酶的来源。发明人从埃氏拟杆菌中克隆获得肝素酶I基因,并对其性质和表达条件进行了大量研究。与普通的肝素酶I基因不同,在使用埃氏拟杆菌来源的肝素酶I基因进行生物工程菌表达的时候,发明人发现表达的肝素酶I的活性有限,于是尝试了使用融合蛋白的方式来提升目的蛋白的活性。并通过大量研究和筛选发现,在制备肝素酶I和麦芽糖结合蛋白形成的融合蛋白时,需要在两个蛋白之间连接上核苷酸序列如SEQ ID NO.11或者SEQ ID NO.12所示的连接肽。由于上述两种连接肽的加入,保证由工程菌表达的融合蛋白可以正确折叠,并保证理想的酶活性。如果在肝素酶I和麦芽糖结合蛋白之间不加入连接肽,获得的融合蛋白的活性显著低于含有连接肽的融合蛋白的活性。如果将本方案的连接肽更换为其他种类的连接肽,获得的融合蛋白的活性也不及本方案制备获得的融合蛋白。由此可见,连接肽的选择对于本方案至关重要,使用核苷酸序列如SEQ ID NO.11或者SEQ ID NO.12所示的连接肽可以较为显著地增加融合蛋白的目的活性,减少包涵体肝素酶I的产生量,进而拓宽肝素酶I的来源,进而可以克服现有的肝素酶I的活性和产量都难以达到工业化水平的技术问题,具有重要的理论和现实意义。
进一步,埃氏拟杆菌肝素酶I融合蛋白的氨基酸序列如SEQ ID NO.13或者SEQ IDNO.14所示。由肝素酶I、连接肽和麦芽糖结合蛋白融合形成的蛋白质序列为SEQ ID NO.13或者SEQ ID NO.14。上述蛋白具有较高的原核表达量和活性,适应于工业化应用和大规模生产。
进一步,埃氏拟杆菌肝素酶I融合蛋白的基因的核苷酸序列如SEQ ID NO.9或者SEQ ID NO.10所示。由肝素酶I基因、连接肽核苷酸序列和麦芽糖结合蛋白基因融合形成的基因序列为SEQ ID NO.9或者SEQ ID NO.10。上述基因可以在原核表达系统中大量表达,形成活性高并适合于工业化应用的融合蛋白。
进一步,埃氏拟杆菌肝素酶I融合蛋白的基因整合在载体pMAL-c2x的多克隆位点上,形成表达载体。载体pMAL-c2x自带麦芽糖结合蛋白标签,为常用的商业化载体,易于获取和操作。
进一步,埃氏拟杆菌肝素酶I融合蛋白的制备方法,包括如下依次进行的步骤:
S1表达载体的构建:
S2工程菌的制备:将表达载体转化进入大肠杆菌中,获得工程菌;所述大肠杆菌的菌种为TB1、BL21、BL21(DE3)、BL21(DE3)pLysS和BL21-CodonPlus(DE3)-RIPL中的一种;
S3融合蛋白的表达:发酵并诱导所述工程菌,获得酶菌体;所述酶菌体中表达有埃氏拟杆菌肝素酶I融合蛋白。
通过转基因感受态细胞,获得工程菌,并诱导工程菌表达目的蛋白,可实现埃氏拟杆菌肝素酶I融合蛋白的大量表达。
进一步,还包括S4融合蛋白的纯化:将所述酶菌体破碎后,离心取上清,获得粗酶;通过亲和柱层析获得纯化后的融合蛋白。通过细胞破碎和后续的纯化步骤,可以获得纯度较高的目的蛋白,高纯度的蛋白将用于后续的制药以及制备检测试剂的应用。
进一步,在S1表达载体的构建步骤中,包括以下步骤:
使用序列如SEQ ID NO.3所示的上游引物P1和序列如SEQ ID NO.4所示的下游引物P2,并以SEQ ID NO.2所示的麦芽糖结合蛋白基因为模板,克隆获得MBP-F;
使用序列如SEQ ID NO.6所示的上游引物P4和序列如SEQ ID NO.7所示的下游引物P5,并以SEQ ID NO.1所示的氏拟杆菌肝素酶I基因为模板,克隆获得F-HEP;
将MBP-F、F-HEP和酶切处理后的载体pMAL-c2x三片段连接,获得表达载体。
进一步,在S1表达载体的构建步骤中,包括以下步骤:
使用序列如SEQ ID NO.3所示的上游引物P1和序列如SEQ ID NO.5所示的下游引物P3,并以SEQ ID NO.2所示的麦芽糖结合蛋白基因为模板,克隆获得MBP-R;
使用序列如SEQ ID NO.8所示的上游引物P6和序列如SEQ ID NO.7所示的下游引物P5,并以SEQ ID NO.1所示的氏拟杆菌肝素酶I基因为模板,克隆获得R-HEP;
将MBP-R、R-HEP和酶切处理后的载体pMAL-c2x三片段连接,获得表达载体。
使用上述引物,通过多步PCR反映以及后续的片段连接过程,可以获得目的基因,并将目的基因连接到空载体中,获得表达载体。
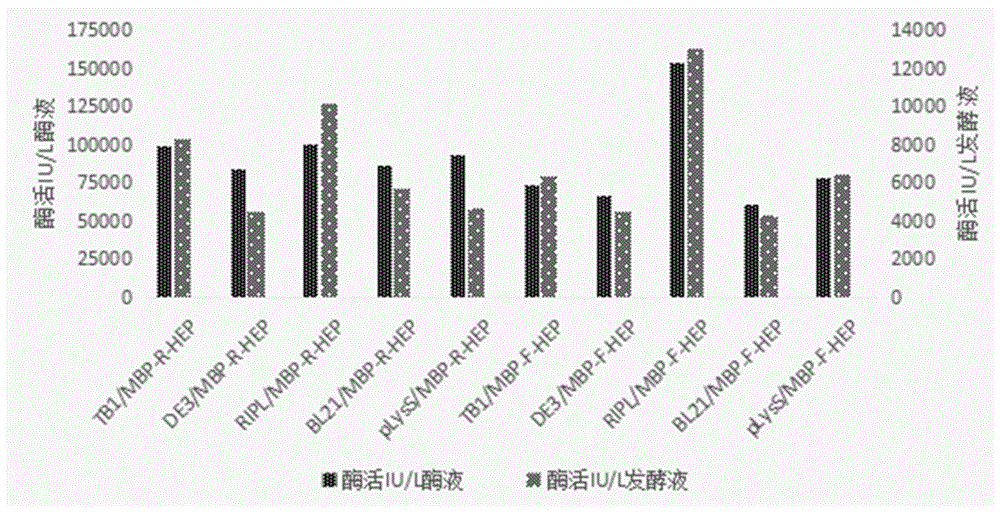
进一步,在S2工程菌的制备中,将表达载体转化进入大肠杆菌BL21-CodonPlus(DE3)-RIPL中,获得工程菌。两种融合蛋白MBP-F-HEP与MBP-R-HEP分别在E.coli BL21-CodonPlus(DE3)-RIPL都具有最高酶活,因此最优宿主菌为E.coli BL21-CodonPlus(DE3)-RIPL。经过第二次筛选实验也验证了E.coli BL21-CodonPlus(DE3)-RIPL是最佳宿主,可以应用于后续的工业化生产。
进一步,在S3融合蛋白的表达中,使用LB培养基30-37℃培养所述工程菌12-16h;然后使用M9YE培养基30-37℃培养工程菌至OD
附图说明
图1为本发明实施例1的带柔性连接肽的MBP的PCR扩增结果。
图2为本发明实施例1的带刚性连接肽的MBP的PCR扩增结果。
图3为本发明实施例1的带柔性连接肽的HEP的PCR扩增结果。
图4为本发明实施例1的带刚性连接肽的HEP的PCR扩增结果。
图5为本发明实施例1的MBP-F-HEP的PCR验证结果。
图6为本发明实施例1的MBP-R-HEP的PCR验证结果。
图7为本发明实施例2的融合蛋白酶活测定结果。
图8为本发明实施例2的MBP-F-HEP融合蛋白的蛋白电泳检测结果。
图9为本发明实施例2的MBP-R-HEP融合蛋白的蛋白电泳检测结果。
图10为本发明实施例3的最优工程菌第二次筛选结果。
图11为本发明实施例4的IPTG添加量的筛选结果。
图12为本发明对比例的连接肽作用研究结果。
具体实施方式
下面结合实施例对本发明做进一步详细的说明,但本发明的实施方式不限于此。若未特别指明,下述实施例所用的技术手段为本领域技术人员所熟知的常规手段;所用的实验方法均为常规方法,并可按照已描述的重组技术(参见分子克隆实验室手册,第2版,冷泉港实验室出版社,冷泉港,纽约)完成;所用的材料、试剂等,均可从商业途径得到。
实施例1:表达载体的构建
采用
1.引物设计
(1)MBP引物对
根据MBP标签序列设计引物对,并在引物序列中添加柔性(Flexibility)或者刚性(Rigidity)连接肽的一部分。其上游引物与pMal-c2x相邻的地方要有重叠的同源序列,下游引物与相邻的埃氏拟杆菌肝素酶I序列要有重叠的同源序列,MBP引物对序列如下:
MBP-F(含有柔性连接肽的MBP引物对):
上游引物P1:
(带边框碱基为Nde I的酶切位点,SEQ ID NO.3)
下游引物P2:
5’-
MBP-R(含有刚性连接肽的MBP引物对):
上游引物P1:
下游引物P3:
5’-
(2)埃氏拟杆菌肝素酶I引物对
根据埃氏拟杆菌肝素酶I序列设计引物对,并在引物序列中添加柔性(Flexibility)或者刚性(Rigidity)连接肽的一部分。其下游引物与pMal-c2x相邻的地方要有重叠的同源序列,埃氏拟杆菌肝素酶I引物对序列如下:
F-HEP(含有柔性连接肽的HEP引物对):
上游引物P4:
5’-
下游引物P5:
(带下划线的部分为与pMal载体同源的部分,带边框碱基为添加的终止密码子,SEQ ID NO.7)
R-HEP(含有刚性连接肽的HEP引物对):
上游引物P6:
5’-
下游引物P5:
(带下划线的部分为与pMal载体同源的部分,带边框碱基为添加的终止密码子,SEQ ID NO.7)
2.MBP和HEP的PCR扩增
PCR反应体系:采用NEB的Q5TM High-Fidelity DNA Polymerase,按照说明书要求配制反应体系,使用“1.引物设计”中的引物分别对MBP和HEP进行体外扩增。扩增程序:98℃预变性30s,98℃变性10s,55-70℃引物退火30s,72℃延伸90s,34个循环后,72℃终延伸5min结束反应。
PCR结果如图1-4所示(图1-4的底部数字表示采用的退火温度,单位℃),表明扩增得到了带刚柔性连接肽的MBP序列与肝素酶序列,测序比对表明分子克隆正确。图1为带柔性连接肽的MBP序列(MBP-F),最优退火温度是55℃;图2为带刚性连接肽的MBP序列(MBP-R),最优退火温度是67℃;图3为带柔性连接肽的肝素酶序列(F-HEP),最优退火温度是68.9-60.7℃;图4为带刚性连接肽的肝素酶序列(R-HEP),最优退火温度是55℃。
3.构建含有目的片段的克隆载体
pMal-c2x空载体(含有Factor Xa和MBP,Factor Xa和MBP之间存在连接序列)用EcoR I与Hind III进行双酶切,后用
MBP-F-HEP片段和MBP-R-HEP片段均连接在pMal-c2x上,获得两种表达载体,分别为表达MBP-F-HEP融合蛋白的表达载体和表达MBP-R-HEP融合蛋白的表达载体。
4.基因转化以及阳性克隆筛选
将“3.构建含有目的片段的克隆载体”中获得的两种连接产物以现有技术的常规方法分别转化进大肠杆菌DH5α感受态细胞中。涂布至含100μg/mL氨苄青霉素的LB抗性平板上进行筛选,37℃培养16h(可选范围:12-20h)。MBP-F-HEP挑选菌落并以此为模板,用引物P1和P5进行菌落PCR鉴定,使用2×Easy Taq SuperMix进行菌落PCR,PCR反应体系及反应条件根据说明书设置。MBP-R-HEP挑菌落提取质粒进行用EcoR I与Hind III酶切验证。反应结束后,对产物进行1%琼脂糖凝胶电泳检测。MBP-F-HEP结果如图5,1-5泳道分别为菌落PCR扩增结果,1、2、4、5泳道阳性,3泳道为阴性,M泳道为Marker;MBP-R-HEP结果如图6,用到1-4泳道分别为酶切验证结果,2、4泳道阳性,1、3泳道为阴性,M泳道为Marker。将筛选得到的阳性克隆菌交由生工生物进行测序。其中MBP-F-HEP的2号菌,MBP-R-HEP的4号菌测序比对结果正确。重组载体命名为MBP-F-HEP表达载体与MBP-R-HEP表达载体。MBP-F-HEP片段和MBP-R-HEP片段均连接在pMal-c2x上,获得两种表达载体,分别为表达MBP-F-HEP融合蛋白的表达载体(MBP-F-HEP表达载体)和表达MBP-R-HEP融合蛋白的表达载体(MBP-R-HEP表达载体)。
实施例2:埃氏拟杆菌肝素酶I融合蛋白的表达和纯化
1.融合蛋白的表达
提取大肠杆菌DH5α中的MBP-F-HEP表达载体和MBP-R-HEP表达载体,按照常规化转方法分别转化大肠杆菌TB1、BL21、BL21(DE3)(简称DE3)、BL21(DE3)pLysS(简称pLysS)、BL21-CodonPlus(DE3)-RIPL(简称RIPL)(厂家均为:唯地生物,货号分别是:EC1030/EC1001/EC1002/EC1003/EC1007),通过氨苄青霉素筛选,得到TB1/MBP-F-HEP、BL21/MBP-F-HEP、DE3/MBP-F-HEP、pLysS/MBP-F-HEP、RIPL/MBP-F-HEP与TB1/MBP-R-HEP、BL21/MBP-R-HEP、DE3/MBP-R-HEP、pLysS/MBP-R-HEP、RIPL/MBP-R-HEP作为表达MBP-F-HEP与MBP-R-HEP的工程菌。
按照以下操作对上面的工程菌进行平行表达:将工程菌分别在含100μg/mL氨苄青霉素抗性的LB培养基37℃(可选范围:30-37℃)培养16h(可选范围:12-16h)。然后按照1%接种量接入M9YE培养基(Yeast Extract 12.5g/L,Na
工程菌都表达除了有活性的可溶性埃氏拟杆菌肝素酶I蛋白,酶活检测结果如图7所示。可以发现MBP-F-HEP与MBP-R-HEP分别在E.coli BL21-CodonPlus(DE3)-RIPL都具有最高酶活,因此最优宿主菌为E.coli BL21-CodonPlus(DE3)-RIPL。
2.融合蛋白纯化
对最优宿主菌E.coli BL21-CodonPlus(DE3)-RIPL表达的MBP-F-HEP与MBP-R-HEP蛋白分别进行纯化。因MBP-F-HEP与MBP-R-HEP均带MBP标签,能够与麦芽糖Dextrin Beads进行吸附实现纯化效果。按照以下步骤分别操作。
将Dextrin Beads装入空层析柱,Tris缓冲液(Tris-HCL 20mM,NaCl 500mM,pH7.4)为柱平衡液,用5倍填料体积的柱平衡液平衡填料,将粗酶倒入填料里,4℃旋转混匀1h,用柱平衡液重力流穿清洗填料2倍填料体积,后用清洗Tris缓冲液(Tris-HCL 20mM,NaCl150mM,pH7.4)清洗至无杂蛋白流出。用洗脱Tris缓冲液(Tris-HCL 20mM,NaCl 150mM,麦芽糖10mM,pH7.4)洗脱目标蛋白,即得纯酶。将粗酶与纯酶分别进行蛋白电泳,MBP-F-HEP融合蛋白如图8所示,M泳道为Marker(由上至下分子量依次均为130kDa、100kDa、70kDa、50kDa、35kDa、25kDa),1-3泳道分别为菌体破碎液、菌体破碎上清(粗酶)、纯酶,箭头所指处为融合蛋白MBP-F-HEP(88kDa,SEQ ID NO.13);MBP-R-HEP融合蛋白(SEQ ID NO.14)如图9所示,M泳道为Marker(由上至下分子量依次均为130kDa、100kDa、70kDa、50kDa、35kDa、25kDa),1-3泳道分别为菌体破碎上清(粗酶)、菌体破碎液、纯酶,箭头所指处为融合蛋白MBP-R-HEP(89kDa)。
实施例3:最优工程菌第二次筛选
经过第一轮筛选后MBP-F-HEP与MBP-R-HEP分别筛选出表现较为优秀的TB1/MBP-F-HEP、RIPL/MBP-F-HEP、TB1/MBP-R-HEP、RIPL/MBP-R-HEP,按照第一轮相同的条件进行发酵表达。结果如图10所示,经过第二轮筛选后MBP-F-HEP与MBP-R-HEP的宿主菌种发酵酶活最高的仍然是RIPL/MBP-F-HEP和RIPL/MBP-R-HEP,进一步验证了我们的结论。
实施例4:IPTG添加量的筛选
按照相同的发酵表达方法,以RIPL/MBP-F-HEP菌株为例,选择添加不同终浓度的IPTG,分别为0.04mM、0.08mM、0.12mM、0.16mM、0.20mM。实验结果如图11所示,可以看出IPTG终浓度为0.2mM发酵酶活与酶液酶活最高。
对比例
为了研究连接肽的加入对于目的蛋白表达的作用,本对比例构建了不含有连接肽的融合基因即将MBP-R-HEP(SEQ ID NO.10)或者MBP-F-HEP(SEQ ID NO.9)中的连接肽序列(SEQ ID NO.12或者SEQ ID NO.11)去掉,获得MBP-HEP基因。按照实施例1和实施例2的方法,构建表达载体和进行蛋白表达,以上过程均为分子克隆的常规技术手段,在此不做赘述。按照常规化转方法转化进大肠杆菌TB1、BL21、BL21(DE3)(简称DE3)、BL21(DE3)pLysS(简称pLysS)、BL21-CodonPlus(DE3)-RIPL(简称RIPL)中,通过氨苄青霉素筛选,得到TB1/MBP-HEP、BL21/MBP-HEP、DE3/MBP-HEP、pLysS/MBP-HEP、RIPL/MBP-HEP作为表达MBP-HEP的工程菌。发酵表达条件得到粗酶,并进行酶活检测,测试结果参见图12(表达MBP-HEP融合蛋白的工程菌为图12右侧5种工程菌,图12左侧10种同图6)。由图12可知MBP-HEP转入各工程菌后,酶活产量均比不上替换连接肽后的MBP-F-HEP与MBP-R-HEP。
以上所述的仅是本发明的实施例,方案中公知的具体技术方案和/或特性等常识在此未作过多描述。应当指出,对于本领域的技术人员来说,在不脱离本发明技术方案的前提下,还可以作出若干变形和改进,这些也应该视为本发明的保护范围,这些都不会影响本发明实施的效果和专利的实用性。本申请要求的保护范围应当以其权利要求的内容为准,说明书中的具体实施方式等记载可以用于解释权利要求的内容。
- 肝素黄杆菌肝素酶Ⅰ的高效制备方法
- 一种高表达高活性多形拟杆菌肝素酶I融合蛋白及其编码基因和应用
- 一种利用SUMO融合表达系统制备重组肝素酶I的方法及其所制备的SUMO_肝素酶I融合蛋白