美登素类衍生物、其偶联物和使用方法
文献发布时间:2023-06-19 18:27:32
本申请要求并享有于2015年3月27日提交的、题为“MAYTANSINOID DERIVATIVES,CONJUGATES THEREOF AND METHODS OF TREATING PROLIFERATIVE DISEASES USING THESAME”的美国临时专利申请号62/139,044的优先权,并且还要求并享有于2015年11月6日提交的、题为“MAYTANSINOID DERIVATIVES,CONJUGATES THEREOF AND METHODS OF TREATINGPROLIFERATIVE DISEASES USING THE SAME”的美国临时专利申请号62/252,239的优先权。每个所述这些临时专利申请的内容通过引用其整体并入本文用于所有目的。
发明领域
本发明涉及美登素类衍生物、其偶联物,和用其治疗或预防增殖性疾病的方法。
发明背景
增殖性疾病,例如癌症,其特征在于异常细胞不受控制的生长。当前增殖性疾病的治疗包括手术、放射疗法、化学疗法、基于激素疗法和/或免疫疗法。许多这些治疗,特别是化学疗法,利用限制异常细胞扩散的抗增殖药物。然而,这些药物通常不加区别地杀伤细胞,而同时影响正常细胞和异常细胞。为了解决此问题,已探索靶向药物递送的各种方法,包括使用肿瘤靶向探针(如抗体或生长因子)与毒素类的偶联物,来选择性地靶向异常细胞。抗体药物偶联物(ADCs)是经由化学连接体与细胞毒类药物连接的抗体构成的化合物。此类化合物利用抗体对其靶标的结合特异性,从而将细胞毒类药物递送至异常细胞。因此,需要抗增殖化合物及其偶联物。
发明摘要
本发明提供了式(I)所示化合物:
或其药学上可接受的盐,
其中:
A是亚芳基或亚杂芳基;
L是连接体;
BA是结合剂;和
k是从1至30的整数。本发明还提供了式(I)所示化合物的立体异构体。
本发明还提供了式(II)所示化合物:
或其药学上可接受的盐,其中A是亚芳基或亚杂芳基。本发明还提供了式(II)所示化合物的立体异构体。
本发明还提供了式PP5所示化合物:
或其盐,其中A是亚芳基或亚杂芳基。本发明还提供了式PP5所示化合物的立体异构体。
本发明还提供了式PT1所示化合物:
或其盐,其中A是亚芳基或亚杂芳基,和L是连接体。本发明还提供了式PT1所示化合物的立体异构体。
此外,本发明提供了治疗增殖性疾病的方法,包括施用本发明所述化合物。
此外,本发明提供了治疗增殖性疾病的方法,包括施用本发明所述偶联物。
此外,本发明提供了制备式(I)所示化合物的方法,包括在转谷氨酰胺酶存在下,使去糖基化抗体或无糖基化(aglycosylated)抗体与式(PT1)所示化合物反应。
附图简要说明
图.1描绘了制备美登素-N-甲基-L-丙氨酸-4-氨基苯甲酰胺基-瓜氨酸-缬氨酸-己酰基-6-马来酰亚胺的合成序列。
图.2描绘了实施例41中测试的某些化合物的细胞活力%与Log
图.3描绘了实施例41中测试的某些化合物的细胞活力%与Log
图.4描绘了实施例41中测试的某些化合物的细胞活力%与Log
图.5描绘了实施例41中测试的某些化合物的细胞活力%与Log
图.6描绘了制备美登素-N-甲基-L-丙氨酸-(4-氨基-2-氟)苯甲酰胺基-瓜氨酸(Cit)-缬氨酸 (Val)-己酰基(Cap)-6-马来酰亚胺(Mal)的合成序列。
图.7描绘了制备美登素-N-甲基-L-丙氨酸-(4-氨基-2-三氟甲基)苯甲酰胺基-瓜氨酸-缬氨酸 -己酰基-6-马来酰亚胺的合成序列。
图.8描绘了制备美登素-N-甲基-L-丙氨酸-(4-氨基-2-甲氧基)苯甲酰胺基-瓜氨酸-缬氨酸- 己酰基-6-马来酰亚胺的合成序列。
图.9描绘了制备美登素-N-甲基-L-丙氨酸-4-氨基苯甲酰胺的合成序列。
图.10描绘了制备美登素-N-甲基-L-丙氨酸-(2-氟-4-氨基)苯甲酰胺的合成序列。
图.11描绘了制备美登素-N-甲基-L-丙氨酸-(2-三氟甲基-4-氨基)苯甲酰胺的合成序列。
图.12描绘了制备美登素-N-甲基-L-丙氨酸-(2-甲氧基-4-氨基)苯甲酰胺的合成序列。
图.13描绘了制备美登素-N-甲基-L-丙氨酸-N-(3-三氟甲基-4-氨基)苯甲酰胺的合成序列。
图.14描绘了制备美登素-N-甲基-L-丙氨酸-N-(2-氯-4-氨基-5-氟)苯甲酰胺的合成序列。
图.15描绘了制备式(II)所示化合物的一般合成序列,其中取代基R具有本发明所述和下述定义。
图.16描绘了制备美登素-N-甲基-L-丙氨酸-N-(2,5-二氟-4-氨基)苯甲酰胺的合成序列。
图.17描绘了制备美登素-N-甲基-L-丙氨酸-(3-氟-4-氨基)苯甲酰胺的合成序列。
图.18描绘了制备美登素-N-甲基-L-丙氨酸-(3-氯-4-氨基)苯甲酰胺的合成序列。
图.19描绘了制备美登素-N-甲基-L-丙氨酸-(5-氨基-8-羧基喹啉)甲酰胺的合成序列。
图.20描绘了制备美登素-N-甲基-L-丙氨酸-(3-溴-4-氨基)苯甲酰胺的合成序列。
图.21描绘了制备美登素-N-甲基-L-丙氨酸-(3-甲氧基-4-氨基)苯甲酰胺的合成序列。
图.22描绘了制备美登素-N-甲基-L-丙氨酸-(2-甲基-4-氨基)苯甲酰胺的合成序列。
图.23描绘了制备美登素-N-甲基-L-丙氨酸-(3-甲基-4-氨基)苯甲酰胺的合成序列。
图.24描绘了制备美登素-N-甲基-L-丙氨酸-(8-氨基-5-羧基喹啉)甲酰胺的合成序列。
图.25描绘了制备美登素-N-甲基-L-丙氨酸-(3-甲氧基-4-氨基)苯甲酰胺基-瓜氨酸-缬氨酸- 己酰基-6-马来酰亚胺的合成序列。
图.26描绘了制备美登素-N-甲基-L-丙氨酸-(2-氟-4-氨基)苯甲酰胺基-瓜氨酸-缬氨酸-己酰基-6-胺的合成序列。
图.27描绘了制备美登素-N-甲基-L-丙氨酸-N-(2-甲氧基-5-氨基)苯甲酰胺的合成序列。
图.28描绘了制备美登素-N-甲基-L-丙氨酸-N-(3-氨基-4-甲氧基)苯甲酰胺的合成序列。
图.29描绘了制备美登素-N-甲基-L-丙氨酸-N-(3-氨基-5-氟)苯甲酰胺的合成序列。
图.30描绘了制备美登素-N-甲基-L-丙氨酸-N-(2-氟-5-氨基)苯甲酰胺的合成序列。
图.31描绘了制备美登素-N-甲基-L-丙氨酸-N-(3-氨基)苯甲酰胺的合成序列。
图.32描绘了制备美登素-N-甲基-L-丙氨酸-N-(3-氨基-4-氟)苯甲酰胺的合成序列。
图.33描绘了制备美登素-N-甲基-L-丙氨酸-N-4-氨基苯甲酰胺-己二酰基-NHS的合成序列。
图.34描绘了制备美登素-N-甲基-L-丙氨酸-4-氨基苯甲酰胺-己酰基-6-马来酰亚胺的合成序列。
图.35描绘了制备美登素-N-甲基-L-丙氨酸-N-(3-甲磺酰基-4-氨基)苯甲酰胺的合成序列。
图.36描绘了制备美登素-N-甲基-L-丙氨酸-N-(3-羟基-4-氨基)苯甲酰胺的合成序列。
图.37描绘了制备美登素-N-甲基-L-丙氨酸-N-(2-氨基)苯甲酰胺的合成序列。
图.38描绘了制备美登素-N-甲基-L-丙氨酸-N-(4-甲氧基-2-氨基)苯甲酰胺的合成序列。
图.39描绘了制备美登素-N-甲基-L-丙氨酸-N-(3-吗啉基-4-氨基)苯甲酰胺的合成序列。
图.40描绘了制备美登素-N-甲基-L-丙氨酸-N-(3-乙酰胺基-4-氨基)苯甲酰胺的合成序列。
图.41描绘了制备美登素-N-甲基-L-丙氨酸-N-4-氨基苯甲酰胺-瓜氨酸-缬氨酸-己酰基-二溴甲基丙烯酰胺的合成序列。
图.42描绘了由实施例43得到的抗体药物偶联物、PRLR-Q-63偶联物的去卷积质谱(MS) 光谱。
图.43描绘了由实施例43得到的同型对照-Q-63偶联物的去卷积MS谱。
图.44描绘了实施例45中测试的某些化合物的细胞活力%与Log
图.45描绘了实施例45中测试的某些化合物的细胞活力%与Log
图.46描绘了实施例45中测试的某些化合物的细胞活力%与Log
图.47描绘了实施例45中测试的某些化合物的细胞活力%与Log
发明详述
A.定义
本发明使用的“烷基”是指一价、饱和的烃基基团部分。烷基是任选取代的,并可以是直链、支链、或环的即环烷基。烷基包括,但不限于,那些具有1-20个碳原子,即C
本发明使用的“卤代烷基”是指上述定义的烷基,其中所述烷基包括至少一个选自卤素,如F、Cl、Br、或I的取代基。
本发明使用的“烯基”是指含有至少两个碳原子和一个或多个非芳香性碳-碳双键的一价烃基基团部分。烯基是任选取代的,并可以是直链、支链、或环的。烯基包括,但不限于,那些具有2-20 个碳原子,即C
本发明使用的“炔基”是指含有至少两个碳原子和一个或多个碳-碳三键的一价烃基基团部分。炔基是任选取代的,并可以是直链、支链、或环的。炔基包括,但不限于,那些具有2-20个碳原子,即C
本发明使用的“烷氧基”是指一价、饱和的烃基基团部分,其中所述烃包括与氧原子连接的单键,和其中所述自由基位于所述氧原子上,例如CH
本发明使用的“卤代烷氧基”是指上述定义的烷氧基,其中所述烷氧基包括至少一个选自卤素,如F、Cl、Br、或I的取代基。
本发明使用的“芳基”是指芳香性化合物的自由基/基团的一价基团部分,其中所述环原子均为碳原子。芳基是任选取代的,并可以是单环或多环,譬如,二环或三环。芳基基团部分的实例包括,但不限于,那些具有6至20个环碳原子,即C
本发明使用的“亚芳基”是指芳香性化合物的二价基团部分,其中所述环原子均仅为碳原子。亚芳基是任选取代的,并可以是单环或多环,譬如,二环或三环。亚芳基基团部分的实例包括,但不限于,那些具有6至20个环碳原子,即C
本发明使用的“烷芳基”是指被至少一个烷基取代的芳基。烷芳基是任选取代的。
本发明使用的“杂烷基”是指其中一个或多个碳原子被杂原子替代的烷基。本发明使用的“杂烯基”是指其中一个或多个碳原子被杂原子替代的烯基。本发明使用的“杂炔基”是指其中一个或多个碳原子被杂原子替代的炔基。合适的杂原子包括,但不限于氮、氧、和硫原子。杂烷基是任选取代的。杂烷基基团部分的实例包括,但不限于氨基烷基、磺酰烷基、亚磺酰烷基。杂烷基基团部分的实例还包括,但不限于甲氨基、甲磺酰基、甲基亚磺酰基。
本发明使用的“杂芳基”是指芳香性化合物的自由基/基团的一价基团部分,其中所述环原子包含碳原子和至少一个氧、硫、氮、或磷原子。杂芳基基团部分的实例包括,但不限于,具有5至20 个环原子;5至15个环原子;和5至10个环原子的那些。杂芳基是任选取代的。
本发明使用的“亚杂芳基”是指其中所述芳环的一个或多个环原子被氧、硫、氮、或磷原子替代的亚芳基。亚杂芳基是任选取代的。
本发明使用的“杂环烷基”是指其中一个或多个碳原子被杂原子替代的环烷基。合适的杂原子包括,但不限于氮、氧、和硫原子。杂环烷基是任选取代的。杂环烷基基团部分的实例包括,但不限于吗啉基、哌啶基、四氢吡喃基、吡咯烷基、咪唑烷基、噁唑烷基、噻唑烷基、二氧戊环烷基(dioxolanyl)、二硫戊环烷基(dithiolanyl)、氧杂环己基(oxanyl)、或硫杂环己基(thianyl)。
本发明使用的“任选取代的”,当用于描述基团部分,譬如,任选取代的烷基,是指此基团部分任选地与一个或多个取代基连接。这些取代基的实例包括,但不限于卤素、氰基、硝基、卤代烷基、叠氮基、环氧基、任选取代的杂芳基、任选取代的杂环烷基、
本发明使用的“结合剂”是指能够与给定结合配偶体特异性结合的任何分子。
本发明使用的“连接体”是指使所述结合剂与本发明所述美登素类衍生物共价连接的二价基团部分。
本发明使用的“酰胺合成条件”是指适于形成酰胺,譬如,通过羧酸、活化的羧酸、或酰卤与胺进行反应的反应条件。在一些实施例,酰胺合成条件是指适于在羧酸和胺之间形成酰胺键的反应条件。在所述这些实施例中的一些,所述羧酸首先在活化羧酸与胺反应形成酰胺之前转化为活化羧酸。适于形成酰胺的条件包括,但不限于,利用试剂来实现羧酸与胺之间反应的那些,包括但不限于,二环己基碳二亚胺(DCC)、二异丙基碳二亚胺(DIC)、(苯并三氮唑-1-基氧基)三(二甲基氨基)磷鎓六氟磷酸盐(BOP)、 (苯并三唑-1-基氧基)三吡咯烷基鏻六氟磷酸盐(PyBOP)、(7-偶氮苯并三氮唑-1-基氧基)三吡咯烷基鏻六氟磷酸盐(PyAOP)、三吡咯烷基溴化鏻六氟磷酸盐(PyBrOP)、O-(苯并三氮唑-1-基)-N,N,N’,N’-四甲基脲六氟磷酸盐(HBTU)、O-(苯并三氮唑-1-基)-N,N,N’,N’-四甲基脲六氟硼酸盐(TBTU)、1-[双(二甲氨基)亚甲基]-1H-1,2,3-三氮唑并[4,5-b]吡啶鎓3-氧化六氟磷酸盐(HATU)、2-乙氧基-1-乙氧羰基-1,2-二氢喹啉(EEDQ),1-乙基-3-(3-二甲氨基丙基)碳二亚胺(EDC)、2-氯-1,3-二甲基咪唑鎓六氟磷酸盐(CIP)、2-氯-4,6-二甲氧基-1,3,5-三嗪(CDMT)、和羰二咪唑(CDI)。在一些实施例,首先将羧酸转化为活化的羧酸酯,然后与胺反应形成酰胺键。在一些实施方案,羧酸与试剂反应。试剂通过使羧酸去质子来活化羧酸,然后由于去质子化的羧酸亲核进攻到质子化试剂上,从而与去质子化的羧酸形成产物复合物。对于某些羧酸,相较羧酸转化之前,所述活化酯更容易受到胺的亲核进攻。此举导致酰胺键形成。因此,所述羧酸被描述为活化的。示例性试剂包括DCC和DIC。
本发明使用的“治疗有效量”是指足以在患者治疗或控制疾病或病症,或延迟或最小化与疾病或病症相关的一种或多种症状时,提供治疗益处的(化合物的)量。
某些基团、部分、取代基、和原子被描述为具有与键相交的波浪线,以指示所述基团、部分、取代基、原子通过其进行结合的原子。譬如,被丙基基团取代的苯基基团可表示为:
具有下述结构:
B.偶联物
本发明提供了式(I)所示化合物:
或其药学上可接受的盐,
其中:
A是亚芳基或亚杂芳基;
L是连接体;
BA是结合剂;和
k是从1至30的整数。
1.“A”部分
在一些实施方案,A是亚芳基。在一些实施方案,A是亚杂芳基。在一些实施方案,所述亚芳基或亚杂芳基被一个或多个吸电子基团和/或一个或多个供电子基团取代。
在一些实施方案,A是苯、吡啶、萘、或喹诺酮的二价基团,所述苯、吡啶、萘、和喹诺酮是任选取代的。
在一些实施方案,A是苯的二价基团,所述苯任选地被选自氨基、酰胺基、烷基、卤素、卤代烷基、烷氧基、和卤代烷氧基组成的组的成员取代。
在一些实施方案,A是:
其中:
R
其中R
n是从0至4的整数;
m是从0至3的整数;
p是从0至6的整数;和
q是从0至5的整数。
在一些实施方案,A是:
其中:
R
其中R
n是从0至4的整数;
m是从0至3的整数;
p是从0至6的整数;和
q是从0至5的整数。
在一些实施方案,A是:
其中:
R
其中R
n是从0至4的整数;
m是从0至3的整数;
p是从0至6的整数;和
q是从0至5的整数。
在一些实施方案,R
在一些实施方案,各R
在一些实施方案,R
在一些实施方案,各R
在一些实施方案,A是:
在一些实施方案,A是:
在一些实施方案,A是:
在一些实施方案,A是:
其中n是0、1或2。
在一些实施方案,A是:
其中n是0、1或2。
在一些实施方案,A是:
其中n是0或1;和R
在一些实施方案,A是:
其中n是0或1;和R
在一些实施方案,A是:
其中n是0或1;和R
在一些实施方案,A是:
其中n是0或1;和R
在一些实施方案,A是:
其中n是0或1;R
在一些实施方案,A是:
其中n是0或1;R
在一些实施方案,A是:
其中n是0、1、2、3、或4。
在一些实施方案,A是:
其中n是0、1、2、3、或4。
在一些实施方案,A是:
其中:
R
n是0、1或2。
在一些实施方案,A是:
其中:
R
n是0、1或2。
在一些实施方案,A是:
其中:
R
n是0、1、2、3、或4。
在一些实施方案,A是:
其中:
R
n是0、1、2、3、或4。
在一些实施方案,A是:
其中R
在一些实施方案,A是:
在一些实施方案,A是:
其中:
R
n是0、1或2。
在一些实施方案,A是:
在一些实施方案,A是:
其中:
X是氢原子、卤素、或三氟甲基。
在一些实施方案,A是:
其中:
X是氢原子、卤素、或三氟甲基;
在一些实施方案,A是:
其中:
R
其中:
R
在一些实施方案,R
在一些实施方案,A是:
其中:
R
在一些实施方案,A是:
在一些实施方案,A是:
在一些实施方案,A是:
在一些实施方案,A是:
在一些实施方案,A是:
在一些实施方案,A是:
在一些实施方案,A是:
其中R
在一些实施方案,A是:
其中
在一些实施方案,A是:
其中R
在一些实施方案,A是:
在一些实施方案,A是:
在一些实施方案,A是:
在一些实施方案,A是:
在一些实施方案,A是:
在一些实施方案,A是:
在一些实施方案,A是:
在一些实施方案,A是:
在一些实施方案,A是:
在一些实施方案,A是:
在一些实施方案,A是:
其中:
在一些实施方案,A是:
其中:
在一些实施方案,A是:
其中:
在一些实施方案,A是:
其中:
在一些实施方案,A是:
在一些实施方案,A是:
其中n是0、1、2、或3。
在一些实施方案,A是:
其中:
R
在一些实施方案,A是:
在一些实施方案,A是:
在一些实施方案,A是:
其中:
各X分别独立地为氢原子、烷基、烷氧基、卤素、卤代烷氧基、卤代烷基、或杂烷基。在一些实施方案,X是氟、氯、溴、碘、二甲氨基、甲氨基、甲氧基、乙氧基、或三氟甲基。
在一些实施方案,A是:
其中:
各X分别独立地为氢原子、烷基、烷氧基、卤素、卤代烷氧基、卤代烷基、或杂烷基。在一些实施方案,X是氟、氯、溴、碘、二甲氨基、甲氨基、甲氧基、乙氧基、或三氟甲基;
2.连接体
本发明所述偶联物的连接体部分是将所述结合剂共价连接至本发明所述美登素类衍生物上的二价基团部分。合适的连接体包括在酶存在下或在特定pH值范围或pH值下释放所述美登素类化合物部分的那些。
在一些实施方案,所述连接体包含可酶切部分。示例性可酶切部分包括,但不限于肽键类、酯键类、腙类、和二硫键类。在一些实施方案,所述连接体包含可进行组织蛋白酶切连接体。
在一些实施方案,所述连接体包含不可裂解部分。在一些实施方案,所述不可裂解连接体是
合适的连接体还包括,但不限于与单一结合剂如抗体的两个半胱氨酸残基进行化学键合的那些。此类连接体可用于摸拟由于偶联过程而被破坏的所述抗体的二硫键。
在一些实施方案,所述连接体包含一个或多个氨基酸。合适的氨基酸包括天然、非天然的、标准、非标准的、蛋白原、非蛋白原的、和L-或D-型α-氨基酸。在一些实施方案,所述连接体包含丙氨酸、缬氨酸、亮氨酸、异亮氨酸、蛋氨酸、色氨酸、苯丙氨酸、脯氨酸、丝氨酸、苏氨酸、半胱氨酸、酪氨酸、天冬酰胺、谷氨酰胺、天冬氨酸、谷氨酸、赖氨酸、精氨酸、组氨酸、或瓜氨酸,或其衍生物。
在一些实施方案,所述连接体包含缬氨酸和瓜氨酸。
在一些实施方案,所述连接体是:
其中:
SP是间隔基团;
AA
AA
所述间隔基团是将所述AA
在一些实施例,合适的间隔基团包括,但不限于伯胺封端的亚烷基或伯胺封端的聚乙二醇。所述间隔基团的伯胺末端可在转谷氨酰胺酶存在下,直接与去糖基化抗体或无糖基化抗体结合。
在一些实施方案,所述间隔基团包含亚烷基。在一些实施方案,所述间隔基团包含C
其中:
b是从2至8的整数。
在一些实施方案,所述间隔基团包含伯胺封端的亚烷基。在一些实施方案,所述间隔基团包含NH
其中:
b是从2至8的整数。
在一些实施方案,所述间隔基团是:
其中:
在一些实施方案,所述间隔基团是:
其中:
在一些实施方案,所述间隔基团是:
其中:
R
R
由
b是从2至8的整数。
在一些实施方案,所述间隔基团是:
其中:
b是从2至8的整数。
在一些实施方案,所述间隔基团是:
其中:
g是从2至20的整数。在一些实施方案,g是2-8。在一些实施方案,g是2、4、6、或8。
在一些实施方案,所述间隔基团是
在一些实施方案,所述间隔基团是:
在一些实施方案,所述间隔基团是
在一些实施方案,所述间隔基团是
在一些实施方案,所述间隔基团是:
在一些实施方案,所述间隔基团是:
在一些实施方案,所述间隔基团是:
在一些实施方案,所述间隔基团是:
在一些实施方案,所述间隔基团是:
在一些实施方案,所述间隔基团是:
在一些实施方案,所述间隔基团是:
在一些实施方案,所述间隔基团是:
其中
X是N或O;R
在一些实施方案,AA
在一些实施方案,AA
在一些实施方案,所述连接体是:
其中:
SP是间隔基团;
R
R
本发明使用的“氨基酸侧链”是指与α-氨基酸包括如天然和非天然氨基酸的α-碳连接的一价、非氢取代基。氨基酸侧链的实例包括,但不限于丙氨酸、缬氨酸、亮氨酸、异亮氨酸、蛋氨酸、色氨酸、苯丙氨酸、脯氨酸、丝氨酸、苏氨酸、半胱氨酸、酪氨酸、天冬酰胺、谷氨酰胺、天冬氨酸、谷氨酸、赖氨酸、精氨酸、组氨酸、和瓜氨酸的α-碳取代基。
在一些实施方案,所述连接体是:
其中:
SP是间隔基团;和
在一些实施方案,所述连接体是:
其中:
b是从2至8的整数。
在一些实施方案,所述连接体是:
其中:
b是从2至8的整数。
在一些实施方案,BA是抗体和所述连接体是:
其中:
由
b是从2至8的整数。
在一些实施方案,BA是抗体和所述连接体是:
其中:
由
b是从2至8的整数。
在一些实施方案,BA是抗体和所述连接体是:
其中:
R
R
由
b是从2至8的整数。
在一些实施方案,所述连接体是:
其中:
b是从2至8的整数。
在一些实施方案,所述连接体是:
其中:
g是从2至20的整数。在一些实施方案,g是2至8。在一些实施方案,g是2、4、6、或 8。
在一些实施方案,所述连接体是:
在一些实施方案,所述连接体是:
在一些实施方案,所述连接体是:
在一些实施方案,所述连接体是:
在一些实施方案,所述连接体是:
在一些实施方案,所述连接体是:
在一些实施方案,所述连接体是:
在一些实施方案,所述连接体是:
3.结合剂
合适的结合剂包括,但不限于抗体类、淋巴因子类、激素类、生长因子类、病毒受体类、白介素类、或任何其他细胞结合或肽结合分子类或物质类。
在一些实施方案,所述结合剂是抗体。在一些实施方案,所述抗体是单克隆抗体,多克隆抗体,抗体片段(Fab、Fab’、和F(ab)2、微抗体、双体抗体、三体抗体等),或双特异性抗体。本发明所述抗体可使用美国专利号6,596,541和美国公开号2012/0096572中所述方法进行人源化,所述此两篇美国专利/申请各自通过引用其全部内容并入本文。
当结合剂是抗体时,其结合作为多肽的抗原结合配偶体,且可以是跨膜分子(譬如,受体) 或可能被糖基化或磷酸化的生长因子。示例性抗原包括,但不限于,分子类如肾素;生长激素,包括人生长激素和牛生长激素;生长激素释放因子;甲状旁腺激素;促甲状腺激素;脂蛋白类;α1-抗胰蛋白酶;胰岛素A链;胰岛素B链;胰岛素原;促卵泡激素;降钙素;促黄体激素;胰高血糖素;凝血因子如因子 vmc,因子IX,组织因子(TF)和血管性血友病因子;抗凝血因子如蛋白C;心房利钠因子;肺表面活性剂;纤溶酶原激活物,如尿激酶或人尿或组织型纤溶酶原激活物(t-PA);蛙皮素;凝血酶;造血生长因子;肿瘤坏死因子-α和-β;脑啡肽酶;RANTES(调节激活正常T细胞表达和分泌);人巨噬细胞炎症蛋白(MlP-I-α);血清白蛋白,如人血清白蛋白;缪勒管(Muellerian)抑制物质;松弛素A链;松弛素B 链;松弛素原(prorelaxin);小鼠促性腺激素相关肽;微生物蛋白,如β-内酰胺酶;脱氧核糖核酸酶;19E;细胞毒性T淋巴细胞相关抗原(CTLA),如CTLA-4;抑制素;激活素;血管内皮生长因子(VEGF);激素或生长因子受体类;蛋白A或D;类风湿因子类;神经营养因子如骨源性神经营养因子(BDNF),神经营养因子-3、-4、-5或-6(NT-3、NT4、NT-5、或NT-6),或神经生长因子如NGF-β;血小板源生长因子(PDGF);成纤维细胞生长因子如aFGF和bFGF;成纤维细胞生长因子受体2(FGFR2),表皮生长因子(EGF);转化生长因子(TGF)如TGF-α和TGF-β,包括TGF-β1、TGF-β2、TGF-β3、TGF-β4、或TGF-β5;胰岛素样生长因子-1和-II(IGF-1和IGF-II);des(I-3)-IGF-l(脑IGF-1),胰岛素样生长因子结合蛋白类,EpCAM,GD3,FLT3,PSMA,PSCA,MUC1,MUC16,STEAP,CEA,TENB2,EphA 受体类,EphB受体类,叶酸受体,FOLRI,间皮素,cripto,αvβ6,整合素类,VEGF,VEGFR,EGFR,转铁蛋白受体,IRTA1,IRTA2,IRTA3,IRTA4,IRTA5;CD蛋白类如CD2,CD3,CD4,CD5,CD6,CD8,CD11,CD14,CD19,CD20,CD21,CD22,CD25,CD26,CD28,CD30,CD33,CD36,CD37, CD38,CD40,CD44,CD52,CD55,CD56,CD59,CD70,CD79,CD80,CD81,CD103,CD105,CD134, CD137,CD138,CD152,或与美国专利公开号2008/0171040或美国专利公开号2008/0305044中公开的一种或多种肿瘤相关抗原或细胞表面受体结合的抗体,并通过引用其全部内容并入本文;促红细胞生成素;骨诱导因子类;免疫毒素类;骨形态发生蛋白(BMP);干扰素,如干扰素-α、β和γ;集落刺激因子(CSFs),例如M-CSF,GM-CSF,和G-CSF;白介素类(ILs),例如,IL-1至IL-10;超氧化物歧化酶;T细胞受体类;表面膜蛋白类;衰变加速因子;病毒抗原,例如,HIV包膜的一部分;转运蛋白类;归巢受体类;禀呈素类(addressins);调节蛋白类;整合素类如CD11a,CD11b,CD11c,CD18,ICAM,VLA-4和 VCAM;肿瘤相关抗原如AFP,ALK,B7H4,BAGE蛋白类,β-连环蛋白,brc-abl,BRCA1,BORIS, CA9(碳酸酐酶IX),半胱天冬酶-8,CD20,CD40,CD123,CDK4,CEA,CLEC12A,c-kit,cMET, CTLA4,细胞周期素-B1,CYP1B1,EGFR,EGFRvIII,内皮糖蛋白,上皮细胞粘附分子(Epcam),EphA2,ErbB2/Her2,ErbB3/Her3,ErbB4/Her4,ETV6-AML,Fra-1,FOLR1,GAGE蛋白类(如GAGE-1、-2), GD2,GD3,GloboH,磷脂酰肌醇聚糖-3,GM3,gp100,Her2,HLA/B-raf,HLA/EBNA1,HLA/k-ras, HLA/MAGE-A3,hTERT,IGF1R,LGR5,LMP2,MAGE蛋白类(例如MAGE-1、-2、-3、-4、-6、和-12), MART-1,间皮素,ML-IAP,Muc1,Muc16(CA-125),MUM1,NA17,NGEP,NY-BR1,NY-BR62,NY-BR85,NY-ESO1,OX40,p15,p53,PAP,PAX3,PAX5,PCTA-1,PDGFR-α,PDGFR-β,PDGF-A,PDGF-B,PDGF-C,PDGF-D,PLAC1,PRLR,PRAME,PSCA,PSGR,PSMA(FOLH1),RAGE蛋白类,Ras,RGS5,Rho,SART-1,SART-3,Steap-1,Steap-2,STn,生存素,TAG-72,TGF-β,TMPRSS2, Tn,TNFRSF17,TRP-1,TRP-2,酪氨酸酶,和尿路上皮特异蛋白-3(uroplakin-3),和任何上述列举的多肽类的片段。
示例性抗原还包括,但不限于BCMA,SLAMF7,B7H4,GPNMB,UPK3A,和LGR5。
在一些实施方案,所述抗原包括催乳素受体(PRLR)或前列腺特异性膜抗原(PSMA)。
结合剂还包括,但不限于锚蛋白重复蛋白类,干扰素类,淋巴因子类如IL-2或IL-3,激素类如胰岛素和糖皮质激素类,生长因子类如EGF、转铁蛋白和纤连蛋白III型。
在一些实施方案,所述结合剂与肿瘤抗原相互作用或与肿瘤抗原结合,包括对特定类型肿瘤共享、过表达或修饰的一类肿瘤或抗原特异的抗原。实例包括,但不限于:具有肺癌的α-辅肌动蛋白-4,具有黑素瘤的ARTC1,具有慢性骨髓性白血病的BCR-ABL融合蛋白,具有黑素瘤的B-RAF、CLPP或 Cdc27,具有鳞状细胞癌的CASP-8和具有肾细胞癌的hsp70-2,以及以下共享的肿瘤特异性抗原,例如: BAGE-1、GAGE、GnTV、KK-LC-1、MAGE-A2、NA88-A、TRP2-INT2。
在一些实施方案,所述结合剂是抗体。在一些实施方案,所述结合剂是单克隆抗体。在一些实施方案,所述结合剂是多克隆抗体。在一些实施方案,所述抗体是抗PSMA、抗MUC16、或抗EGFRvIII、或抗STEAP-2抗体。
所述连接体可通过抗体或抗原结合分子内特定氨基酸的附接物与结合剂例如抗体或抗原结合分子结合。可在本发明公开的上下文中使用的示例性氨基酸附接物包括,譬如,赖氨酸(参见,如US 5,208,020;US 2010/0129314;Hollander et al.,BioconjugateChem.,2008,19:358-361;WO 2005/089808;US 5,714,586;US 2013/0101546;和US 2012/0585592),半胱氨酸(参见,如US 2007/0258987;WO 2013/055993; WO 2013/055990;WO2013/053873;WO 2013/053872;WO 2011/130598;US 2013/0101546;和US 7,750,116),硒代半胱氨酸(参见,如WO 2008/122039;和Hofer et al.,Proc.Natl.Acad.Sci.,USA,2008,105:12451-12456),甲酰甘氨酸(参见,如Carrico et al.,Nat.Chem.Biol.,2007,3:321-322;Agarwal et al.,Proc. Natl.Acad.Sci.,USA,2013,110:46-51,和Rabuka et al.,Nat.Protocols,2012,10:1052-1067),非天然氨基酸类(参见,如WO 2013/068874,和WO2012/166559),和酸性氨基酸类(参见,如WO 2012/05982)。连接体可通过谷氨酰胺通过基于转谷氨酰胺酶的化学酶偶联进行偶联(参见,如Dennler et al.,BioconjugateChem.2014,25,569-578)。连接体还可通过连接至碳水化合物(参见,如US 2008/0305497,WO 2014/065661,和Ryan et al.,Food&Agriculture Immunol.,2001,13:127-130)和二硫化物连接体(参见,如WO 2013/085925,WO 2010/010324,WO 2011/018611,WO 2014/197854,和Shaunak et al.,Nat.Chem.Biol.,2006, 2:312-313),从而与抗原结合蛋白偶联。
在一些实施方案,所述结合剂是抗体,和所述抗体是通过赖氨酸残基与所述连接体结合。在一些实施方案,所述抗体是通过半胱氨酸残基与所述连接体结合。
4.说明性实施方案/实施例
在一些实施方案,
A是:
其中:
R
其中R
n是从0至4的整数;
m是从0至3的整数;
p是从0至6的整数;和
q是从0至5的整数;和
L是:
其中:
SP是间隔基团;
AA
AA
在一些实施方案,
A是:
其中:
R
n是从0至4的整数;
m是从0至3的整数;
p是从0至6的整数;和
q是从0至5的整数;和
L是:
其中:
SP是间隔基团;
AA
AA
在一些实施方案,A是:
其中:
R
其中R
n是从0至4的整数;
m是从0至3的整数;
p是从0至6的整数;和
q是从0至5的整数;和
L是:
其中:
SP是间隔基团;
AA
在一些实施方案,
A是:
其中:
R
n是从0至4的整数;
m是从0至3的整数;
p是从0至6的整数;和
q是从0至5的整数;和
L是:
其中:
SP是:
其中:
b是从2至8的整数;和
AA
AA
在一些实施方案,A是:
其中:
R
其中R
n是从0至4的整数;
m是从0至3的整数;
p是从0至6的整数;和
q是从0至5的整数;和
L是:
其中:
SP是:
其中:
b是从2至8的整数;和
AA
AA
在一些实施方案,
A是:
其中:
R
n是从0至4的整数;
m是从0至3的整数;
p是从0至6的整数;和
q是从0至5的整数;和
L是:
其中:
SP是间隔基团;
R
R
在一些实施方案,A是:
其中:
R
其中R
m是从0至3的整数;
p是从0至6的整数;和
q是从0至5的整数;和
L是:
其中:
SP是间隔基团;
R
R
在一些实施方案,
A是:
其中:
R
n是从0至4的整数;
m是从0至3的整数;
p是从0至6的整数;和
q是从0至5的整数;和
L是:
其中:
SP是间隔基团;和
在一些实施方案,A是:
其中:
R
其中R
n是从0至4的整数;
m是从0至3的整数;
p是从0至6的整数;和
q是从0至5的整数;和
L是:
其中:
SP是间隔基团;和
在一些实施方案,
A是:
其中:
R
n是从0至4的整数;
m是从0至3的整数;
p是从0至6的整数;和
q是从0至5的整数;和
L是:
其中:
SP是:
其中:
b是从2至8的整数。
在一些实施方案,A是:
其中:
R
其中R
n是从0至4的整数;
m是从0至3的整数;
p是从0至6的整数;和
q是从0至5的整数;和
L是:
其中:
SP是:
其中:
b是从2至8的整数。
在一些实施方案,
A是:
其中:
R
n、m、p、和q为0、1、或2;和
L是
其中:
b是从2至8的整数。
在一些实施方案,A是:
其中:
R
其中R
n是从0至4的整数;
m是从0至3的整数;
p是从0至6的整数;和
q是从0至5的整数;和
L是
其中:
b是从2至8的整数。
在一些实施方案,
A是:
其中
R
n是0、1、或2;和
L是:
其中:
SP是间隔基团;
R
R
在一些实施方案,A是:
其中:
R
其中R
其中n是从0至4的整数;
L是:
其中:
SP是间隔基团;
R
R
在一些实施方案,
A是:
其中
R
n是0、1、或2;和
L是:
其中:
b是从2至8的整数。
在一些实施方案,A是:
其中:
R
其中R
L是:
其中:
其中n是从0至4的整数;和
b是从2至8的整数。
在一些实施方案,
A是:
其中
R
n是0、1、或2;和
L是:
其中
在一些实施方案,A是:
其中:
R
其中R
其中n是从0至4的整数;
L是:
其中
在一些实施方案,
A是:
L是
在一些实施方案,A是:
其中:
R
在一些实施方案,A是:
其中:
R
在一些实施方案,
A是:
L是
在一些实施方案,
BA是抗体,
A是:
其中
R
n是0、1、或2;和
L是:
其中
在一些实施方案,
A是:
其中
R
q是从0至5的整数;和
L是:
其中:
SP是间隔基团;
R
R
在一些实施方案,A是:
其中:
R
其中q是从0至5的整数;
L是:
其中:
SP是间隔基团;
R
R
在一些实施方案,
A是:
其中:
R
其中q是从0至5的整数;和
L是:
其中:
b是从2至8的整数。
在一些实施方案,
A是:
其中:
R
q是从0至5的整数;和
L是:
其中
在一些实施方案,A是:
其中:
R
其中q是从0至5的整数;
L是:
其中
在一些实施方案,
A是:
L是
在一些实施方案,式I所示化合物是:
其中,A是亚芳基或亚杂芳基,L
在一些实施方案,式I所示化合物是:
其中,A是亚芳基或亚杂芳基,L
在一些实施方案,式I所示化合物是:
其中,A是亚芳基或亚杂芳基,L
在一些实施方案,A是:
其中:
R
其中R
n是从0至4的整数;
m是从0至3的整数;
p是从0至6的整数;和
q是从0至5的整数。
在一些实施方案,所述连接体是:
其中:
SP是间隔基团;
AA
AA
所述间隔基团是使所述AA
在一些实施方案,所述间隔基团包含亚烷基。在一些实施方案,所述间隔基团包含C
其中:
b是从2至8的整数。
在一些实施方案,所述间隔基团是:
其中:
在一些实施方案,所述间隔基团是:
其中:
R
R
由
b是从2至8的整数。
在一些实施方案,所述间隔基团是:
其中:
b是从2至8的整数。
在一些实施方案,所述间隔基团是:
其中:
g是从2至20的整数。在一些实施方案,g是2-8。在一些实施方案,g是2、4、6、或8。
在一些实施方案,所述间隔基团是:
在一些实施方案,所述间隔基团是:
在一些实施方案,所述间隔基团是:
在一些实施方案,所述间隔基团是:
在一些实施方案,所述间隔基团是:
在一些实施方案,所述间隔基团是:
在一些实施方案,所述间隔基团是:
在一些实施方案,所述间隔基团是:
在一些实施方案,所述间隔基团是:
其中,
X是N或O;R
在一些实施方案,AA
在一些实施方案,AA
在一些实施方案,式I所示化合物是:
其中X是N或O,
R
b是从1至8的整数,
A是芳基或杂芳基,和
t是1-8的整数。
在一些实施方案,式I所示化合物是:
其中:
Ab是抗体;
k是从1至30的整数;和
t是从1至8的整数。在一些实施例,k是从1至8的整数。在一些实施例,t是从1至4的整数。在一些实施例,当
在一些实施方案,式I所示化合物是:
在一些实施方案,k是从1至30的整数。在一些实施方案,k是从1至8的整数。在一些实施方案,k是从1至6的整数。在一些实施方案,k是从1至4的整数。在一些实施方案,k是从1至3 的整数。在一些实施方案,所述偶联物的药物-抗体比率(DRA)是1.0至3.0。
C.美登素类衍生物
本发明提供了式(II)所示化合物:
或其药学上可接受的盐,
其中A是亚芳基或亚杂芳基。
在一些实施方案,所述这些化合物代表本发明所述偶联物的有效负载部分,并且在将所述偶联物内化入细胞后,例如通过酶蛋白水解进行释放。本发明提供的方法包括治疗增殖性疾病(例如癌症) 的方法,包括向患者施用治疗有效量的偶联物,例如在所述偶联物内化入所述患者中的细胞后,释放式(II) 所示化合物的抗体-药物偶联物。
在一些实施方案,所述这些化合物表示本发明所述偶联物的代谢产物,例如酶蛋白水解产物。在一些实施方案,所述这些化合物表示本发明所述偶联物的分解代谢产物。在一些实施方案,所述这些化合物表示本发明所述偶联物的细胞产物。
在一些实施方案,A是苯、吡啶、萘、或喹诺酮的二价基团,所述苯、吡啶、萘、或喹诺酮是任选取代的。
在一些实施方案,A是亚芳基。
在一些实施方案,A是:
其中:
R
其中R
n是从0至4的整数;
m是从0至3的整数;
p是从0至6的整数;和
q是从0至5的整数。
在一些实施方案,式(II)所示化合物是式(IIA)的化合物:
其中R
在一些实施方案,式(II)所示化合物是式(IIB)的化合物:
其中R
在一些实施方案,式(II)所示化合物是式(IIB2)的化合物:
其中R
在一些实施方案,式(II)所示化合物是式(IIB3)的化合物:
其中R
在一些实施方案,式(II)所示化合物是式(IIC)的化合物:
其中R
在一些实施方案,式(II)所示化合物是式(IID)的化合物:
其中R
在一些实施方案,式(II)所示化合物是式(IIE)的化合物:
其中R
在一些实施方案,式(II)所示化合物是式(IIF)的化合物:
其中R
在一些实施方案,式(II)所示化合物是式(IIG)的化合物:
其中R
在一些实施方案,各R
在一些实施方案,各R
在一些实施方案,式(II)所示化合物是式(IIA)的化合物:
其中:
R
n是0、1、或2。
在一些实施方案,式(II)所示化合物是式(IIB)的化合物:
其中:
R
q是0、1、或2。
在一些实施方案,式(II)所示化合物是:
在一些实施方案,式(II)所示化合物选自下述化合物:
在一些实施方案,式(II)所示化合物是:
在一些实施方案,所述这些化合物表示本发明所述偶联物的有效负载部分,并且在将所述偶联物内化入细胞后,例如通过酶蛋白水解进行释放。本发明提供的方法包括治疗增殖性疾病(例如癌症) 的方法,包括向患者施用治疗有效量的偶联物,例如在所述偶联物内化入所述患者中的细胞后,释放式(II) 所示化合物的抗体-药物偶联物。
在一些实施方案,所述这些化合物表示本发明所述偶联物的代谢产物,例如酶蛋白水解产物。
在一些实施方案,A是苯、吡啶、萘、或喹诺酮的二价基团,所述苯、吡啶、萘、或喹诺酮是任选取代的。
在一些实施方案,A是亚芳基。
在一些实施方案,A是:
其中:
R
其中R
n是从0至4的整数;
m是从0至3的整数;
p是从0至6的整数;和
q是从0至5的整数。
在一些实施方案,式(II)所示化合物是式(IIA)的化合物:
其中R
在一些实施方案,式(II)所示化合物是式(IIB)的化合物:
其中R
在一些实施方案,式(II)所示化合物是式(IIB2)的化合物:
其中R
在一些实施方案,式(II)所示化合物是式(IIB3)的化合物:
其中R
在一些实施方案,各R
在一些实施方案,式(II)所示化合物是:
在一些实施方案,式(II)所示化合物是式(IIH)的化合物:
其中R
在一些实施方案,式(II)所示化合物是选自下述化合物:
在一些实施方案,式(II)所示化合物是选自下述化合物:
D.化合物的制备
式I所示化合物可通过使式P1的化合物与结合剂如抗体在标准偶联条件下(参见如 Doronina et al.,Nature Biotechnology 2003,21,7,778,其通过引用并入本文)进行偶联合成得到。当所述结合剂是抗体时,所述抗体可通过所述抗体的一个或多个半胱氨酸或赖氨酸残基与式P1的化合物偶联。式 P1的化合物可与半胱氨酸残基偶联,譬如,通过使抗体经还原剂,如二硫苏糖醇,来裂解抗体的二硫键,通过例如凝胶过滤来纯化还原的抗体,随后使抗体与含有活性部分如马来酰亚胺基基团的式P1的化合物反应。适宜的溶剂包括,但不限于水、DMA、DMF、和DMSO。含有活性部分如活化酯或酰卤基团的式P1的化合物,可与赖氨酸残基偶联。适宜的溶剂包括,但不限于水、DMA、DMF、和DMSO。式I所示化合物可使用已知蛋白技术,包括譬如体积排阻色谱法、透析法、和超滤/渗滤法进行纯化。
其中RL是活性连接体,A是亚芳基或亚杂芳基,L是连接体,和BA是结合剂。
在一些实施方案,式P1所示化合物包括A,其中A是:
其中n是0或1;和R
在一些实施方案,式P1所示化合物包括A,其中A是:
其中n是0或1;和R
在一些实施方案,式P1所示化合物包括A,其中A是:
其中n是0或1;R
活性连接体是包含其上能够与结合剂反应(例如,与抗体在其半胱氨酸或赖氨酸残基处进行反应)以形成式I所示化合物的结构部分的基团部分。与所述结合剂偶联之后,所述活性连接体成为式I所示化合物的连接体(L)部分。示例性活性连接体包括,但不限于包含卤代乙酰基,异硫氰酸酯,或能够与所述结合剂反应的马来酰亚胺部分的那些。活性部分还包括具有以下结构的基团部分:
其中X是–O–或–NH–,和LG是离去基团,例如Br。
在一些实施方案,所述活性连接体是:
其中:
SP
AA
AA
所述活性间隔基团是包含上述活性连接体部分的基团部分,所述活性连接体部分能够与结合剂反应,并使所述部分与AA
在一些实施方案,所述活性间隔基团包含选自下述的不可分裂基团部分:
在一些实施方案,所述活性间隔基团是:
其中b是从2至8的整数。
在一些实施方案,所述活性间隔基团是:
在一些实施方案,所述间隔基团是
在一些实施方案,所述间隔基团是
在一些实施方案,所述活性间隔基团是:
其中b是从2至8的整数,和g是从2至20的整数。
在一些实施方案,所述活性间隔基团是:
在一些实施方案,AA
在一些实施方案,AA
在一些实施方案,所述活性连接体是:
其中:
SP
R
R
在一些实施方案,所述活性连接体是:
其中:
SP是活性间隔基团。
在一些实施方案,所述活性连接体是:
其中b是从2至8的整数。
在一些实施方案,所述活性连接体是:
其中b是从2至8的整数。
在一些实施方案,所述活性连接体是:
其中b是从2至8的整数。
在一些实施方案,所述活性连接体是:
其中b是从2至8的整数。
在一些实施方案,所述活性连接体是:
其中b是从2至8的整数,R
在一些实施方案,所述活性连接体是:
其中b是从2至8的整数。
在一些实施方案,所述活性连接体是:
其中b是从2至8的整数。
在一些实施方案,所述活性连接体是:
其中b是从2至8的整数;R
在一些实施方案,所述活性连接体是:
其中b是从2至8的整数。
在一些实施方案,所述活性连接体是:
其中g是从2至8的整数。
在一些实施方案,所述活性连接体是:
在一些实施方案,所述活性连接体是:
在一些实施方案,所述活性连接体是:
在一些实施方案,所述活性连接体是:
在一些实施方案,所述活性连接体是:
在一些实施方案,所述活性连接体是:
在一些实施方案,所述活性连接体是:
在一些实施方案,所述活性连接体是:
在一些实施方案,所述活性连接体是:
在一些实施方案,所述活性连接体是:
在一些实施方案,所述活性连接体是:
在一些实施方案,所述活性连接体是:
在一些实施方案,式P1所示化合物是式P1A的化合物:
其中:
A是:
其中:
R
n是从0至4的整数;
m是从0至3的整数;
p是从0至6的整数;和
q是从0至5的整数;
SP
AA
AA
在一些实施方案,式P1A的化合物是包括A的化合物,其中A是:
其中n是0或1;和R
在一些实施方案,式P1A的化合物是包括A的化合物,其中A是:
其中n是0或1;和R
在一些实施方案,式P1A的化合物是包括A的化合物,其中A是:
其中q是从0至5的整数;和R
在一些实施方案,式P1A的化合物是包括A的化合物,其中A是:
其中q是从0至5的整数;和R
在一些实施方案,式P1A的化合物是包括A的化合物,其中A是:
其中n是0或1;和R
在一些实施方案,式P1所示化合物是式P1B的化合物:
其中
A是:
其中:
R
n是从0至4的整数;
m是从0至3的整数;
p是从0至6的整数;和
q是从0至5的整数;和
SP
在一些实施方案,式P1所示化合物是式P1C的化合物:
其中:
SP
AA
AA
R
n是0、1、或2。
在一些实施方案,式P1所示化合物是式P1D的化合物:
其中:
SP
AA
AA
R
n是0、1、或2。在一些实施方案,n是0。在一些实施方案,n是1。在一些实施方案,n是2。
在一些实施方案,式P1所示化合物是式P1D的化合物,其中R
在一些实施方案,式P1所示化合物是式P1D的化合物,其中R
在一些实施方案,式P1所示化合物是式P1E的化合物:
其中:
SP
R
R
R
n是0、1、或2。
在一些实施方案,式P1所示化合物是式P1F的化合物:
其中:
SP
R
n是0、1、或2。
在一些实施方案,式P1所示化合物是式P1G的化合物:
其中:
SP
R
在一些实施方案,式P1所示化合物是式P1H的化合物:
其中:
R
b是从2至8的整数。
在一些实施方案,式P1所示化合物是式P1I的化合物:
其中:
R
b是从2至8的整数。
在一些实施方案,式P1所示化合物是式P1J的化合物:
其中:
R
b是从2至8的整数。
在一些实施方案,式P1所示化合物是式P1K的化合物:
其中R
在一些实施方案,式P1所示化合物是式P1L的化合物:
其中R
在一些实施方案,式P1所示化合物是式P1M的化合物:
其中R
在一些实施方案,式P1所示化合物是式P1N的化合物:
其中R
在一些实施方案,式P1所示化合物是式P1O的化合物:
其中R
在一些实施方案,式P1所示化合物是式P1P的化合物:
其中R
在一些实施方案,式P1所示化合物是式P1Q的化合物:
其中R
在一些实施方案,式P1所示化合物是式P1R的化合物:
其中R
在一些实施方案,式P1所示化合物是式P1S的化合物:
其中R
在一些实施方案,式P1所示化合物是式P1T的化合物:
其中R
在一些实施方案,式P1所示化合物是式P1U的化合物:
其中R
在一些实施方案,式P1所示化合物是式P1V的化合物:
其中R
在一些实施方案,式P1所示化合物是式P1W的化合物:
其中R
在一些实施方案,式P1所示化合物是式P1X的化合物:
其中:
R
在一些实施方案,式P1所示化合物是式P1Y的化合物:
其中,R
在一些实施方案,式P1所示化合物是式P1Z的化合物:
其中R
在一些实施方案,式P1所示化合物是具有以下结构之一的化合物:
在一些实施方案,式P1所示化合物是具有以下结构之一的化合物:
式P1所示化合物可在酰胺合成条件下,使式P2的化合物与式P3的化合物反应,从而合成得到。合适的酰胺合成条件包括,但不限于,在羧酸活化剂和碱存在下,使与式P2的化合物接触。适宜的活化剂包括,但不限于EDC、HATU、HBTU、DCC、BOP、和EEDQ。适宜的碱包括,但不限于DIEA、 DBU、三丁胺、和2,6-二甲基吡啶。
式P2的化合物可使用已知技术(参见,如美国专利号4,308,269,其通过引用并入本文) 直接由美登醇与丙氨酸合成得到。
式I所示化合物可通过在酰胺合成条件下,使式PP3所示化合物:
与式PP4所示化合物进行偶联,合成得到:
其中:
BA是结合剂;
SP是间隔基团;
R
R
A是亚芳基或亚杂芳基;和
k是从1至10的整数。
式PP3所示化合物可通过使式PP5所示化合物与合适的还原剂接触,合成得到:
其中A是亚芳基或亚杂芳基。
在一些实施方案,所述合适的还原剂包括金属、金属箔、金属粉、金属汞合金、或金属屑。在一些实施方案,所述金属选自锌、铁、铝、钯、或雷尼镍。
譬如,在一些实施方案,可采用下述还原剂条件。关于化合物PP5的量,譬如,在本发明的一些方法中,将约20当量的锌粉和40当量的乙酸进行组合。在一些实施例,还原反应是在室温下进行约1至20小时。在所述这些实施例中的一些,前述乙酸可被另一合适的弱酸或质子供体替换。合适的弱酸或质子供体的实例包括,但不限于甲酸、pTsOH、和NH
合适的酸包括,但不限于乙酸。
在一些实施方案,A是:
其中:
R
其中R
n是从0至4的整数;
m是从0至3的整数;
p是从0至6的整数;和
q是从0至5的整数。
在一些实施方案,式PP5所示化合物是式PP5A的化合物:
其中R
在一些实施方案,各R
在一些实施方案,式PP5所示化合物是式PP5A的化合物:
其中:
R
n是0、1或2。
在一些实施方案,式PP5所示化合物是式PP5A2的化合物:
其中:
R
q是从0至5的整数。
在一些实施方案,式PP5所示化合物是式PP5A3的化合物:
其中:
R
q是从0至5的整数。在一些实施方案,R
在一些实施方案,式PP5所示化合物是式PP5A4的化合物:
其中R
式PP5所示化合物可通过在酰胺合成条件下,使式P2所示化合物与式PP6所示化合物接触,合成得到:
合适的式PP6的化合物包括,但不限于,3-硝基-苯甲酸,3-氯-5-硝基-苯甲酸,3-氟-5-硝基-苯甲酸,3-硝基-1-萘羧酸,2-氟-5-硝基-苯甲酸,3-(二甲氨基)-5-硝基-苯甲酸,3-乙氧基-5-硝基-苯甲酸, 2-甲氧基-5-硝基-苯甲酸,4-甲氧基-3-硝基-苯甲酸,2,6-二氟-3-硝基-苯甲酸,2-氯-6-氟-3-硝基-苯甲酸,6- 氯-2-氟-3-硝基-苯甲酸,2-氯-4-氟-5-硝基-苯甲酸,4-氯-2-氟-5-硝基-苯甲酸,2-乙氧基-5-硝基-苯甲酸,2-(甲氨基)-3-硝基-苯甲酸,6-硝基-8-喹啉羧酸,4-(二甲氨基)-3-硝基-苯甲酸盐酸盐(1:1),2-甲基-硝基-苯甲酸,3-甲基-4-硝基-苯甲酸,4-硝基-1-萘羧酸,2,6-二甲基-4-硝基-苯甲酸,3-氟-4-硝基-苯甲酸,3-氯-4-硝基-苯甲酸,3-溴-4-硝基-苯甲酸,3-氰基-4-硝基-苯甲酸,3-环丙基-4-硝基-苯甲酸,3-甲氧基-4-硝基-苯甲酸,2-甲氧基-4-硝基-苯甲酸,5-氯-2-甲基-4-硝基-苯甲酸,8-硝基-5-异喹啉羧酸,5-硝基-8-喹啉羧酸,8- 硝基-5-喹啉羧酸,2,5-二氟-4-硝基-苯甲酸,2-(二甲氨基)-4-硝基-苯甲酸,2-氯-5-氟-4-硝基-苯甲酸,3-(二甲氨基)-4-硝基-苯甲酸,2-[(1-甲基乙基)巯基]-4-硝基-苯甲酸,4-硝基-3-(三氟甲基)-苯甲酸,4-硝基-2-(三氟甲基)-苯甲酸,3,5-二甲氧基-4-硝基-苯甲酸,4-硝基-2-(丙氨基)-苯甲酸,3-(二氟甲氧基)-4-硝基-苯甲酸, 2-(2-氟-苯基)-4-硝基-苯甲酸,4-硝基-2-(4-吡啶基)-苯甲酸,4-硝基-3-(4-吡啶基)-苯甲酸,或4-硝基-2-(1- 吡咯烷基)-苯甲酸。
合适的式PP6的化合物包括具有任一以下结构通式的化合物:
其中,R
在一些实施方案,R
在一些实施方案,本发明提供了式PP5所示化合物:
其中A是亚芳基或亚杂芳基。
在一些实施方案,式PP5所示化合物是选自下述化合物:
在一些实施方案,式PP5所示化合物是:
式III所示化合物:
可通过在偶联条件下,使式PP1所示化合物:
与结合剂接触,从而合成得到,
其中:
BA是结合剂;
SP是间隔基团;
SP
R
R
A是亚芳基或亚杂芳基;和
k是从1至30的整数。
式PP1所示化合物可通过使式PP2所示化合物与式P2所示化合物接触制备得到:
其中:
SP
R
R
A是亚芳基或亚杂芳基。
式PP1所示化合物可通过使式PP3所示化合物与式PP7所示化合物接触制备得到:
其中:
SP
R
R
A是亚芳基或亚杂芳基。
式PP2所示化合物可通过使式PP8所示化合物与双官能团间隔基团接触制备得到:
式PP7所示化合物可通过使式PP9所示化合物与双官能团间隔基团接触制备得到:
双官能团间隔基团是与式PP3所示化合物反应,从而附加存在于式PP2所示化合物中的SP
式PP8所示化合物可通过使式PP10所示化合物与式PP11所示化合物接触,然后除去保护基团,制备得到:
其中PG是胺保护基团,和Y是使其连接至亲电羰基的基团部分。式PP10所示化合物可通过使用标准氨基酸偶联技术,包括譬如,在DIEA、DBU、或三丁胺存在下,使用HATU、BOP/HOBt、或EDC/N-羟基琥珀酰胺形成活性酯,偶联其相应氨基酸制备得到。
双官能团间隔基团是与式PP9所示化合物反应,从而附加存在于式PP7所示化合物中的SP
式(I)所示的抗体-药物-偶联物化合物还可通过在转谷氨酰胺酶存在下,使合适的抗体如去糖基化抗体或无糖基化抗体,与式(PT1)所示化合物反应制备得到:
其中:
A是亚芳基或亚杂芳基;和
L是连接体。
在一些实施方案,A是:
其中:
R
其中R
n是从0至4的整数;
m是从0至3的整数;
p是从0至6的整数;和
q是从0至5的整数。
在一些实施方案,A是:
其中:
R
其中R
n是从0至4的整数;
m是从0至3的整数;
p是从0至6的整数;和
q是从0至5的整数。
在一些实施方案,各R
在一些实施方案,R
在一些实施方案,所述连接体包含一个或多个氨基酸。合适的氨基酸包括天然、非天然的、标准、非标准的、蛋白原、非蛋白原的、和L-或D-型的α-氨基酸。在一些实施方案,所述连接体包含丙氨酸、缬氨酸、亮氨酸、异亮氨酸、蛋氨酸、色氨酸、苯丙氨酸、脯氨酸、丝氨酸、苏氨酸、半胱氨酸、酪氨酸、天冬酰胺、谷氨酰胺、天冬氨酸、谷氨酸、赖氨酸、精氨酸、组氨酸、或瓜氨酸,或其衍生物。
在一些实施方案,所述连接体包含缬氨酸和瓜氨酸。
在一些实施方案,所述连接体是:
其中:
一个
另一个
AA
AA
所述连接体可进一步包含将所述AA
PT1包括伯胺封端的亚烷基或伯胺封端的聚乙二醇。所述伯胺末端基团可在转谷氨酰胺酶存在下,直接与去糖基化抗体或无糖基化抗体结合。
在一些实施方案,所述化合物包含伯胺封端的亚烷基。在一些实施方案,所述化合物包含 NH
其中:
b是从2至8的整数。
在一些实施方案,所述化合物包含:
其中:
在一些实施方案,PT1的化合物是
在一些实施方案,式(I)所示化合物是通过在转谷氨酰胺酶存在下,在适合于转谷氨酰胺酶反应的条件下使结合剂与PT1接触,从而制备得到。在一些实施方案,所述转谷氨酰胺酶反应是在pH 值为约7和约8之间进行4小时。在一些实施例,所述pH值是7.2、7.3、7.4、7.5、7.6、7.8、或8。
在一些实施方案,式(I)所示化合物是通过转谷氨酰胺酶反应制备得到,其中式(PT1) 所示化合物的浓度与去糖基化抗体或无糖基化抗体相比,是至少30摩尔当量的浓度。在一些实施方案,式(I)所示化合物是通过转谷氨酰胺酶反应制备得到,其中式(PT1)所示化合物的浓度与去糖基化抗体或无糖基化抗体相比,是30至150摩尔当量的浓度。
在一些实施方案,式(I)所示化合物是通过转谷氨酰胺酶反应制备得到,其中式(PT1) 所示化合物的浓度是1至30U每毫克的去糖基化抗体或无糖基化抗体。
在一些实施方案,在转谷氨酰胺酶反应之前,所述抗体用肽N-糖苷酶F(PNGaseF)进行去糖基化。
在一些实施方案,所述抗体是无糖基化的。无糖基化抗体可通过诱变技术,以除去所述抗体糖基化所需的一个或多个氨基酸序列,从而制备得到。在一些实施方案,所述抗体包含具有将另一氨基酸替换为N180的突变的重链。在一些实施方案,所述无糖基化抗体包含一个或多个N180Q重链多肽。
在一些实施方案,式(I)所示化合物是通过在选自由水、缓冲水、盐水、缓冲盐水、和有机物组成的组中的一种或多种溶剂中进行的转谷氨酰胺酶反应制备得到。
在一些实施方案,式(I)所示化合物是通过在用磷酸盐、HEPES、或MOPS缓冲的水中进行的转谷氨酰胺酶反应制备得到。
在一些实施方案,式(I)所示化合物是通过转谷氨酰胺酶反应制备得到,所述转谷氨酰胺酶反应包括使谷氨酰胺酰基修饰的抗体与活性间隔基团化合物反应,以形成抗体-间隔基团偶联物;然后使所述抗体-间隔基团偶联物与活性有效负载化合物反应,以形成抗体-间隔基团-有效负载偶联物。
在一些实施方案,本发明提供了由本发明所述方法生产得到的谷氨酰胺酰基修饰的抗体。
在一些实施方案,本发明提供了药物组合物,其包含由本发明所述方法生产得到的谷氨酰胺酰基修饰的抗体。
在一些实施方案,本发明提供了在有需要的受试者中治疗疾病/病症的方法,其包含对所述受试者施用药学上可接受的量的本发明所述抗体或抗体-药物-偶联物。
在一些实施方案,本发明提供了本发明所述抗体或抗体-药物-偶联物用于治疗。
在一些实施方案,本发明提供了本发明所述抗体或抗体-药物-偶联物用于治疗癌症。
E.使用方法和药物组合物
本发明包括治疗或预防疾病、病情、或病症,譬如,增殖性疾病如癌症的方法,其包含施用治疗有效量的一种或多种本发明化合物,譬如,一种或多种式(I)或(II)所示化合物。疾病、病症、和/或病情包括,但不限于与本发明所列抗原相关的那些。在一些实施例,所述抗原是PSMA、MUC16、或EGFRvIII。
本发明公开化合物可用于治疗脑和脑膜,口咽,肺和支气管树,胃肠道,男性和女性生殖道,肌肉,骨骼,皮肤及附属物,结缔组织,脾脏,免疫系统,造血细胞和骨髓,肝脏和尿道,以及特殊的感觉器官如眼中产生的原发性和/或转移性肿瘤。在一些实施方案,本发明所述化合物用于治疗一种或多种下述癌症:肾细胞癌,胰腺癌,头颈癌,前列腺癌,恶性胶质瘤,骨肉瘤,结直肠癌,胃癌(例如,具有MET扩增的胃癌),恶性间皮瘤,多发性骨髓瘤,卵巢癌,小细胞肺癌,非小细胞肺癌,滑膜肉瘤,甲状腺癌,乳腺癌,或黑素瘤。在一些实施方案,所述癌症是乳腺癌。
本发明所述化合物可单独给药或与一种或多种附加治疗剂一同给药。所述一种或多种附加治疗剂可在本发明所述化合物给药之前、同时给药,或给药不久之后进行给药。本发明还包括药物组合物,其包含本发明所述的任一化合物与一种或多种附加治疗剂组合使用/联用,以及治疗方法,其包含将此类组合施用于有需要的患者。
合适的附加治疗剂包括,但不限于:EGFR拮抗剂(例如,抗EGFR抗体[如西妥昔单抗或帕尼单抗]或EGFR的小分子抑制剂[例如,吉非替尼或厄洛替尼]),另一EGFR家族成员如Her2/ErbB2、 ErbB3或ErbB4的拮抗剂(例如,抗ErbB2[如曲妥单抗或T-DM1{
合适的治疗剂还包括,但不限于化学治疗剂类,包括烷化剂如噻替派和环磷酰胺(Cytoxan
本发明所述化合物还可与抗病毒剂类、抗生素类、镇痛剂类、皮质类固醇类、类固醇类、氧、抗氧化剂类、COX抑制剂类、心脏保护剂类、金属螯合剂类、IFN-γ、和/或NSAID类组合/联合施用和/或共同配制。
在本发明所述方法的一些实施方案中,多剂量的本发明化合物(或包含本发明所述化合物与本发明提及的任何附加治疗剂的组合的药物组合物)可在规定时间内对受试者进行给药。本发明此方面的方法包括向受试者顺序施用多剂量的本发明所述化合物。本发明使用的“依次施用/给药”是指每种剂量的所述化合物在不同的时间点,例如在以预定间隔(如几小时、几天、几周、或几个月)分开的不同天数,对受试者进行给药。本发明包括方法,其包含对患者依次施用单个初始剂量的本发明所述化合物,随后施用一个或多个二次剂量的所述化合物,以及任选地后续一个或多个三次剂量的化合物。
术语“初始剂量”,“二次剂量”,和“三次剂量”是指本发明所述化合物的给药时间顺序。因此,“初始剂量”是在治疗方案开始时给药的剂量(也称为“基线剂量”);“二次剂量”是在所述初始剂量给药后施用的剂量;和“三次剂量”是在所述二次剂量给药后施用的剂量。所述初始、二次和三次剂量均可含有相同量的本发明化合物,但通常可在给药频率方面彼此不同。在一些实施方案,在治疗过程中,所述初始、二次和/或三次剂量中所含的化合物的量彼此变化(例如,酌情向上或向下调整)。在一些实施方案,在治疗方案开始时,将两种或更多种(例如,2、3、4或5种)剂量施用为“负荷剂量”,随后以较不频繁的基础施用后续剂量(例如“维持剂量”)。
在本发明所述的一些示例性实施方案中,每个二次和/或三次剂量在紧随前述剂量之后进行给药1至26周(例如,1、1
本发明此方面的方法可包含向患者施用任何数量的二次和/或三次剂量的本发明化合物。譬如,在一些实施方案,仅向患者施用单次二次剂量。在其他实施方案,向患者施用两次或更多次(例如,2、3、4、5、6、7、8或更多次)二次剂量。同理,在一些实施方案,仅向患者施用单次的三次剂量。在其他实施方案,向患者施用两次或更多次(例如,2、3、4、5、6、7、8或更多次)三次剂量。给药方案可在特定受试者的寿命期内无限期地进行,或者直至此种治疗不再是治疗需要或有利的。
在涉及多个二次剂量的实施方案中,每个二次剂量可以与其他二次剂量相同的频率施用/ 给药。譬如,每个二次剂量可在紧随前述剂量之后,对患者进行给药1至2周或1至2个月。同理,在涉及多个三次剂量的实施方案中,每个三次剂量可以与其他三次剂量相同的频率施用/给药。譬如,每个三次剂量可在紧随前述剂量之后,对患者进行给药2至12周。在本发明的一些实施方案中,向患者施用二次和/或三次剂量的频率可在治疗方案的过程中变化。也可根据临床检查后的个体患者的需要,由医师在治疗过程中调整给药频率。
本发明包括其中以第一频率(例如,每周一次,每两周一次,每三周一次,每月一次,每两个月一次等)向患者施用2至6次负荷剂量,然后以较不频繁的方式向患者施用两次或更多次维持剂量的给药方案。例如,根据本发明的此方面,如果以每月一次的频率施用负荷剂量,那么维持剂量可以每6 周施用一次,每两个月一次,每三个月一次等。
本发明包括本发明所述化合物和/或偶联物如式(I)和(II)所示化合物的药物组合物,譬如,组合物包含本发明所述化合物、其盐、立体异构体、多晶型物,和药学上可接受的载体、稀释剂、和 /或辅料。合适的载体、稀释剂和辅料的实例包括,但不限于:用于维持适当组合物pH值的缓冲剂类(例如,柠檬酸盐缓冲液、琥珀酸盐缓冲液、乙酸盐缓冲液、磷酸盐缓冲液、乳酸盐缓冲液、草酸盐缓冲液等),载体蛋白类(如人血清白蛋白),盐水,多元醇类(例如,海藻糖、蔗糖、木糖醇、山梨糖醇等),表面活性剂类(例如,聚山梨醇酯20、聚山梨醇酯80、聚氧乙烯酸酯(polyoxolate)等),抗微生物剂类,和抗氧化剂类。
F.实施例
质子NMR光谱是在Varian Inova 300或500MHz仪器上获得,而质谱是在Agilent1100或 1200系列LC/MSD上使用电喷雾离子源和单四极或离子阱分析仪采集得到。酶测定中的某些连接体有效负载是通过Waters Xevo TQ-S质谱仪进行分析。除非另有说明,所有原料和溶剂均是商业购买的,且不经纯化即可使用。
实施例1
化合物10是由化合物1通过如下所述和如图.1所示方法合成得到。
步骤A:向圆底烧瓶中称取Boc-L-缬氨酸(1.03g,4.74mmol),N-羟基琥珀酰亚胺(1.22 g,10.6mmol)和EDC(1.60g,8.35mmol)。将试剂溶解在干燥DCM(30mL)中,烧瓶通过橡胶隔片密封,通入氩气排气,然后将反应物在环境温度下搅拌反应。3天后,通过TLC(用茚三酮着色后)检测分析发现没有Boc-缬氨酸剩余,反应物用水和饱和NaHCO
将前述步骤得到的Boc-L-缬氨酸-琥珀酸酯(1)(1.50g,4.77mmol)溶于乙腈(MeCN, 15mL)中,然后用L-瓜氨酸(2,1.07g,6.11mmol)的水(9mL)溶液和饱和NaHCO
步骤B:向圆底烧瓶中称取前述步骤所得产物(3,152mg,0.406mmol),4-氨基苯甲酸叔丁酯(4,150mg,0.776mmol),和1-[双(二甲氨基)亚甲基]-1H-1,2,3-三氮唑并[4,5-b]吡啶鎓3-氧化六氟磷酸盐(HATU,488mg,1.28mmol),并溶于无水N,N-二甲基甲酰胺(DMF,3mL)。向所述反应中加入N,N-二异丙基乙胺(DIEA,0.25mL,1.44mmol),烧瓶通过橡胶隔片密封,用氩气排气,反应物在环境温度下搅拌反应。18小时后,反应物通过ISCO方法(梯度洗脱:20-80%MeCN的水溶液,0.05%乙酸,洗脱20分钟)直接在100g C18 RediSep金色(Gold)柱上纯化。合并含产物的级分,部分真空浓缩,残留物在干冰上冷冻,冻干过夜,得到不纯的白色固体(115mg)。将其溶于DCM中,并通过ISCO (梯度洗脱:用0-10%甲醇的DCM洗脱12分钟)在12g硅胶RediSep柱上再次纯化,并将较慢洗脱得到的产物级分蒸发,残留物真空干燥,得到标题化合物为浅黄色固体(65mg,29%)。MS(ESI,pos.):计算值C
步骤C:标题化合物是采用Mehta等(Tet.Lett.1992,33,5441-5444)报道的方法制备得到。在圆底烧瓶中,将前述步骤所得产物(5,61mg,0.111mmol)溶于干燥DCM(3mL),并用三氟乙酸 (TFA,110uL,1.44mmol)和三乙基硅烷[TES(Et
步骤D:将前述步骤所得产物(6,55mg,0.108mmol)溶于水(3mL)中,用饱和NaHCO
步骤E:向圆底烧瓶中称取前述步骤所得产物(8,33mg,0.056mmol),HATU(33mg,0.087mmol),和美登素-N-甲基-L-丙氨酸(9,使用美国专利申请US 2007/0037972 A1中所述方法由美登醇制备得到为金色固体,25mg,0.038mmol),溶于无水DMF(2mL)中,并用DIEA(20uL,0.115mmol) 处理。烧瓶通过橡胶隔片密封,用氩气排气,反应物在环境温度下搅拌20小时。将反应体系用水(1mL) 稀释,并通过ISCO(梯度洗脱:20-80%MeCN的水溶液,0.05%乙酸,洗脱12分钟)直接在50g C18 Aq RediSep金色柱上纯化。将产物级分合并,部分真空浓缩,残留物在干冰上冷冻,并冻干过夜,得到标题化合物为白色固体(8mg,17%)。MS(ESI,pos.):计算值C
实施例2
化合物15是由如下所述和如图.6所示方法合成得到。
步骤A:按照Wipf&Heimgartner(Helv.Chim.Acta,1998,71,140-154)的方法,将Boc-L- 缬氨酸-L-瓜氨酸(3,155mg,0.414mmol)和二环己基碳二亚胺(DCC,95mg,0.460mmol)溶于干燥二氯甲烷(DCM,3mL)中,所得混合物冷却至0℃,搅拌5分钟。然后加入(+)-樟脑-10-磺酸(CSA,15 mg,0.065mmol)和4-氨基-2-氟苯甲酸叔丁酯(99mg,0.469mmol),反应物在搅拌反应3天的过程中缓慢升温至环境温度。LCMS分析显示在m/z566(ESI,neg.)处有较大峰。反应物用DCM稀释,并用10%v/v HCl、水和饱和NaHCO
步骤B:标题化合物是由前述步骤所得产物(12,94mg,0.166),采用实施例1中步骤C 所述方法制备得到的类白色固体(112mg)。MS(ESI,pos.):计算值C
步骤C:标题化合物是由前述步骤所得产物(13,106mg,0.166mmol),采用实施例1中步骤D所述方法制备得到的白色固体(92mg),其通过LCMS分析,纯度仅为70%,但无需进一步纯化,直接用于下一步反应。MS(ESI,pos.):计算值C
步骤D:标题化合物是由前述步骤所得产物(14,50mg,0.077mmol)和美登素-N-甲基 -L-丙氨酸(9,50mg,0.077mmol),采用实施例1中步骤E所述方法制备得到的白色固体(18mg),其通过LCMS分析,纯度仅为55%。使用Phenomenex Gemini C185u,30x150mm柱(20-80%,随后40-60%的MeCN的水溶液,0.1%HOAc两相,洗脱20分钟,30mL/min)通过HPLC纯化两次,得到标题化合物为白色固体(3mg,3%)。MS(ESI,pos.):计算值C
实施例3
化合物20是由如下所述和如图.7所示方法合成得到。
步骤A:标题化合物是由Boc-L-缬氨酸-L-瓜氨酸(3,175mg,0.467mmol)和4-氨基-2- 三氟甲基苯甲酸叔丁酯(150mg,0.574mmol),采用Wipf&Heimgartner(Helv.Chim.Acta,1998,71,140-154) 的方法制备得到的白色固体(77mg,27%)。MS(ESI,neg.):计算值C
步骤B:标题化合物是由前述步骤所得产物(17,67mg,0.108mmol),采用实施例1中步骤C所述方法制备得到的类白色固体(77mg)。MS(ESI,pos.):计算值C
步骤C:标题化合物是由前述步骤所得产物(18,75mg,0.108mmol),采用实施例1中步骤D所述方法制备得到的白色固体(47mg,66%)。MS(ESI,pos.):计算值C
步骤D:标题化合物是由前述步骤所得产物(19,34mg,0.052mmol)和美登素-N-甲基 -L-丙氨酸(9,34mg,0.052mmol),采用实施例1中步骤E所述方法制备,之后使用ISCO(100g C18 Aq金色柱,30-70%MeCN的水溶液,0.05%HOAc两相,洗脱15min,50mL/min)进行二次纯化得到的白色固体(11mg,16%)。MS(ESI,pos.):计算值C
实施例4
化合物25是由如下所述和如图.8所示方法合成得到。
步骤A:标题化合物是由Boc-L-缬氨酸-L-瓜氨酸(3,143mg,0.382mmol)和4-氨基-2- 甲氧基苯甲酸叔丁酯(109mg,0.488mmol),采用Wipf&Heimgartner(Helv.Chim.Acta,1998,71,140-154) 的方法制备得到的白色固体(92mg,42%)。MS(ESI,neg.):计算值C
步骤B:标题化合物是由前述步骤所得产物(22,90mg,0.155),采用实施例1中步骤C 所述方法制备得到的浅白色固体(99mg),将其用DCM研磨两次,然后溶于MeCN和THF中,过滤,真空蒸发溶剂,得到标题化合物为类白色固体(79mg,95%)。MS(ESI,pos.):计算值C
步骤C:标题化合物是由前述步骤所得产物(23,76mg,0.141mmol),采用实施例1中步骤D所述方法制备得到的白色固体(50mg,57%)。MS(ESI,pos.):计算值C
步骤D:标题化合物是由前述步骤所得产物(24,49mg,0.079mmol)和美登素-N-甲基 -L-丙氨酸(9,34mg,0.052mmol),采用实施例1中步骤E所述方法制备得到的白色固体(34mg,34%)。 MS(ESI,pos.):计算值C
实施例5
化合物27是由化合物26通过如下所述和如图.9所示方法合成得到。
向干燥的圆底烧瓶中称取美登素-N-甲基-L-丙氨酸(9,96mg,0.15mmol),4-硝基苯甲酸(26)(42mg,0.25mmol)和HATU(0.12g,0.31mmol)。将试剂溶于无水DMF(3.0mL)中,并用DIEA(0.10mL,0.57mmol)处理,烧瓶用氩气排气并用橡胶隔片密封。反应物在环境温度下搅拌反应 3天,之后LCMS分析显示美登素-N-甲基-L-丙氨酸(Maytan-NMA)转化完毕,因此用几毫升水稀释,直接在100g C18 Aq金色柱上(梯度洗脱:20-80%MeCN的水溶液,0.05%乙酸,洗脱15分钟)纯化。合并最纯净的产物级分,部分真空浓缩,残留物在干冰上冷冻,并冻干过夜,得到标题化合物为黄色固体(64 mg,54%)。MS(ESI,pos.):计算值C
将前述步骤所得产物(63mg,0.079mmol)和锌粉(<10um,98+%纯度,108mg,1.65mmol)溶于/悬浮于THF(4mL)和水(1mL)的混合物中。向所得混合物中加入乙酸(0.180mL,3.14 mmol),烧瓶用橡胶隔片密封,并将反应物在环境温度下搅拌1小时。粗品混合物的LCMS显示已完全转化,因此将反应物经硅藻土过滤,用MeCN洗,真空浓缩滤液。粗产物直接在50g C18Aq金色柱上(梯度洗脱液:20-80%MeCN的水溶液,0.05%乙酸,洗脱12分钟)纯化。合并最纯净的产物级分,部分真空浓缩,残留物在干冰上冷冻,并冻干过夜,得到46mg白色固体,LCMS显示纯度仅为88%。将其溶于 1:1MeCN/水(3mL)中,并使用PhenomenexGemini C18 5u,30x150mm柱进行两次注射(40-80%和30-70%MeCN的水溶液,0.05%HOAc两相,洗脱20分钟,30mL/min),将最纯净的级分浓缩,所得产物冷冻,并如上所述冻干,得到标题化合物为白色固体(31mg,48%)。MS(ESI,pos.):计算值C
实施例6
化合物29是由化合物28通过如下所述和如图.10所示方法合成得到。
标题化合物是由美登素-N-甲基-L-丙氨酸(9,40mg,0.056mmol)和2-氟-4-硝基苯甲酸 (28)(26mg,0.140mmol),采用实施例5中步骤A所述方法制备得到的浅黄色固体(16mg,35%)。 MS(ESI,pos.):计算值C
标题化合物是由前述步骤所得产物(15mg,0.018mmol),采用实施例5中步骤B所述方法制备得到的白色固体(8mg,50%)。MS(ESI,pos.):计算值C
1H),2.83(s,3H),2.66(dd,1H,J=15Hz,13Hz),2.19(dd,1H,J=15Hz,3Hz),1.67(s,3H),1.63(m,1H), 1.51-1.45(m,2H),1.43(d,3H,J=7Hz),1.30(d,3H,J=7Hz),1.27(m,1H),0.84(s,3H)。
实施例7
化合物31是由如下所述和如图.11所示方法合成得到。
标题化合物是由美登素-N-甲基-L-丙氨酸(9,68mg,0.105mmol)和2-三氟甲基-4-硝基苯甲酸(30)(37mg,0.157mmol),采用实施例5中步骤A所述方法制备得到的浅黄色固体(82mg, 90%)。MS(ESI,pos.):计算值C
标题化合物是由前述步骤所得产物(79mg,0.091mmol),采用实施例5中步骤B所述方法制备得到的白色固体(29mg,35%)。MS(ESI,pos.):计算值C
实施例8
化合物33是由如下所述和如图.12所示方法合成得到。
标题化合物是由美登素-N-甲基-L-丙氨酸(9,68mg,0.105mmol)和2-甲氧基-4-硝基苯甲酸(32)(32mg,0.162mmol),采用实施例5中步骤A所述方法制备得到的浅黄色固体(78mg,90%)。 MS(ESI,pos.):计算值C
标题化合物是由前述步骤所得产物(75mg,0.090mmol),采用实施例5中步骤B所述方法制备得到的白色固体(62mg,79%)。MS(ESI,pos.):计算值C
实施例9
化合物35是由如下所述和如图.13所示方法合成得到。
标题化合物是由美登素-N-甲基-L-丙氨酸(9,46mg,0.071mmol)和3-三氟甲基-4-硝基苯甲酸(34)(25mg,0.106mmol),采用实施例5中步骤A所述方法制备得到的浅黄色固体(37mg, 61%)。MS(ESI,pos.):计算值C
标题化合物是由前述步骤所得产物(36mg,0.042mmol),采用实施例5中步骤B所述方法制备得到的白色固体(17mg,46%)。MS(ESI,pos.):计算值C
实施例10
化合物37是由如下所述和如图.14所示方法合成得到。
标题化合物是由美登素-N-甲基-L-丙氨酸(9,46mg,0.071mmol)和
标题化合物是由前述步骤所得产物(36mg,0.042mmol),采用实施例5中步骤B所述方法制备得到的白色固体(17mg,49%)。MS(ESI,pos.):计算值C
实施例11
化合物39是由如下所述和如图.16所示方法合成得到。
标题化合物是由美登素-N-甲基-L-丙氨酸(9,48mg,0.074mmol)和2,5-二氟-4-硝基苯甲酸(38,27mg,0.133mmol),采用实施例5中步骤A所述方法制备得到的黄色固体(37mg,60%)。 MS(ESI,pos.):计算值C
标题化合物是由前述步骤所得产物(36mg,0.043mmol),采用实施例5中步骤B所述方法制备得到的白色固体(22mg,59%)。MS(ESI,pos.):计算值C
实施例12
化合物41是由如下所述和如图.17所示方法合成得到。
标题化合物是由美登素-N-甲基-L-丙氨酸(9,47mg,0.072mmol)和3-氟-4-硝基苯甲酸 (40)(24mg,0.130mmol),采用实施例5中步骤A所述方法制备得到的黄色固体(40mg,68%)。 MS(ESI,pos.):计算值C
标题化合物是由前述步骤所得产物(39mg,0.048mmol),采用实施例5中步骤B所述方法制备得到的白色固体(24mg,60%)。MS(ESI,pos.):计算值C
实施例13
化合物43是由如下所述和如图.18所示方法合成得到。
标题化合物是由美登素-N-甲基-L-丙氨酸(9,45mg,0.069mmol)和3-氯-4-硝基苯甲酸 (42)(26mg,0.129mmol),采用实施例5中步骤A所述方法制备得到的黄色固体(36mg,62%)。 MS(ESI,pos.):计算值C
标题化合物是由前述步骤所得产物(35mg,0.042mmol),采用实施例5中步骤B所述方法制备得到的白色固体(24mg,67%)。MS(ESI,pos.):计算值C
实施例14
化合物45是由如下所述和如图.19所示方法合成得到。
标题化合物是由美登素-N-甲基-L-丙氨酸(9,45mg,0.069mmol)和5-硝基-8-羧基喹啉(44)(24mg,0.110mmol),采用实施例5中步骤A所述方法制备得到的浅黄色固体(26mg,44%)。 MS(ESI,pos.):计算值C
标题化合物是由前述步骤所得产物(25mg,0.029mmol),采用实施例5中步骤B所述方法制备得到的浅黄色固体(8mg,31%)。MS(ESI,pos.):计算值C
实施例15
化合物47是由如下所述和如图.20所示方法合成得到。
标题化合物是由美登素-N-甲基-L-丙氨酸(9,49mg,0.075mmol)和3-溴-4-硝基苯甲酸 (46)(30mg,0.122mmol),采用实施例5中步骤A所述方法制备得到的黄色固体(38mg,58%)。 MS(ESI,pos.):计算值C
标题化合物是由前述步骤所得产物(37mg,0.042mmol),采用实施例5中步骤B所述方法制备得到的白色固体(28mg,74%)。MS(ESI,pos.):计算值C
实施例16
化合物49是由如下所述和如图.21所示方法合成得到。
标题化合物是由美登素-N-甲基-L-丙氨酸(9,49mg,0.075mmol)和3-甲氧基-4-硝基苯甲酸(48)(23mg,0.117mmol),采用实施例5中步骤A所述方法制备得到的黄色固体(34mg,55%)。 MS(ESI,pos.):计算值C
标题化合物是由前述步骤所得产物(33mg,0.040mmol),采用实施例5中步骤B所述方法制备得到的白色固体(25mg,74%)。MS(ESI,pos.):计算值C
实施例17
化合物51是由如下所述和如图.22所示方法合成得到。
标题化合物是由美登素-N-甲基-L-丙氨酸(9,49mg,0.075mmol)和2-甲基-4-硝基苯甲酸(50)(24mg,0.132mmol),采用实施例5中步骤A所述方法制备得到的浅黄色固体(32mg,52%)。 MS(ESI,pos.):计算值C
标题化合物是由前述步骤所得产物(30mg,0.037mmol),采用实施例5中步骤B所述方法制备得到的白色固体(17mg,55%)。MS(ESI,pos.):计算值C
实施例18
化合物53是由如下所述和如图.23所示方法合成得到。
标题化合物是由美登素-N-甲基-L-丙氨酸(9,49mg,0.075mmol)和3-甲基-4-硝基苯甲酸(52)(26mg,0.143mmol),采用实施例5中步骤A所述方法制备得到的黄色固体(34mg,56%)。 MS(ESI,pos.):计算值C
标题化合物是由前述步骤所得产物(33mg,0.041mmol),采用实施例5中步骤B所述方法制备得到的白色固体(24mg,71%)。MS(ESI,pos.):计算值C
实施例19
化合物55是由如下所述和如图.24所示方法合成得到。
标题化合物是由美登素-N-甲基-L-丙氨酸(9,47mg,0.072mmol)和8-硝基-5-羧基喹啉 (54)(35mg,0.160mmol),采用实施例5中步骤A所述方法制备得到的浅黄色固体(30mg,49%)。 MS(ESI,pos.):计算值C
标题化合物是由前述步骤所得产物(29mg,0.034mmol),采用实施例5中步骤B所述方法制备得到的浅黄色固体(18mg,60%)。MS(ESI,pos.):计算值C
实施例20
化合物60是由如下所述和如图.25所示方法合成得到。
标题化合物是由Boc-L-缬氨酸-L-瓜氨酸(3,100mg,0.267mmol)和4-氨基-3-甲氧基苯甲酸叔丁酯(56,61mg,0.273mmol),采用实施例2中步骤A所述方法制备得到的白色固体(74mg,48%)。 MS(ESI,pos.):计算值C
标题化合物是由前述步骤所得产物(57,72mg,0.124mmol),采用实施例1中步骤C所述方法制备得到的类白色固体(68mg,100%)。MS(ESI,pos.):计算值C
标题化合物是由前述步骤所得产物(58,67mg,0.124mmol),采用实施例1中步骤D所述方法制备得到的白色固体(45mg,59%)。MS(ESI,pos.):计算值C
标题化合物是由前述步骤所得产物(59,44mg,0.071mmol)和美登素-N-甲基-L-丙氨酸 (9,49mg,0.075mmol),采用实施例1中步骤E所述方法制备得到的白色固体(14mg,16%)。MS(ESI, pos.):计算值C
实施例21
化合物63是由如下所述和如图.26所示方法合成得到。
标题化合物是由Boc-6-氨基己酸(64,502mg,2.17mmol),采用实施例1中步骤A所述方法制备得到的白色固体(712mg,99%)。MS(ESI,pos.):计算值C
标题化合物是由前述步骤所得产物(710mg,2.16mmol)和L-缬氨酸-L-瓜氨酸TFA盐(970 mg,2.51mmol),采用实施例1中步骤D所述方法制备得到的淡金色固体(720mg,69%)。MS(ESI,pos.): 计算值C
标题化合物是由前述步骤所得产物(62,25mg,0.051mmol)和美登素-N-甲基-L-丙氨酸 -(2-氟-4-氨基)苯甲酰胺(29,35mg,0.041mmol),采用实施例2中步骤A所述方法制备得到的白色固体(17mg,33%)。MS(ESI,pos.):计算值C
将前述步骤所得产物(16mg,0.013mmol)溶于乙腈(MeCN,3mL)和水(1mL)中,然后用三氟乙酸(TFA,1.0mL,13.0mmol)处理,烧瓶用橡胶隔片密封,并用氩气排气,反应物在环境温度下搅拌反应。24h之后,将反应物部分真空浓缩,残留物用水(约1mL)稀释,然后使用ISCO方法 (20-80%MeCN的水溶液,0.1%TFA,两相)在C18 Aq RediSep金色柱上纯化两次。将由LCMS分析得到的最纯级分合并,环境温度下部分真空浓缩,在-78℃下冷冻,并冻干得到标题化合物为白色固体(9mg, 56%)。MS(ESI,pos.):计算值C
实施例22
化合物65是由如下所述和如图.27所示方法合成得到。
标题化合物是由美登素-N-甲基-L-丙氨酸(9,50mg,0.077mmol)和2-甲氧基-5-硝基苯甲酸(64)(25mg,0.127mmol),采用实施例5中步骤A所述方法制备得到的白色固体(51mg,80%)。 MS(ESI,pos.):计算值C
标题化合物是由前述步骤所得产物(50mg,0.060mmol),采用实施例5中步骤B所述方法制备得到的白色固体(19mg,40%)。MS(ESI,pos.):计算值C
Example 23
化合物67是由如下所述和如图.28所示方法合成得到。
标题化合物是由美登素-N-甲基-L-丙氨酸(9,50mg,0.077mmol)和3-硝基-4-甲氧基苯甲酸(66)(25mg,0.127mmol),采用实施例5中步骤A所述方法制备得到的白色固体(46mg,72%)。 MS(ESI,pos.):计算值C
标题化合物是由前述步骤所得产物(45mg,0.054mmol),采用实施例5中步骤B所述方法制备得到的白色固体(23mg,53%)。MS(ESI,pos.):计算值C
实施例24
化合物69是由如下所述和如图.29所示方法合成得到。
标题化合物是由美登素-N-甲基-L-丙氨酸(9,50mg,0.077mmol)和3-硝基-5-氟苯甲酸(68)(24mg,0.127mmol),采用实施例5中步骤A所述方法制备得到的黄色固体(37mg,59%)。 MS(ESI,pos.):计算值C
标题化合物是由前述步骤所得产物(37mg,0.045mmol),采用实施例5中步骤B所述方法制备得到的白色固体(9.1mg,24%)。MS(ESI,pos.):计算值C
实施例25
化合物71是由如下所述和如图.30所示方法合成得到。
标题化合物是由美登素-N-甲基-L-丙氨酸(9,50mg,0.077mmol)和2-氟-5-硝基苯甲酸(70)(24mg,0.127mmol),采用实施例5中步骤A所述方法制备得到的黄色固体(37mg,59%)。 MS(ESI,pos.):计算值C
标题化合物是由前述步骤所得产物(37mg,0.045mmol),采用实施例5中步骤B所述方法制备得到的白色固体(2.2mg,6%)。MS(ESI,pos.):计算值C
实施例26
化合物73是由如下所述和如图.31所示方法合成得到。
标题化合物是由美登素-N-甲基-L-丙氨酸(9,50mg,0.077mmol)和3-硝基苯甲酸(72) (21mg,0.127mmol),采用实施例5中步骤A所述方法制备得到的白色固体(34mg,56%)。MS(ESI, pos.):计算值C
标题化合物是由前述步骤所得产物(34mg,0.043mmol),采用实施例5中步骤B所述方法制备得到的白色固体(9.3mg,27%)。MS(ESI,pos.):计算值C
实施例27
化合物75是由如下所述和如图.32所示方法合成得到。
标题化合物是由美登素-N-甲基-L-丙氨酸(9,50mg,0.077mmol)和3-硝基-4-氟苯甲酸 (74)(24mg,0.127mmol),采用实施例5中步骤A所述方法制备得到的白色固体(34mg,54%)。 MS(ESI,pos.):计算值C
标题化合物是由前述步骤所得产物(34mg,0.042mmol),采用实施例5中步骤B所述方法制备得到的白色固体(12mg,37%)。MS(ESI,pos.):计算值C
实施例28
化合物77是由如下所述和如图.33所示方法合成得到。
标题化合物是通过下述方法制备得到:向圆底烧瓶中称取27(20mg,0.026mmol)和己二酸酐(17mg,0.133mmol),并溶于吡啶(1.5mL)。烧瓶用橡胶隔片密封,并用氮气排气,反应物在环境温度下搅拌反应。4小时后,反应物通过ISCO方法(梯度洗脱:20-90%MeCN的水溶液,0.05%乙酸,洗脱20分钟)直接在30g C18 RediSep金色Aq柱上进行纯化。将含产物的级分合并,部分真空浓缩,残留物在干冰上冷冻,并冻干得到类白色固体(16mg,67%)。MS(ESI,pos.):计算值C
标题化合物是由前述步骤所得产物(76,16mg,0.017mmol),采用实施例1中步骤A所述方法制备得到的白色固体(10mg,58%)。MS(ESI,pos.):计算值C
实施例29
化合物78是由如下所述和如图.34所示方法合成得到。
标题化合物是由27(15mg,0.019mmol)和6-马来酰亚胺基己酸(6mg,0.029mmol),采用实施例5中步骤A所述方法制备得到的类白色固体(9.8mg,52%)。MS(ESI,pos.):计算值 C
实施例30
化合物80是由如下所述和如图.35所示方法合成得到。
标题化合物是由美登素-N-甲基-L-丙氨酸(9,40mg,0.062mmol)以及3-甲磺酰基-4-硝基苯甲酸(79)(25mg,0.102mmol),采用实施例5中步骤A所述方法制备得到的黄色固体(37mg, 69%)。MS(ESI,pos.):计算值C
标题化合物是由前述步骤所得产物(36mg,0.041mmol),采用实施例5中步骤B所述方法制备得到的白色固体(28mg,76%)。MS(ESI,pos.):计算值C
实施例31
化合物82是由如下所述和如图.36所示方法合成得到。
标题化合物是由美登素-N-甲基-L-丙氨酸(9,47mg,0.072mmol)和3-羟基-4-硝基苯甲酸(81)(20mg,0.109mmol),采用实施例5中步骤A所述方法制备得到的黄色固体(29mg,49%)。 MS(ESI,pos.):计算值C
标题化合物是由前述步骤所得产物(28mg,0.034mmol),采用实施例5中步骤B所述方法制备得到的淡黄色固体(20mg,69%)。MS(ESI,pos.):计算值C
实施例32
化合物84是由如下所述和如图.37所示方法合成得到。
标题化合物是由美登素-N-甲基-L-丙氨酸(9,30mg,0.046mmol)和2-硝基苯甲酸(83) (15mg,0.092mmol),采用实施例5中步骤A所述方法制备得到的黄色固体(26mg,71%)。MS(ESI, pos.):计算值C
标题化合物是由前述步骤所得产物(24mg,0.038mmol),采用实施例5中步骤B所述方法制备得到的白色固体(13mg,54%)。MS(ESI,pos.):计算值C
实施例33
化合物86是由如下所述和如图.38所示方法合成得到。
标题化合物是由美登素-N-甲基-L-丙氨酸(9,30mg,0.046mmol)和4-甲氧基-2-硝基苯甲酸(85)(18mg,0.092mmol),采用实施例5中步骤A所述方法制备得到的黄色固体(18mg,47%)。 MS(ESI,pos.):计算值C
标题化合物是由前述步骤所得产物(18mg,0.022mmol),采用实施例5中步骤B所述方法制备得到的白色固体(15mg,84%)。MS(ESI,pos.):计算值C
实施例34
化合物88是由如下所述和如图.39所示方法合成得到。
标题化合物是由美登素-N-甲基-L-丙氨酸(9,30mg,0.046mmol)和3-吗啉基-4-硝基苯甲酸(87)(23mg,0.092mmol),采用实施例5中步骤A所述方法制备得到的黄色固体(28mg,70%)。 MS(ESI,pos.):计算值C
标题化合物是由前述步骤所得产物(28mg,0.032mmol),采用实施例5中步骤B所述方法制备得到的类白色固体(12mg,52%)。MS(ESI,pos.):计算值C
实施例35
化合物90是由如下所述和如图.40所示方法合成得到。
标题化合物是由美登素-N-甲基-L-丙氨酸(9,50mg,0.077mmol)和3-乙酰胺基-4-硝基苯甲酸(89)(29mg,0.129mmol),采用实施例5中步骤A所述方法制备得到的蓬松淡黄色固体(36mg, 54%)。MS(ESI,pos.):计算值C
标题化合物是由前述步骤所得产物(35mg,0.042mmol),采用实施例5中步骤B所述方法制备得到的白色固体(19mg,57%)。MS(ESI,pos.):计算值C
实施例36
化合物100是由如下所述和如图.41所示方法合成得到。
向配备有磁力搅拌器和氮气入口的10mL圆底烧瓶中加入3-溴-2-溴甲基-丙酸(9,1.1g, 4.1mmol)和亚硫酰氯(3.0mL)。将所得溶液加热回流3小时,浓缩得到0.90g(84%收率)棕色油状物。
向配备有磁力搅拌器和氮气入口的50mL圆底烧瓶中加入6-氨基己酸(93,2.0g,15mmol) 和亚硫酰氯(5.0mL,69mmol,4.5当量)。所得溶液在低于30℃下搅拌反应2小时,真空浓缩至干。向棕褐色半固体浆体中加入碳酸氢钠(2.6g,30mmol,2.0当量)的t-BuOH(5.0mL,87mmol,5.7当量)混合物,所得浆体在环境温度下再搅拌2h。在40℃下真空浓缩除去丁醇。所得浓的白色浆体用乙酸乙酯稀释,并分别用4份1N NaOH、3份H
向配备有磁力搅拌器和氮气入口的50mL圆底烧瓶中加入6-氨基己酸叔丁酯(94,0.50g, 2.7mmol)和二甲基氨基吡啶(0.03g,0.27mmol,0.10当量)的DCM(5.0mL)溶液。所得溶液通过冰浴冷却至0℃。然后将3-溴-2-溴甲基-丙酰氯(92,0.90g,3.4mmol,1.2当量)溶于DCM(5mL),并在0℃下缓慢加至反应混合物中。将反应物搅拌,并升温至环境温度过夜。用乙酸乙酯稀释反应混合物,有机混合物用H
向配备有磁力搅拌器和氮气入口的50mL圆底烧瓶中加入6-(3-溴-2-溴甲基-丙酰基氨基)- 己酸叔丁酯(95,0.26g,0.62mmol)和三氟乙酸(0.70mL,9.3mmol,15当量)的DCM(10mL)溶液。所得溶液在环境温度下搅拌过夜,将反应物浓缩至干,残留物溶于乙腈和H
标题化合物是由实施例1中步骤D所得产物(6,100mg,0.254mmol)和二碳酸二叔丁酯(61mg,0.279mmol)制备得到,即将其称取至圆底烧瓶中,并用1MNaOH(2mL)的THF(3mL)和H
标题化合物是由前述步骤所得产物Boc-缬氨酸-瓜氨酸-4-氨基苯甲酸(97,76mg,0.154 mmol)和美登素-N-甲基-L-丙氨酸(9,50mg,0.077mmol),采用实施例5中步骤A所述方法制备得到的白色固体(22mg,26%)。MS(ESI,pos.):计算值C
标题化合物是由前述步骤所得产物(98,20mg,0.018mmol)制备得到,即将其称取至圆底烧瓶中,并溶于ACN(乙腈,3mL)和H
标题化合物是由前述步骤所得产物(99,8mg,0.008mmol)和6-(3-溴-2-溴甲基-丙酰基氨基)-己酸(96),采用实施例5中步骤A所述方法制备得到的白色固体(8mg,75%)。MS(ESI,pos.): 计算值C
实施例37
五种抗体采用下述方法与本发明所述连接体-有效负载化合物进行偶联。在所述这些实验中使用的四种靶向抗体是:(1)WO2007002222A2中所述的具有克隆AB-PG1-XG1-006的重链和轻链可变结构域的PSMA抗体,(2)WO2007001851中所述的具有衍生自克隆3A5的可变区的抗MUC16抗体,和(3)WO2015026907A1中所述的具有克隆H1H6765P和H1H6958N2的重链和轻链可变结构域的两种 PRLR抗体。所有的单克隆抗体均在CHO细胞中表达,并通过蛋白A进行纯化。还使用衍生自与肿瘤学无关的抗原的非结合同型对照。
实施例38
马来酰亚胺类的偶联方法
在50mM HEPES、150mMNaCl、pH值为7.5中的抗体(1-10mg/ml),用1mM二硫苏糖醇在37℃下处理30分钟。凝胶过滤之后(G-25,pH值为4.5的乙酸钠),将马来酰亚胺基连接体有效负载衍生物10、15、20、25、60、和78(1.2当量/SH组)的DMSO溶液(10mg/ml)加至还原的抗体中,并将混合物用1M HEPES(pH 7.4)调节pH值为7.0。1小时后,反应体系用过量的N-乙基马来酰亚胺淬灭。偶联物通过体积排阻色谱法进行纯化,并进行无菌过滤。蛋白浓度和有效负载与抗体比值是通过紫外 (UV)光谱分析进行测定。体积排阻HPLC确定所使用的所有偶联物均为>95%单体,和RP-HPLC确定存在<0.5%未偶联的连接体有效负载。所有偶联的抗体用于连接体有效负载的负载值是根据Hamblett et al, Cancer Res 2004107063,通过UV进行分析。收率和有效负载与抗体比值如表1所示。
实施例39
环境温度下,使50mM HEPES、150mMNaCl、pH值为8.0、和15%(v/v)DMA中的抗体(1-10mg/ml)与6倍过量的化合物77进行偶联1-2小时。所得偶联物通过体积排阻色谱法进行纯化,并进行无菌过滤。蛋白和连接体有效负载的浓度是通过UV光谱分析进行测定。体积排阻HPLC确定所使用的所有偶联物均为>95%单体,和RP-HPLC确定存在<0.5%未偶联的连接体有效负载。基于蛋白的收率如表1所示。所有偶联的抗体用于连接体有效负载的负载值是根据Hamblett et al,Cancer Res 2004107063,通过UV进行分析。收率和有效负载与抗体比值如表1所示。
实施例40
体外无细胞酶法测定方法是采取Dubowchik,et al.BioconjugateChem.200213855中所述方法。连接体有效负载10是在25mM乙酸钠缓冲液、1mM EDTA、pH值为5.0中最终设定为100μg/mL,并在37℃下进行预孵育。室温下,组织蛋白酶B(Sigma#C8571)用1当量的30mM DTT、15mM EDTA 至2当量的组织蛋白酶B原液活化15分钟。将活化的组织蛋白酶B溶液以1:20的摩尔比(纯化的H
将样品以12,000g离心5分钟。回收上层清液,并通过液相色谱-质谱(ThermoQuantiva) 经联合灌注0.3ml/min的30:70流动相B:A(流动相A:0.1%FA的H
本实施例的结果在一定程度上是因为组织蛋白酶B蛋白水解10应仅在存在酶的细胞中内化ADC之后发生。应降低目标效应,因为抗体将细胞毒性有效负载直接递送至靶细胞。
实施例41
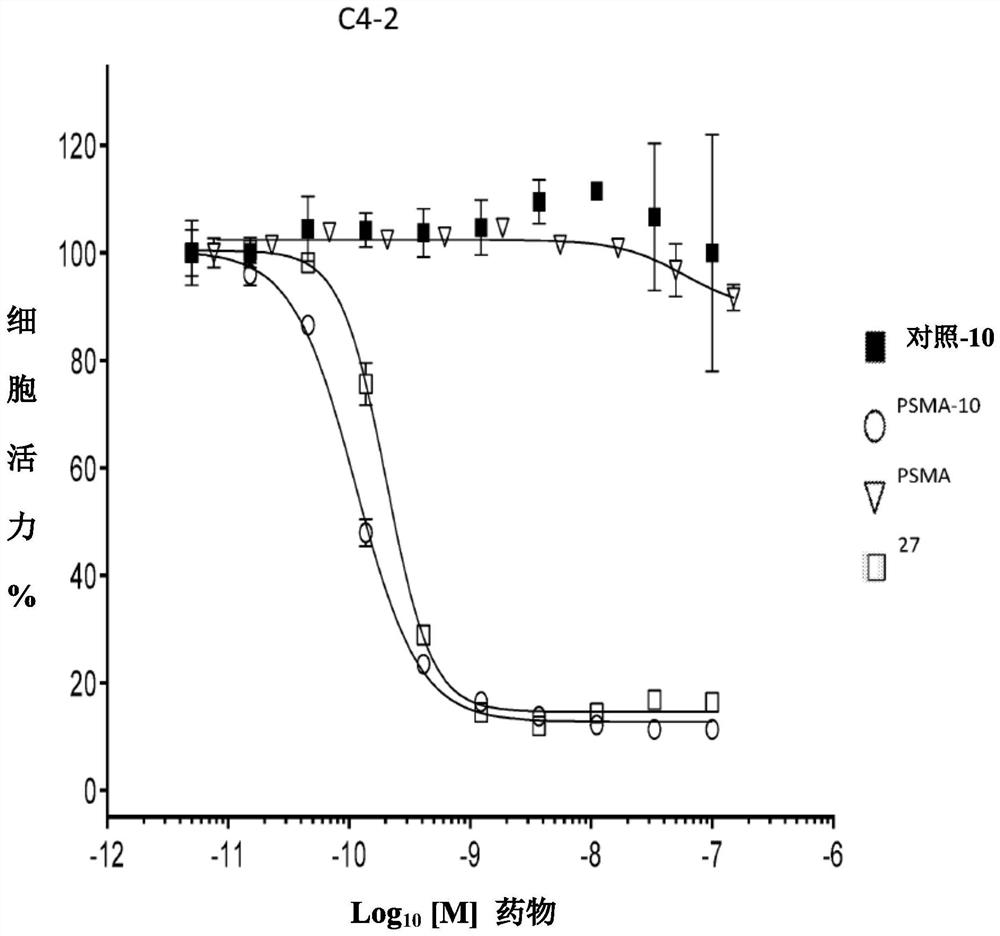
在本实施例中,评估了各种抗体-药物偶联物或其相关有效负载在体外杀伤抗原表达肿瘤细胞的能力。
Ovcar3(Muc16+)或C4-2(PSMA+)细胞在96孔板中以3000(C42)细胞每孔,在完全生长培养基中进行接种,并生长过夜。对于细胞活力曲线,将连续稀释的偶联物或有效负载以范围为300nM 至5pM的终浓度加至细胞中并孵育3天。为了测定活力,将细胞与CCK8(Dojindo)一起孵育最后1-3 小时,并在Victor(PerkinElmer)上测定450nm(OD
在C4-2细胞(前列腺癌细胞系)中,天然表达PSMA是上述同型对照结合的271倍,美登素类偶联物PSMA-10和PSMA-25的IC
在Ovcar-3细胞(卵巢癌细胞系)中,天然表达MUC16是同型对照结合的320倍,美登素类偶联物MUC16-10和MUC16-25的IC
表2列出了有效负载仅在Ovcar3(Muc16+)或C4-2(PSMA+)细胞中的抗增殖能力。所有化合物均具有次纳摩尔活性,化合物35和37的IC
表2
实施例42
抗体表达
试验/实验类型:经修饰以含有位点特异性偶联基序的抗体的克隆、表达和纯化。
本实施例提供了具有使通过转谷氨酰胺酶反应进行位点特异性偶联的氨基酸序列的抗体的产生。
为了产生抗体,使用快速闪电多定点诱变试剂盒(QuikChange Lightning MultiSite-Directed Mutagenesis Kit)(Agilent,#210516),对编码人IgG1(UniprotKB登录号为P01857的氨基酸1至330) 的CH1、CH2和CH3结构域的质粒进行诱变,以在位点180产生N至Q突变。选择两个抗体可变区重链 (VH)片段,其中一个编码抗人PRLR抗体,H1H6958N2(国际专利申请WO 2015026907 A1)的VH,和另一个编码识别外源抗原的同型对照抗体的VH。设计引物(idtdna.com)以使用Kapa HiFi DNA聚合酶 (Kapa Biosciences;#KK2102)扩增这两种抗体的VH区。在进行PCR扩增的同时,将含有人IgG1 N180Q 突变的质粒用LguI酶(Fermentas,#FD1934)在37℃下消化30分钟。一旦扩增完毕,消化的人IgG1质粒和两个PCR产物在含有SYBR Safe(安全)染色(Life Technologies,#S33102)的1%琼脂糖凝胶上用完。使用干净的剃刀刀片,确定适当尺寸的条带并从凝胶上切下。使用凝胶提取试剂盒(GelExtraction kit) (Qiagen,#28704)纯化所有切离的产物。然后使用消化的人IgG1载体与VH PCR产物为3:1比例进行融合后克隆反应(Clontech,#638911),然后在50℃下孵育15分钟。孵育后,将每个反应转化至Mix和Go 感受态细胞(Zymo,#T3007)中,在冰上孵育5分钟,将感受态细胞接种在LB+羧苄青霉素孔板(Teknova, VWR,#101320-126)上,在37℃下孵育过夜。
第二天,将单个菌落从板中接种至含有100μg/mL羧苄青霉素的LB液体培养基中,并在 37℃的台式培养箱中振荡生长过夜。然后通过离心沉淀细胞,并使用PureLinkHiPure质粒(Plasmid)微量化试剂盒(miniprep kit)(Thermo Fisher,#K210003)在Hamilton Starlet(汉密尔顿新星)机器人上进行微量化。对纯化的DNA进行测序,并使用Sequencher软件(GeneCodes)分析结果。挑选每个连接反应的克隆重新转化至Mix和Go感受态细胞中,在冰上孵育5分钟,然后将感受态细胞接种在含有羧苄青霉素的LB板上,在37℃下孵育过夜。
其后的一天,从每个板挑取单个菌落并在含有100μg/mL羧苄青霉素的LB液体培养基中生长3至4小时。然后将样品稀释至含有10μg/ml羧苄青霉素的LB液体培养基中,并转移到37℃振荡培养箱中生长过夜。然后在两个质粒上进行大量质粒提取(maxiprep)DNA提取,并对完整的DNA开放阅读框进行测序。一旦序列得到证实,则将重链DNA连同先前克隆的附属轻链DNA一起稳定转染到CHO 细胞系中以产生每种抗体。
将含有N180Q Fc突变的H1H6958N2(国际专利申请WO 2015026907 A1)称为PRLR-Q,将识别同样含有N180Q Fc突变的外源性抗原的同型对照抗体称为同型对照-Q(ISOTYPECONTROL-Q)。
实施例43
PRLR-Q(MW 145438Da)和同型对照-Q(MW 144602Da)抗体以1-10mg/mL在PBS、pH值为7.4中进行偶联。连接体有效负载63以超过抗体的10至25倍摩尔过量加入,通过加入1-5单位的细菌转谷氨酰胺酶(Zedira,T1001)每毫克抗体引发酶反应,并在37℃下振荡培养4-16小时。偶联物是通过体积排阻色谱法进行纯化,并进行无菌过滤。通过紫外(UV)光谱分析测定蛋白和连接体有效负载的浓度。体积排阻HPLC确定所使用的所有偶联物均为>95%单体。基于蛋白的收率如表1所示。所有偶联的抗体用于连接体有效负载的负载值是根据Hamblett et al,Cancer Res 2004 10 7063,通过UV进行分析。此外,偶联物用于连接体有效负载的载量是使用Waters Synapt G2-Si QTOF质谱联用AcquityUPLC(超高效液相色谱),通过ESI-MS进行分析。色谱分离是在C4柱(Waters蛋白BEH C4,50mm X 1mm,1.7μm) 上以25分钟梯度(分钟:流动相B的百分比为0:20%,1:20%,18:40%,18.1:90%,20:95%,20.8:95%, 20.9:20%,25:20%)完成的。流动相A为0.1%甲酸的水溶液,和流动相B为0.1%甲酸的乙腈溶液。流速设定为100μl/min。检测器TOF扫描从m/z 700-5000设置25分钟,主要参数如所列举的(毛细管电压3.2kV;取样锥150;源偏移(Source Offset)为80;源温度120℃;去溶剂化温度500℃;陷阱碰撞能量30(Trap collision Energy);转移碰撞能量关闭(Transfer Collision Energy Off);气体控制关闭;解析四极(ResolvingQuadrupole):LM分辨率为4.7)。使用MassLynx软件中的MaxEnt函数对组合的光谱进行去卷积。当根据强度加权时产生的分子离子对应于表3所列的载量。图42和43示出了实际质谱图。
表3.化合物63的PRLR-Q和同型对照-Q偶联物中强度加权的平均连接体-有效负载载量的总结。
实施例44
采用Biacore3000仪器使用实时表面等离子体共振生物传感器测定人PRLR与未修饰的 H1H6958N2、PRLR-Q或PRLR-Q-63的纯化抗PRLR抗体结合的平衡解离常数(K
在25℃下,hPRLR-MMH与不同抗PRLR抗体结合的结合动力学参数如表4所示。25℃下, hPRLR-MMH与亲本抗体H1H6958N2结合,其K
表4:25℃下抗PRLR抗体结合hPRLR-MMH的结合动力学参数
实施例45
本发明实施例提供了本发明所述偶联物的细胞毒性试验。为了评估与63、60、77、和78 偶联的抗PRLR抗体杀伤PRLR表达细胞系的能力,采用使用T47D导管癌细胞系(ATCC,#HTB-133) 的体外细胞毒性试验,所述T47D导管癌细胞系先前被确定为在其细胞表面表达>27,000份副本的人 PRLR。
对于所述试验,将T47D细胞以2,000细胞/孔接种在含有补充有10%FBS、NEAA和青霉素/链霉素(pencillin/streptomycinin,完全培养基)的DMEM的培养基中的白色96孔板上。其在37℃、 5%CO
如图.44所示,与63特异性偶联的抗PRLR抗体(PRLR-Q-63)显示了对T47D细胞的细胞毒性,其IC
如图.45所示,与60偶联的抗PRLR抗体(PRLR-60)显示了对T47D细胞的细胞毒性,其IC
如图.46所示,与77偶联的抗PRLR抗体(PRLR-77)显示了对T47D细胞的细胞毒性,其IC
如图.47所示,与78偶联的抗PRLR抗体(PRLR-78)显示了对T47D细胞的细胞毒性,其IC
表5列出了有效负载仅在Ovcar3(Muc16+)、C4-2(PSMA+)、和T47D(PRLR+)细胞中的抗增殖能力。
表5
上述实施方案和实施例旨在仅仅是示例性的而非限制性的。本领域技术人员将认识到或将能够使用仅常规实验,来确定具体化合物、材料/物料和程序/方法的许多等同物。所有这些等同物被认为在本发明范围之内,并被所附权利要求所涵盖。
- 过渡金属催化的C-H卡宾体偶联反应合成C-C键及N杂环类衍生物的绿色新方法
- 美登素类衍生物、其偶联物和使用方法
- 美登素类化合物衍生物、其偶联物和使用方法