一种环状RNA和疾病关联预测方法
文献发布时间:2023-06-19 19:30:30
技术领域
本发明涉及生物信息学技术领域,具体涉及一种基于图嵌入和动态卷积自编码器来预测circRNA和疾病关联的方法。
背景技术
环状RNA(circRNAs)是一类客观存在于生物体中的非编码RNA分子,没有50-cap和30-多聚腺苷酸化尾,通过共价键形成环状结构。CircRNAs含有比线性mRNAs更丰富的转录本,可以在转录或转录后水平调控多种生命活动。此外,circRNAs还可以作为竞争性内源性RNAs(ceRNAs)的成分来抑制miRNAs的活性,从而控制基因的转录、翻译等功能。许多研究也证明circRNA存在于各种生物体中,具有重要的调控作用,也表明circRNA与疾病之间有着密切的联系,circRNA在多种疾病中发挥着重要作用,circRNAs可以作为新的疾病诊断生物标志物,在药物研发和疾病诊治中具有良好的应用前景。
由于circRNA与疾病之间有着密不可分的联系,把握这种联系对于疾病的研究和治疗具有重要价值。然而,普通的生物实验投入了大量的人力物力,只能确认其中的一小部分联系。快速有效的计算方法是解决这一问题的关键。目前,有许多模型可以预测circRNA与疾病的关联。2019年,Wang等人提出了一种基于多源信息融合和卷积神经网络(CNN)的预测circRNA-疾病关联的方法。2019年,Li等人提出了一种基于网络共识投影的circRNA-疾病关联预测方法。2020年,K.Deepthi等人提出了一种基于自动编码器(AE)和深度神经网络的方法来预测circRNA-疾病关联。2022年,Zhang等人提出了一种基于图表示学习的方法来预测circRNA-疾病关联。但这些模型预测在生物信息的融合上并没有最大化利用生物信息的相关性,如基因序列的相似度或表达相似度等,且某些疾病或circRNA的深层特征难以提取,计算力复杂度较高,导致目前存在的预测的关联结果仍存在很大偏差。
发明内容
用以解决或者提高现有技术中的方法存问题和性能,本发明提出以下技术方案:
一种环状RNA和疾病关联预测方法,包括以下步骤:
S1)数据组织
从公共数据库获取已知的circRNA和疾病关联信息、circRNA表达谱数据、疾病语义相似度信息形成原始数据集;
S2)数据融合
根据疾病语义相似度、circRNA表达谱数据以及circRNA和疾病的关联矩阵,计算出疾病语义相似度、疾病和circRNA的Jaccard相似度和GIP(高斯交互表达谱相似度)以及circRNA表达谱相似度,并按照不同疾病之间是否有语义相似性和不同circRNA之间是否有表达谱数据来进行融合,最后通过矩阵拼接和归一化操作融合数据形成统一的融合标识符;
S3)特征提取
通过AANE和DCAEs组成的深度学习模型提取融合标识符的低维特征和深层特征,AANE通过特定的损失函数,当损失函数的值达到设定值后,即完成训练和提取,DCAEs则使用MSE作为损失函数并通过Adadelta算法对模型参数和网络结构进行优化;
S4)分类器预测
将最终提取的特征放入随机森林分类器,对分类器进行训练,并优化关键的参数,优化完毕后,将测试集放入分类器进行circRNA和疾病的关联预测,并得到最后的预测分数矩阵。
进一步的,融合标识符的形成方法为:若疾病或者circRNA之间有语义相似度,对其最终的融合数据进行取平均值的运算;若没有,则取高斯交互核相似度来补充不同数据库之间的数据差异,最后将疾病和circRNA的融合相似度降维后进行拼接形成最后的融合标识符。
进一步的,疾病语义相似度的具体计算方式为:
疾病e与疾病d为关联的两种疾病,根据疾病之间的有向无环图计算疾病的语义价值
其次,考虑疾病编号对疾病贡献的影响,采用下式计算疾病e在疾病数量影响下的贡献
其中,num(DAGd(e))是指与疾病d相关的疾病数量,num(diseases)指所有疾病的个数;
得到疾病d(i)和疾病d(j)的疾病语义相似度DSS2(d(i),d(j))
进一步的,circRNA表达谱相似度的具体计算方式为:将带有表达谱数据的circRNA表示为一个32维的特征向量,并使用降序的方法对不同circRNA的表达谱数据进行重新排序,使用Spearman相关系数得到circRNA之间的表达谱相似性。
进一步的,Jaccard相似度的具体计算方式为:
使用以下公式计算疾病d(i)和疾病d(j)的Jaccard相似度JD(d(i),d(j))
/>
其中,CA(d(i))是指与疾病d(i)相关的circRNA组;
根据上式可以推断出circRNA的Jaccard相似模型JC(c(i),c(j))
其中,DA(c(i))是指与circRNA c(i)相关的疾病组,通过两个circRNA关联疾病的交集比上两个circRNA关联疾病的并集,就可以得到两个circRNA的JC相似度。
进一步的,融合方法的具体实现为:
将疾病相似性多源信息DS和疾病Jaccard相似度JD进行拼接形成疾病相似度模型DM=[DS,JD];
将circRNA相似性多源信息CS和circRNA Jaccard相似度JC进行拼接形成circRNA相似度模型CM=[CS,JC];
融合过程中,使用PCA降维归一化后将CM与DM进行拼接后得到融合标识符FM(c(i),d(i))
FM(c(i),d(j))=[CM(c(i)),DM(d(j))]
其中,CM(c(i))表示CM的第i行向量,DM(d(j))表示DM的第j列向量。
进一步的,通过AANE算法,得到与余弦相似度矩阵差值最小的图嵌入表达矩阵,所得到的图嵌入表达矩阵即为低维特征,利用AANE提取低维特征的具体步骤包括:
对于网络N=(V,E,W),V为N中的节点集,W为N中的边集,E为边所代表值的集合,W中的边eij表示连接节点i和节点的边j,其大小与两个节点之间的相似度密切相关,如果eij的值较大,则节点i与节点j更相似;根据实对称矩阵可以正交相似对角化的推论,可以得到如下公式:
A=HΛH
其中A指的是半定对称矩阵,A可以用一个正交矩阵H和一个对角矩阵Λ来表示,B是定义的新矩阵,即Λ中的元素,应用该算法时,只需给出待输入的属性矩阵A,通过余弦相似度计算相似度矩阵S,可以推出:S=QQ
在以下两种情况下,节点最有可能具有相似的向量表示,一种是拓扑更相似的节点,另一种是连接权重更高的节点,因此定义目标函数L如下:
这里的S=QQT只是理论上可以得到,但是实际中,他们两个是具有差值的,我们这个算法就是为了得到与S最小的差值的Q,即L的值最小;其中,λ为平衡参数,F为为(),ω
定义参数Z=Q,目标函数也可以写成如下形式:
ρ代表惩罚参数,u
使用乘法器的交替方向法解决目标函数的优化问题,在连续求导的情况下,使用如下迭代公式:
其中t表示第t次迭代,si表示余弦相似度矩阵中的值,I是固定参数,从1开始增加,每次迭代I+1。
进一步的,对模型参数和网络结构进行优化的步骤为:
在训练每一层的过程中,计算解码有的重构向量x'和输入向量x的损失函数,并将损失函数优化到设定值,重复这个动作直到所有层都训练完毕,编码公式如下:
y=subsampling(x)
其中t为通过动态卷积编码后得到的中间值,解码的具体公式如下:
其中,π
进一步的,随机森林的生成方法为:
S41)使用Bootstrap从有放回的C个样本集中随机选择c个样本,选取的c个样本作为决策树根节点的样本用于训练决策树;
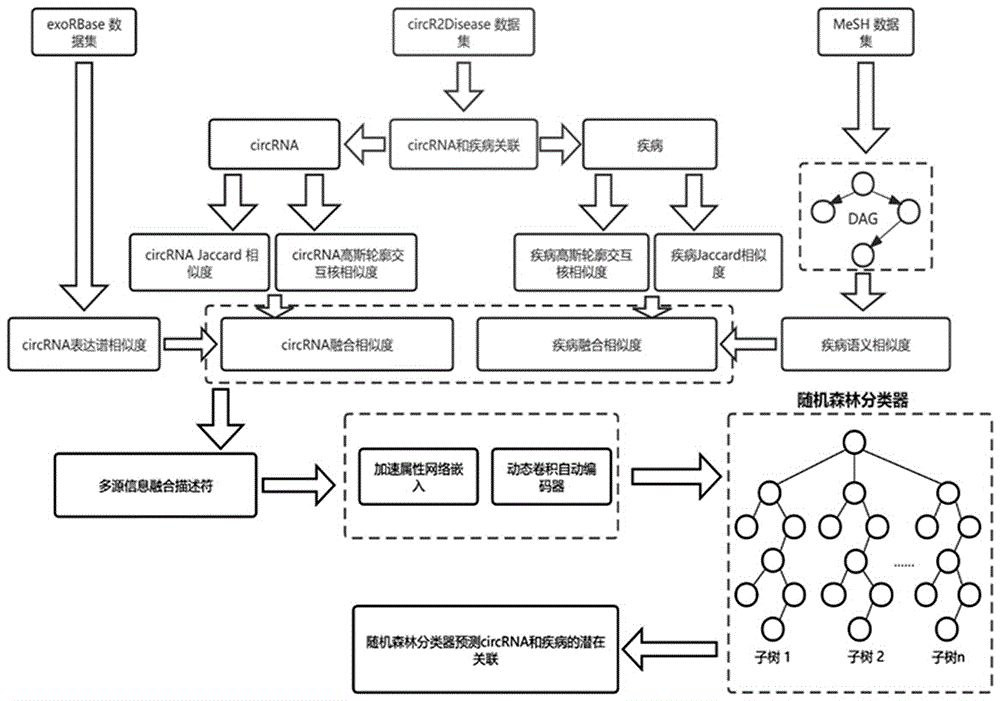
S42)从样本的M个特征中随机选择m个特征,满足条件m< S43)在形成决策树的过程中,每个节点都必须按照步骤S42进行分裂,直到不能再分裂,在整个决策树形成过程中不进行剪枝; S44)按照步骤S41-S43构建多个决策树,形成随机森林。 优选的,本方法基于PyTorch和Python及其辅助库编写。 本发明提出了一种基于图嵌入和动态卷积自编码器来预测circRNA和疾病关联的方法,本方法采用的模型集合了更多的生物学信息并且创新了提取能力更强和计算成本更低的深度学习模型。与现有技术中融合的生物学信息数量相比,本发明可以将Jaccard和circRNA表达谱相似度融合到传统的生物信息中,充分利用有限的生物信息并创新了性能更好的深度学习模型,所以本发明可以更为有效地应用到circRNA和疾病关联进而提升预测精度。同时,本发明提供的方法不仅融合了circRNA和疾病的网络结构信息,还有效地利用circRNA和疾病的各种特征信息,因而本发明不仅仅可以推断未知的circRNA和疾病关联,而且能很高效地预测当前没有与任何疾病关联的新circRNA,以及预测当前没有与任何circRNA关联的新的疾病,同时,动态卷积自编码器编码器可以在提高提取特征效果的同时控制计算成本的增长,推动模型性能的进一步提升。通过实验验证,我们的方法在预测的circRNA和疾病关联分数前20的关联中,有16种得到了文献验证,这也足以证明我们的方法可以为生物实验提供可靠的验证对象。 附图说明 图1为本发明实施例1的总流程图。 图2为本发明实施例1的AANE算法流程图。 图3为本发明实施例1的DCAEs算法流程图。 图4为本发明实施例1不同分类器的ROC曲线图 具体实施方式 为了使本发明的目的、技术方案及优点更加清楚明白,以下结合附图及实施例,对本发明进行进一步详细说明。 本实施例公开了一种环状RNA和疾病关联预测方法,其数学模型全部基于PyTorch和Python及其辅助库编写,请参照图1,具体步骤如下。 步骤1:数据组织 从公共数据库获取已知的circRNA和疾病关联信息、circRNA表达谱数据、疾病语义相似度信息形成原始数据集,本实施例中将以数据circR2Disease数据集为基准数据集,进行完成的多源数据的融合过程以及特征提取过程,但应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定单一特例数据集。circR2Disease数据集是包含circRNA和疾病关联最全的数据集,最新的circR2Disease数据库包含661个circR2Disease、100个疾病和739个circRNA和疾病关联,本发明以circR2Disease的739个已确认关联作为阳性样本,但是如果将其他未知关联都作为阴性样本,阴性样本的数量会远大于正样本的数量,并且数据集将变得非常不均匀,这可能导致结果具有欺骗性。因此,此处随机选择了739个未知关联作为负样本来平衡数据集(在661×100=66100个总关联中,有739个未知关联可以忽略不计),完成平衡数据集的构建。另外定义了一个m×n的邻接矩阵AM,其中m是circRNA的个数,取值为661,n是疾病的个数,取值为100,AM存储circRNA与疾病的关联信息。如果AM(i,j)=1,则表示circRNA c(i)与疾病d(j)相关联,否则AM(i,j)=0。 步骤2:数据融合 在从MeSH数据库、exoRBase数据库以及circR2Disease数据集中所需要的生物学信息提取出来后,根据circRNA和疾病的关联邻接矩阵等信息计算出疾病语义相似度、疾病和circRNA的Jaccard相似度和GIP以及circRNA表达谱相似度,并按照不同疾病之间是否有语义相似性和不同circRNA之间是否有表达谱数据来进行不同方式的融合,最后通过矩阵拼接和归一化操作融合数据形成统一的融合标识符。 MeSH数据库是我们疾病语义相似度的主要信息来源,circR2Disease数据集中的疾病并未全部包含在MeSH中,导致疾病相似度信息无法全面表达,因此引入使用高斯交互轮廓核相似度来细化疾病相似度信息。高斯交互轮廓核相似度依赖于以下假设:如果circRNA C1与疾病D1相关,那么与D1相似的疾病也倾向于具有与C1功能相似的circRNA,反之亦然。疾病d(i)和疾病d(j)的疾病高斯交互轮廓核相似度为: GD(d(i),d(j))=exp(-μ‖V(d(i))-V(d(j))‖ d(i)表示第i个疾病的行向量,μ是GIP的带宽参数,计算方式为 同理可得circRNA之间的高斯交互轮廓核相似度。 在本实施例中,提出以下方法计算步骤2中各类相似度矩阵。 步骤2.1:疾病语义相似度的计算 根据MeSH数据库中的疾病有向无环图,如果疾病e与疾病d有关联,那么疾病e对于疾病d的贡献有如下公式: e′表示疾病e的关联疾病,μ=0.5表示疾病贡献因子,计算疾病的语义价值 仅使用疾病语义相似度DSS1(d(i),d(j))并不能涵盖疾病之间联系的全部情况,还应考虑疾病编号对疾病贡献的影响,采用下式计算疾病e在疾病数量影响下的贡献 其中,num(DAGd(e))是指与疾病d相关的疾病数量,num(diseases)指所有疾病的个数; 得到疾病d(i)和疾病d(j)的疾病语义相似度DSS2(d(i),d(j)) 步骤2.2:circRNA表达谱相似度的计算 将带有表达谱数据的circRNA表示为一个32维的特征向量,并使用降序的方法对不同circRNA的表达谱数据进行重新排序,使用Spearman相关系数 得到circRNA之间的表达谱相似性,d 步骤2.3:Jaccard相似度计算 J(A,B)是A和B的交集大小与A和B的并集大小之比,Jaccard值越大,相似度越大,反之越低。将Jaccard加入到数据融合可以挖掘疾病与circRNA的相似性,更全面地了解circR2Disease数据集的信息并充分利用它。对于疾病Jaccard模型,使用以下公式计算疾病d(i)和疾病d(j)的Jaccard相似度: 其中,CA(d(i))是指与疾病d(i)相关的circRNA组; 同理可以推断出circRNA的Jaccard相似模型JC(c(i),c(j))。 步骤2.4:融合 将疾病相似性多源信息DS和疾病Jaccard相似度JD进行拼接形成疾病相似度模型DM=[DS,JD],其中,DS通过以下公式计算: 其中GD是疾病高斯交互表达谱相似度矩阵; 将circRNA相似性多源信息CS和circRNA Jaccard相似度JC进行拼接形成circRNA相似度模型CM=[CS,JC],CS通过以下公式计算: 其中GC是circRNA高斯交互表达谱相似度矩阵; 融合过程中,使用PCA降维归一化得到融合标识符FM(c(i),d(i)) FM(c(i),d(j))=[CM(c(i)),DM(d(j))] 其中,CM(c(i))表示CM的第i行向量,DM(d(j))表示DM的第j列向量。 步骤3:特征提取 在融合形成统一的融合标识符后,将融合标识符通过AANE和DCAEs组成的深度学习模型之中,提取融合标识符的低维特征和深层特征,AANE通过特定的损失函数,当损失函数的值达到设定值后,即完成训练和提取,DCAEs则使用MSE作为损失函数并通过Adadelta算法对模型参数和网络结构进行优化。 AANE算法流程请参照图2,对于网络N=(V,E,W),V为N中的节点集,W为N中的边集,W中的边eij表示连接节点i和节点的边j,其大小与两个节点之间的相似度密切相关,如果eij的值较大,则节点i与节点j更相似;根据实对称矩阵可以正交相似对角化的推论,可以得到如下公式: A=HΛH 其中A指的是半定对称矩阵,A可以用一个正交矩阵H和一个对角矩阵Λ来表示,B是定义的新矩阵,即Λ中的元素,应用该算法时,只需给出待输入的属性矩阵A,通过余弦相似度计算相似度矩阵S,可以推出:S=QQ 在以下两种情况下,节点最有可能具有相似的向量表示,一种是拓扑更相似的节点,另一种是连接权重更高的节点,因此定义目标函数L如下: 其中,λ为平衡参数,F为为(),ω 定义参数Z=Q,目标函数也可以写成如下形式: q代表惩罚参数,u 使用乘法器的交替方向法解决目标函数的优化问题,在连续求导的情况下,使用如下迭代公式: /> DCAEs算法流程请参照图3,在训练每一层的过程中,计算解码有的重构向量x'和输入向量x的损失函数,并将损失函数优化到设定值,重复这个动作直到所有层都训练完毕,编码公式如下: y=subsampling(x) 解码的具体公式如下: 其中,π 步骤4:分类器预测 将最终提取的特征放入随机森林分类器,对分类器进行训练,并优化关键的参数,优化完毕后,将测试集放入分类器进行circRNA和疾病的关联预测,并根据Rank排名,为生物学提供有效的关联信息。 随机森林的生成方法为: S41)使用Bootstrap从有放回的N个样本集中随机选择N个样本,选取的N个样本作为决策树根节点的样本用于训练决策树; S42)从样本的M个特征中随机选择m个特征,满足条件m< S43)在形成决策树的过程中,每个节点都必须按照步骤S42进行分裂,直到不能再分裂,在整个决策树形成过程中不进行剪枝; S44)按照步骤S41-S43构建多个决策树,形成随机森林。 步骤5:得到预测结果。 通过评估计算,本发明提出的基于图嵌入和动态卷积自编码器来预测circRNA和疾病关联的新型计算模型在circR2Disease数据集上使用5折交叉验证的AUC值0.928。Acc、Sen、F1和MCC的平均得分分别为0.9273、0.9165、0.8939和0.8261。与大多数模型的结果相我们的发明具有良好的预测性能,可以帮助预测潜在的circRNA和疾病关联。同时,为了进一步评测我们发明的性能,我们替换了不同的分类器并在其他数据集上也取得了好的实验效果,不同分类器的实验结果请参照图4,在最终的预测结果中我们的发明在预测的circRNA和疾病关联分数前20的关联中,有16种得到了文献和数据集验证,这也足以证明我们的发明可以为生物实验提供可靠的验证对象。 以上所述仅为本发明以circR2Disease数据集为基准数据集上的实施例而已,并不用以限制本发明,凡在本发明精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
- 一种基于矩阵分解的食物-疾病关联预测方法
- 一种基于自回避随机游走的疾病关联miRNA预测方法及系统
- 一种新型的环状RNA-疾病关联预测方法
- 一种基于深度矩阵分解的环状RNA疾病关联预测方法