一种用于血清中农、兽药及化学污染物残留的筛查方法
文献发布时间:2024-04-18 19:52:40
技术领域
本发明涉及分析化学领域,是一种针对血清中强极性和非极性农,兽药及化学污染物同时筛查的基于二维液相色谱-高分辨质谱技术的高覆盖分析方法
背景技术
目前,食用农产品中的农兽药,化学污染物残留超标,已成为世界各地广泛关注的话题。据文献报道,农兽药等污染物残留在人体的蓄积,会产生急性或慢性毒性作用,对人体的神经系统,免疫系统等产生危害。基于国家对高覆盖检测技术及膳食慢性暴露与慢性病关联研究的需求,实现对农兽药等污染物在人体中残留形态的侦测,一种针对人体血清基质的高覆盖的农,兽药及化学污染物残留筛查技术时必不可少的。
基于LC-MS的分析技术被广泛应用到食品及人体的农、兽药及化学污染物残留筛查研究中。目前检索到的基于LC-MS的残留筛查研究已超过一千篇,其中大多数检测手段只针对某一类或某几类农兽药进行靶向选择性离子监测分析,如有机磷类,有机氯类等。近年来,学者们基于超高效液相色谱及高分辨质谱分析方法,不断发展针对不同基质的农兽药污染物非靶标筛查方法。而针对人体基质的残留筛查技术受限于样品量少,提取难度大,农、兽药及化学污染物残留丰度低等多方面困难,难以突破瓶颈。此外,10.常用的农兽药及污染物的log P范围大概维-4到10,一根色谱柱难以满足log P范围过宽的物质的同时保留和分离。
面对众多的log P相差较大的农、兽药及化学污染物品种,目前的检测技术仍不能满足高通量高分辨的要求。基于以上需求,本发明开发针对血清基质的高覆盖高分辨率的农、兽药及化学污染物非靶向筛查技术,通过引入捕集柱将进行高效浓缩预处理后的血清样品中的化学性质各异的农兽药及化学污染物分离并转移,结合后续分别针对强极性组和非极性组的平行柱分析,实现一次进样同时有效筛查强极性组和非极性组外源农、兽药及化学污染物。
发明内容
本发明是针对血清中可能存在的化学性质各异的农、兽药及化学污染物,提供一种基于2D LC-MS的非靶向筛查技术,通过引入捕集柱将进行高效浓缩预处理后的血清样品中的农兽药及化学污染物分离并转移,结合后续分别针对强极性组和非极性组的平行柱分析,实现一次进样同时有效筛查强极性组和非极性组外源农、兽药及化学污染物。
为实现上述目的,本发明采用的技术方案如下:
(1)取90μL血清样本,加入360μL乙腈溶液,然后旋转1分钟,将混合物在10000g,4℃条件下离心10分钟;然后移取上清液400μL,冷冻干燥;在二维液相色谱串联-高分辨质谱(2D LC-MS)残留分析之前,冻干样品在30μL的H
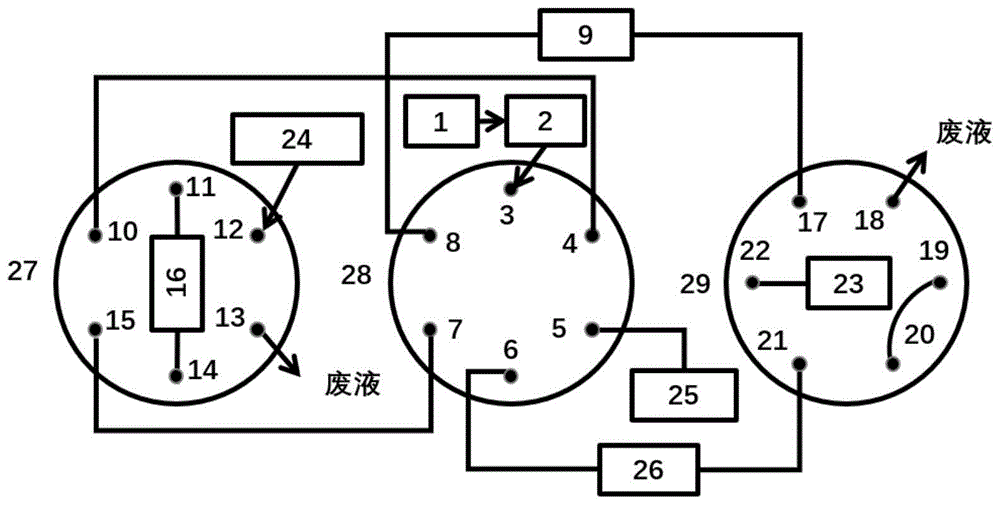
(2)将重新复溶的样品注入2D LC-MS系统进行分析。采用的2D LC装置包括液相色谱泵1(1)、液相色谱泵2(25)、稀释液泵(24)、自动进样器(2)、液相色谱柱1(9)、液相色谱柱2(26)、捕集柱(16)、六通阀1(27)、六通阀2(28)、六通阀3(29)、和检测器(23);主要包括强极性组份转移、强极性组份分离、非极性组份分离三个过程;
初始时,六通阀2(28)的3号位与4号位相连,六通阀1(27)的12号位与13号位相连,六通阀3(29)的19号位与20号位相连;液相色谱泵1(1)的输出端经过自动进样器(2)与六通阀2(28)的3号位相连,六通阀(28)的4号位通过管线与六通阀2(27)的10号位相连,六通阀1(27)的11号位与捕集柱(16)的输入端相连,捕集柱(16)的输出端与六通阀1(27)的14号位相连,六通阀1(27)的15号位通过管线与六通阀2(28)的7号位相连,六通阀2(28)的8号位与液相色谱柱1(9)的输入端相连,液相色谱柱1(9)的输出端与六通阀3(29)的17号位相连,六通阀3(29)的22号位与检测器相连;液相色谱泵2(25)与六通阀2(28)的5号位相连,六通阀2(28)的6号位和与色谱柱2(26)的输入端相连,色谱柱2(26)的输出端与六通阀3(29)的21号位相连,六通阀3(29)的20号位与19号位通过管线相连,19号位接入废液;在该位置,自动进样器(2)将复杂样品提取液进样到捕集柱(16),样品中的强极性组组份首先流出并转移至液相色谱柱1(9)中;
待强极性组组份从捕集柱(16)完全流出后,控制六通阀1(27)切换,六通阀1(27)的10号位与15号位相连,六通阀1(27)的10号位和11号位堵死;在该位置实现强极性组组份在液相色谱柱1(9)中的分离以及液相色谱柱2(26)的平衡;
待强极性组组份在液相色谱柱1(9)中分离完成后,控制六通阀1(27)、六通阀2(28)、和六通阀3(29)同时切换,在该位置,捕集柱(16)中的非极性组组份转移到液相色谱柱2(26)中并进行分离;与此同时,完成液相色谱柱1(9)的平衡;
待非极性组组份分离完成后,控制六通阀1(27)切换;稀释液泵(24)与六通阀1(27)的12号位相连,六通阀1(27)的11号位与捕集柱(16)的输入端相连,捕集柱(16)的输出端与六通阀1(27)的14号位相连,六通阀1(27)的13号位接入废液;在该位置,实现捕集柱(16)中残留溶剂的替换。
(3)基于2D LC-MS的农、兽药及化学污染物筛查方法如下:
液相色谱分离方法:液相色谱柱1(9)采用反相Discovery(R)HS F5,2.1mm×50mm,3.0μm色谱柱用于强极性组的分离;液相色谱柱2(26)采用反相BEH C18,2.1mm×50mm,1.7μm色谱柱用于非极性组份的分离;捕集柱(16)采用反相BEH C18,2.1mm×5mm,1.7μm色谱柱用于强极性组和非极性组组份的转移。液相色谱泵1(1)的流动相A为水、流动相B为甲醇,液相色谱泵2(25)的流动相A为水、流动相B为体积比为3/2的乙腈/异丙醇。正离子模式均添加5mmol/L的甲酸铵和0.1%的甲酸,负离子模式添加5mmol/L的甲酸铵;稀释液泵(24)的流动相为体积比1/10的甲醇/水溶液。柱温为55℃。
质谱参数:采用All MS质谱数据采集方法,具体实施方法:选择MS扫描模式,扫描窗口1:碰撞能(CE)设置为0V;扫描窗口2:CE设置为30V,以实现所有一级母离子都打二级的All MS采集模式。扫描速度:5张谱/秒;扫描离子范围m/z=50-1050(正负离子模式)
(4)所述液相色谱分析方法可以用于血清样品中强极性组和非极性组份农、兽药及化学污染的同时筛查。强极性组份包含油水分配系数(log P)小于等于3.52的常用农、兽药及化学污染物,即在色谱柱1(9)上洗脱;非极性组组份包括log P大于3.52的常用农、兽药及化学污染物,即在色谱柱2(26)上洗脱。
(5)建立常用农、兽药及化学污染物定性数据库并验证数据库定性结果的可靠性:含有常用农、兽药及化学污染物的标准品溶液稀释至浓度为1μg/mL,直接进入高分辨质谱进行分析,采用全扫(Full Scan)扫描模式采集一级质谱信息和保留时间。根据获得的m/z和t
质谱参数:一级MS采集方法:MS扫描模式,扫描窗口1:碰撞能(CE)设置为0V;扫描速度:5张谱/秒;扫描离子范围m/z=50-1050(正负离子模式);获取标准品m/z和t
将血清筛查结果与数据库进行匹配,质量精度为±10ppm,保留时间偏差设为±0.2min,二级质谱碎片匹配数量大于等于2。满足上述条件即可判断为该残留组份的阳性筛查结果。
本发明通过将血清样品提取物进行浓缩富集,使其在进样后保留在捕集柱上,根据log P的差异将血清样品中的强极性组和非极性组组份分离,通过控制六通阀的位置分别将强极性组和非极性组组份转移到液相色谱柱1和液相色谱柱2上,结合后续的高效液相色谱分离系统,实现一次进样同时分析血清中强极性和非极性农、兽药及化学污染物。
本发明特点在于通过引入捕集柱将进行高效浓缩预处理后的血清样品中的化学性质各异的农兽药及化学污染物分离并转移,结合后续分别针对强极性组和非极性组的平行柱分析,实现一次进样同时有效筛查强极性组和非极性组外源农、兽药及化学污染物。本发明方法适用于复杂样品中强极性组和非极性组农、兽药及化学污染物残留的同时筛查,具有通量高、残留筛查覆盖度广、操作简便、重复性好等优势。
本发明具有如下优点:本发明方法适用于血清中强极性和非极性农、兽药及化学污染物的同时筛查。与传统的一维液相色谱筛查方法相比,使用本发明方法只需一次进样就可实现log P=-4~10的可能外源残留组份的同时分析,缩短了分析时间,提高了分析通量;此外,降低了血清样品的使用量,对于十分有限的血清样本特别重要;筛查覆盖度广,对于极性较强和非极性较强组分均可得到良好的分离效果;具有操作简便、重复性好等优势。
附图说明
图1为本发明提供的液相色谱分析方法的装置图;
图2a为本发明提供的液相色谱分析方法的流路图之一;
图2b为本发明提供的液相色谱分析方法的流路图之二;
图2c为本发明提供的液相色谱分析方法的流路图之三;
图2d为本发明提供的液相色谱分析方法的流路图之四;
图1-图2d中数字代表:1为液相色谱泵1,2为自动进样器,27为六通阀1,10-15为六通阀1(27)的接口,9为液相色谱柱1,26为液相色谱柱2,25为液相色谱泵2,28为六通阀2,3-8为六通阀(28)的接口,16为捕集柱,24为稀释液泵,29为六通阀3,17-22为六通阀(28)的接口,23为检测器。
图3为本发明对强极性和非极性进行分析的总提取离子流图。图中横坐标为保留时间(min),纵坐标为响应强度/10
图4为本发明对血清样品进行分析的总离子流图。图中横坐标为保留时间(min),纵坐标为响应强度/10
表1为本发明2D LC分离过程的具体梯度条件。
表2为发明对血清样品进行分析的阳性筛查结果。
具体实施方式
下面结合实施例对本发明进行进一步描述。
实施例1:强极性和非极性农、兽药及化学污染物同时分析的2D LC-MS方法建立。
常用的农、兽药及化学污染物涉及的农药包括有机氯类、有机氟类、有机氮类、有机磷类、氨基甲酸酯类和三唑类,兽药包括头孢菌素类、磺胺类、喹诺酮类、大环内酯类、苯并咪唑类和非甾体抗炎药类,污染物包括苯胺类、硝基苯类、邻苯二甲酸酯类和全氟化合物类,共计1205种。将含有以上常用农、兽药及化学污染物的标准品用乙腈稀释至总浓度为1μg/mL的混合标准品溶液,直接进入2D LC-MS进行分析,采用全扫(Full Scan)扫描模式采集一级质谱信息和保留时间。根据获得的m/z和t
本发明方法采用的2D LC装置包括液相色谱泵1、液相色谱泵2、稀释液泵、自动进样器、液相色谱柱1、液相色谱柱2、捕集柱、六通阀1、六通阀2、六通阀3和检测器;主要包括强极性组份转移、强极性组份分离、非极性组份分离三个过程;
初始时(图2a),六通阀2(28)的3号位与4号位相连,六通阀1(27)的12号位与13号位相连,六通阀3(29)的19号位与20号位相连;液相色谱泵1(1)的输出端经过自动进样器(2)与六通阀2(28)的3号位相连,六通阀(28)的4号位通过管线与六通阀2(27)的10号位相连,六通阀1(27)的11号位与捕集柱(16)的输入端相连,捕集柱(16)的输出端与六通阀1(27)的14号位相连,六通阀1(27)的15号位通过管线与六通阀2(28)的7号位相连,六通阀2(28)的8号位与液相色谱柱1(9)的输入端相连,液相色谱柱1(9)的输出端与六通阀3(29)的17号位相连,六通阀3(29)的22号位与检测器相连;液相色谱泵2(25)与六通阀2(28)的5号位相连,六通阀2(28)的6号位和与色谱柱2(26)的输入端相连,色谱柱2(26)的输出端与六通阀3(29)的21号位相连,六通阀3(29)的20号位与19号位通过管线相连,19号位接入废液;在该位置,自动进样器(2)将复杂样品提取液进样到捕集柱(16),样品中的强极性组组份首先流出并转移至液相色谱柱1(9)中;
待强极性组组份从捕集柱(16)完全流出后,控制六通阀1(27)切换,六通阀1(27)的10号位与15号位相连,六通阀1(27)的10号位和11号位堵死;在该位置实现强极性组组份在液相色谱柱1(9)中的分离以及液相色谱柱2(26)的平衡(图2b);
待强极性组组份在液相色谱柱1(9)中分离完成后,控制六通阀1(27)、六通阀2(28)、和六通阀3(29)同时切换,在该位置,捕集柱(16)中的非极性组组份转移到液相色谱柱2(26)中并进行分离;与此同时,完成液相色谱柱1(9)的平衡(图2c);
待非极性组组份分离完成后,控制六通阀1(27)切换;稀释液泵(24)与六通阀1(27)的12号位相连,六通阀1(27)的11号位与捕集柱(16)的输入端相连,捕集柱(16)的输出端与六通阀1(27)的14号位相连,六通阀1(27)的13号位接入废液;在该位置,实现捕集柱(16)中残留溶剂的替换(图2d)。
液相色谱分离方法:液相色谱柱1(9)采用反相Discovery(R)HS F5,2.1mm×50mm,3.0μm色谱柱用于强极性组的分离;液相色谱柱2(26)采用反相BEH C18,2.1mm×50mm,1.7μm色谱柱用于非极性组份的分离;捕集柱(16)采用反相BEH C18,2.1mm×5mm,1.7μm色谱柱用于强极性组和非极性组组份的转移。液相色谱泵1(1)的流动相A为水、流动相B为甲醇,液相色谱泵2(25)的流动相A为水、流动相B为体积比为3/2的乙腈/异丙醇。正离子模式流动相A和流动相B中均分别添加5mmol/L的终浓度甲酸铵和终体积浓度0.1%的甲酸,负离子模式流动相A和流动相B中均分别添加终浓度5mmol/L的甲酸铵;
稀释液泵(24)的流动相为体积比1/10的甲醇/水溶液。柱温为55℃。
梯度洗脱的具体过程和切阀时间见表1。
质谱参数:一级MS采集方法:MS扫描模式,扫描窗口1:碰撞能(CE)设置为0V;扫描速度:5张谱/秒;扫描离子范围m/z=50-1050(正负离子模式);获取标准品m/z和t
在本实施例中,通过图2a-2d过程,便实现了强极性和非极性农、兽药及化学污染物标准品的同时分析,并建立了包含1205种常用农、兽药及化学污染物色谱质谱数据库。
实施例2:血清样品体系强极性和非极性农、兽药及化学污染物的同时分析
样品:基质加标血清提取物
具体提取过程如下:取90μL血清样本,加入330μL乙腈溶液,再加入30μL含总浓度为1μg/mL的常用强极性和非极性农、兽药及化学污染物的混合标准品乙腈溶液,包括强极性标准品2-甲基-5-硝基咪唑、环丙氨嗪、磺胺吡啶、地昔尼尔、替硝唑、噻虫嗪、灭害威、可待因、利妥特灵、萎锈灵、甲氧苄啶、多菌灵、乙氧酰胺苯甲酯、左旋咪唑、脱叶磷、黄曲霉毒素B1、抗蚜威、氨基甲苯哒唑、磺胺硝苯、氯丙那林、溴布特罗、克林霉素、卡拉洛尔、马来酸溴苯那敏、阿普洛尔和非极性标准品碱性橙14、艾地那非、泰乐菌素、盐酸苄达明、酮康唑、酮咯酸、血根碱、甲霜灵、孔雀石绿、去烷基氟拉西泮、盐酸氯丙嗪、喷布洛尔、草乃敌、玉米赤霉醇、氟啶酮、芬苯达唑、敌稗、氟尼辛葡甲胺、醋酸氟轻松、百克敏、全氟十一烷酸、扑虱灵、马杜霉素等共1205种农、兽药及化学污染物。然后旋转1分钟,将混合物在10000g,4℃条件下离心10分钟;然后移取上清液400μL,冷冻干燥;在二维液相色谱串联-高分辨质谱(2DLC-MS)残留分析之前,冻干样品在30μL的H
基于2D LC-MS的农、兽药及化学污染物筛查方法如下:
液相色谱分离方法:用于强极性组分离的色谱柱采用反相Discovery(R)HS F5,2.1mm×50mm,3.0μm色谱柱;用于非极性组的分离采用反相BEH C18,2.1mm×50mm,1.7μm色谱柱用于非极性组份的分离;捕集柱采用反相BEH C18,2.1mm×5mm,1.7μm色谱柱用于强极性组和非极性组组份的转移。于强极性组分离的色谱流动相A为水、流动相B为甲醇,强极性组分离的色谱流动相A为水、流动相B为体积比为3/2的乙腈/异丙醇。正离子模式均添加5mmol/L的甲酸铵和0.1%的甲酸,负离子模式添加5mmol/L的甲酸铵;稀释液泵的流动相为体积比1/10的甲醇/水溶液。柱温为55℃。
梯度洗脱的具体过程和切阀时间见表1。
质谱参数:采用All MS质谱数据采集方法,具体实施方法:选择MS扫描模式,扫描窗口1:碰撞能(CE)设置为0V;扫描窗口2:CE设置为30V,以实现所有一级母离子都打二级的All MS采集模式。扫描速度:5张谱/秒;扫描离子范围m/z=50-1050(正负离子模式)
使用本发明方法同时分析血清样品体系强极性和非极性农、兽药及化学污染物,质谱采集的总提取离子流图如附图3所示,将加标血清筛查结果与实施例1所建立的1205种农、兽药及化学污染物数据库进行匹配,质量精度为±5-10ppm,保留时间偏差设为±0.2-0.5min,若提取的色谱峰采集到的质谱信号与数据库中的t
实施例3:血清样品残留非靶向筛查
样品:血清提取物
具体提取过程如下:取90μL血清样本,加入360μL乙腈溶液,然后旋转1分钟,将混合物在10000g,4℃条件下离心10分钟;然后移取上清液400μL,冷冻干燥;在二维液相色谱串联-高分辨质谱(2D LC-MS)残留分析之前,冻干样品在30μL的H
基于2D LC-MS的农、兽药及化学污染物筛查方法如下:
液相色谱分离方法:用于强极性组分离的色谱柱采用反相Discovery(R)HS F5,2.1mm×50mm,3.0μm色谱柱;用于非极性组的分离采用反相BEH C18,2.1mm×50mm,1.7μm色谱柱用于非极性组份的分离;捕集柱采用反相BEH C18,2.1mm×5mm,1.7μm色谱柱用于强极性组和非极性组组份的转移。于强极性组分离的色谱流动相A为水、流动相B为甲醇,强极性组分离的色谱流动相A为水、流动相B为体积比为3/2的乙腈/异丙醇。正离子模式均添加5mmol/L的甲酸铵和0.1%的甲酸,负离子模式添加5mmol/L的甲酸铵;稀释液泵的流动相为体积比1/10的甲醇/水溶液。柱温为55℃。
梯度洗脱的具体过程和切阀时间见表1。
质谱参数:采用All MS质谱数据采集方法,具体实施方法:选择MS扫描模式,扫描窗口1:碰撞能(CE)设置为0V;扫描窗口2:CE设置为30V,以实现所有一级母离子都打二级的All MS采集模式。扫描速度:5张谱/秒;扫描离子范围m/z=50-1050(正负离子模式)
使用本发明方法对血清样品进行农、兽药及化学污染物筛查,质谱采集的总离子流图如附图4所示,将采集的色谱质谱信息与实施例1所建立的1205种农、兽药及化学污染物血清筛查结果与数据库进行匹配,质量精度为±5-10ppm,保留时间偏差设为±0.2-0.5min,若实际样品中采集到的质谱信号与数据库中的t
表1 2D LC分离过程的具体梯度条件
表2对血清样品进行分析的阳性筛查结果
/>
/>
- 一种用于果蔬中农药残留的非靶向快速筛查方法
- 一种用于多种兽药残留快速筛查的检测方法