一种3-苄氧甲基吲哚-2-羧酸衍生物及其制备方法与应用
文献发布时间:2024-04-18 19:58:26
技术领域
本发明涉及医药技术领域,具体是一种3-苄氧甲基吲哚-2-羧酸衍生物及其制备方法与应用。
背景技术
自从20世纪80年代早期,获得性免疫缺陷综合症(AIDS)艾滋病在全世界传播以来,已经给人类健康和生命带来了巨大的威胁。迄今为止,由艾滋病导致的死亡已超过百万,而目前艾滋病病毒(HIV-1)携带者的人数已达数千万。当前AIDS治疗的主要策略是联合使用逆转录酶抑制剂和蛋白酶抑制剂的高效抗逆转录病毒疗法(Highly active anti-retroviral therapy,HAART),又称“鸡尾酒疗法”。尽管该策略在AIDS治疗上发挥了重要作用,但两类抑制剂存在较大的抗药性、高毒性及高价格,临床上迫切需要寻找新的抗AIDS药物靶点。
在AIDS生命周期中,病毒整合过程至关重要。整合过程是将病毒的DNA整合到人宿主DNA的过程,是HIV-1复制周期的不可缺少的过程。HIV-1整合酶(integrase)参与整合全过程,催化整个整合反应,是HIV-1复制过程必不可少的酶,也是病毒稳定感染必不可缺的酶。另外,由于作用靶点不一样,它不受目前的因化疗药使用所产生的耐药性影响,对目前的耐药变异株仍有作用。其次,在人类细胞中没有的对应物,使得靶向整合酶的抑制剂具有较低的毒性。因此,整合酶已成为抗HIV-1研究中极具吸引力的新靶点。
尽管目前已有5个整合酶抑制剂上市,但是由于病毒耐药株的出现,整合酶抑制剂的疗效有所降低;另外,虽然公开号为CN1560035A的专利公开了5-羟基吲哚-3-羧酸酯类衍生物,其具有抗HIV病毒的活性,但其显示的是抗HIV-1蛋白酶活性效果,安全性和抗HIV-1整合酶活性效果未知,且该方案的制备工艺繁杂,需要采用催化剂、氧化剂等多种试剂,同时还需要通过控制氧化剂的配比和反应时间来控制目标产物,这直接提高了控制要求,导致目标产物难控制,此外,由于该化合物为蛋白酶抑制剂,与整合酶是完全不同的抗病毒靶点,作用机制也完全不同。因此,探索一种作用模式新颖、高效、低毒、合成控制简单的新一代整合酶抑制剂十分有必要。
发明内容
为了解决现有技术中存在的上述技术问题,本发明提供一种3-苄氧甲基吲哚-2-羧酸衍生物及其制备方法与应用,具体如下:
一种3-苄氧甲基吲哚-2-羧酸衍生物,其结构通式如式(I)所示:
优选的,所述通式(Ⅰ)中,R
一种3-苄氧甲基吲哚-2-羧酸衍生物,包括但不限于以下化合物:
本发明的另一个目的是提供3-苄氧甲基吲哚-2-羧酸衍生物的制备方法,以吲哚-2-羧酸类化合物为原料,经酯化反应保护羧基后,在吲哚环上的2、3或6位上引入不同取代基,最后经碱水解释放出羧基,即得3-苄氧甲基吲哚-2-羧酸衍生物。
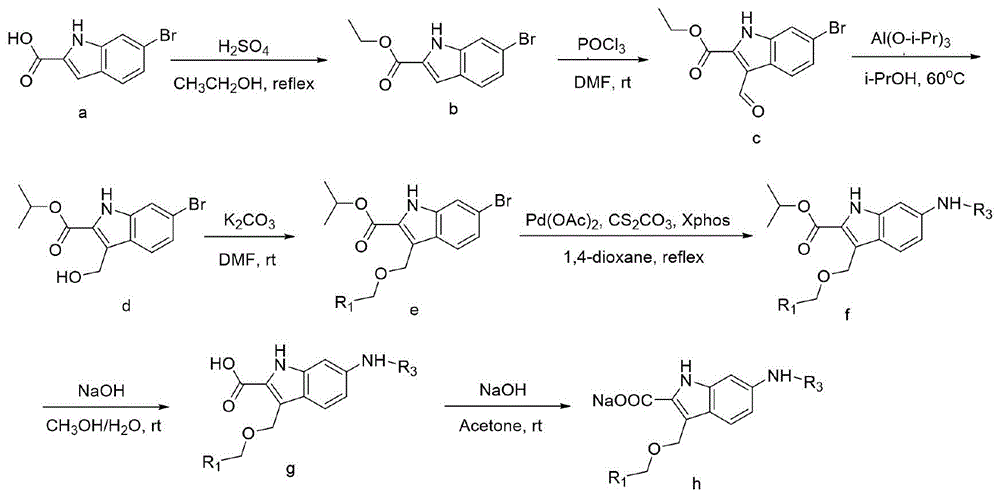
具体地,所述的3-苄氧甲基吲哚-2-羧酸衍生物的制备方法,包括如下步骤:
(1)合成6-溴-1H-吲哚-2-羧酸乙酯:将6-溴-1H-吲哚-2-羧酸(即化合物a)溶于乙醇中,加入浓硫酸,油浴条件下80℃回流搅拌反应,薄层色谱检测反应,反应完全后,取出反应液进行后处理,即得6-溴-1H-吲哚-2-羧酸(即化合物b);
(2)合成3-甲酰基-6-溴-1H-吲哚-2-羧酸乙酯:将6-溴-1H-吲哚-2-羧酸(化合物b)溶于N,N-二甲基甲酰胺中,再缓慢加入三氯氧磷,室温反应,薄层色谱检测反应,反应完全后取出反应液,将反应液缓慢滴加到冰水中,进行后处理,即得3-甲酰基-6-溴-1H-吲哚-2-羧酸乙酯(即化合物c);
(3)合成3-(羟甲基)-6-溴-1H-吲哚-2-羧酸异丙酯(即化合物d):将3-甲酰基-6-溴-1H-吲哚-2-羧酸乙酯(化合物c)溶于异丙醇中,加入异丙醇铝,60℃条件下油浴反应,薄层色谱检测反应,反应完全后取出反应液,旋干,用硅胶柱色谱分离纯化即得化合物d;
(4)合成中间化合物e:将3-(羟甲基)-6-溴-1H-吲哚-2-羧酸异丙酯(化合物d)、取代苄溴或取代溴甲基吡啶、碳酸钾溶于N,N-二甲基甲酰胺中,常温反应,薄层色谱检测反应,反应完全后取出反应液,加水,用乙酸乙酯萃取,旋干萃取液,用硅胶柱色谱分离纯化中间化合物e;
(5)合成目标化合物f:将步骤(4)合成的中间化合物e、取代苯胺类或取代吡啶、醋酸钯、2-二环己基膦-2',4',6'-三异丙基联苯、以及碳酸铯溶于1,-4-二氧六环中,110℃条件下油浴反应,薄层色谱检测反应,反应完全后取出反应液,过滤反应液,旋干滤液,用硅胶柱色谱分离纯化目标化合物f。
进一步地,一种3-苄氧甲基吲哚-2-羧酸衍生物的制备方法,在上述步骤(1)-步骤(5)的基础上,还包括(6)合成目标化合物g,即将步骤(5)合成的目标化合物f及氢氧化钠溶于甲醇与水的混合溶剂中,常温搅拌反应,薄层色谱检测反应,反应完全后用乙酸调节pH至弱酸性,加水,用乙酸乙酯萃取,旋干萃取液,装柱,用硅胶柱色谱分离纯化目标化合物B;所述甲醇与水的混合溶剂中甲醇:水=3:1。
再进一步地,一种3-苄氧甲基吲哚-2-羧酸衍生物的制备方法,在上述步骤(1)-步骤(6)的基础上,还包括(7)合成目标化合物h,即将步骤(6)合成的目标化合物B溶解于丙酮中,向该溶液中逐步滴加氢氧化钠水溶液至大量白色固体析出,少量冷水洗涤滤饼,干燥,即得目标化合物h。
优选的,在步骤(1)、步骤(2)中,所述后处理是先调反应液pH,再依次经乙酸乙酯萃取、浓缩、薄层色谱分离纯化。进一步优选的,所述调反应液pH、用乙酸乙酯萃取、浓缩、分离等操作,具体是调pH至弱碱性,用乙酸乙酯萃取,浓缩萃取液,装柱,用薄层色谱分离纯化目标产物。
所述调反应液pH是采用无水碳酸钠调节反应液pH至弱碱性。
所述薄层色谱分离为硅胶柱色谱分离纯化。
在步骤(4)中,所述取代苄溴为对三氟甲基苄溴、邻氟苄溴中任一种;所述取代吡啶为4-三氟甲基-3羟甲基吡啶。
在步骤(5)中,所述取代胺类为3-氟-4-甲氧基苯胺、2,4-二氟苯胺、对氟苄胺中任一种;所述取代吡啶为2-甲基-5氨基吡啶。
本发明的再一方面是提供一种3-苄氧甲基吲哚-2-羧酸衍生物在制备抗HIV的药物中的应用。
进一步优选为,提供一种3-苄氧甲基吲哚-2-羧酸衍生物在制备HIV-1型整合酶抑制剂中的应用。
更进一步地,一种HIV-1型整合酶抑制剂包括具有通式(I)所示化合物,或其立体异构体、几何异构体、水合物、溶剂化物、药学上可接受的盐,或包含通式(I)所示化合物的药物组合物。
有益效果:
本发明的3-苄氧甲基吲哚-2-羧酸衍生物经实验验证,具有优异的HIV-1整合酶抑制活性和高安全性。
本发明的3-苄氧甲基吲哚-2-羧酸衍生物的制备方法简单、步骤简便、结构新颖;制备时间短,总反应时间少于24h;反应条件温和;利用TLC的方法,具有简单、快速、灵敏,可实时监测的优势;本发明的工艺控制简单,无须复杂地控温和时间控制。
附图说明
图1为3-苄氧甲基吲哚-2-羧酸衍生物的合成路线;
图2为3-苄氧甲基吲哚-2-羧酸衍生物与整合酶的结合模式图。
具体实施方式
实施例1
(1)合成6-溴-1H-吲哚-2-羧酸乙酯(化合物b1):将100mg化合物a1(6-溴-1H-吲哚-2-羧酸)溶于10mL乙醇中,加入0.5mL浓硫酸,80℃油浴搅拌反应1.5h,待反应结束后,取出反应液,加入无水碳酸钠调反应液pH至弱碱性,用乙酸乙酯萃取、浓缩萃取液,用硅胶柱色谱分离纯化出目标化合物b1;
(2)合成3-甲酰基-6-溴-1H-吲哚-2-羧酸乙酯(化合物c1):将100mg化合物b1溶于10mLN,N-二甲基甲酰胺中,再缓慢加入1.9mL三氯氧磷,室温反应2h,待反应结束后,取出反应液,加入无水碳酸钠调反应液pH至弱碱性,用乙酸乙酯萃取、旋干萃取液,用硅胶柱色谱分离纯化出目标化合物c1;
(3)合成3-(羟甲基)-6-溴-1H-吲哚-2-羧酸异丙酯(化合物d1):将100mg化合物c1溶于15mL异丙醇中,加入150mg异丙醇铝,油浴锅60℃下反应约5h,待反应结束后,取出反应液,旋干反应液,用硅胶柱色谱分离纯化出目标化合物d1;
(4)合成6-溴-3-(4-三氟甲基苄氧甲基)-1H-吲哚-2-羧酸异丙酯(化合物e1):将100mg化合物d1、78mg对三氟甲基苄溴、160mg碳酸钾溶于10mL N,N-二甲基甲酰胺中,常温反应5h,待反应结束后,取出反应液,加适量水,用乙酸乙酯萃取、旋干萃取液,用硅胶柱色谱分离纯化出目标化合物e1;
(5)合成6-(3-氟-4-甲氧基苯氨基)-3-(4-三氟甲基苄氧甲基)-1H-吲哚-2-羧酸异丙酯(化合物f1):将100mg化合物e1,115mg 3-氟-4甲氧基苯胺,20mg醋酸钯,38mg 2-二环己基膦-2',4',6'-三异丙基联苯,110mg碳酸铯溶于10mL 1,-4-二氧六环中,油浴锅110℃下反应约4h,待反应结束后,取出反应液,过滤反应液,旋干滤液,用硅胶柱色谱分离纯化出目标化合物f1;
具有HIV-1整合酶抑制活性的化合物f1
(6)合成6-((3-氟-4-甲氧基苯氨基)-3-(4-三氟甲基苄氧甲基)-1H-吲哚-2-羧酸(化合物g1):将100mg化合物f1及60mg氢氧化钠溶于4mL甲醇与水的混合溶剂中(甲醇:水=3:1)中,常温搅拌反应。待反应结束后进行后处理,然后分离纯化出目标化合物g1;
具有HIV-1整合酶抑制活性的化合物g1
(7)合成6-((3-氟-4-甲氧基苯氨基)-3-(4-三氟甲基苄氧甲基)-1H-吲哚-2-羧酸钠(化合物h1):将100mg的化合物g1溶解于5mL丙酮中,向该溶液中逐步滴加氢氧化钠水溶液至大量白色固体析出,过滤,少量冷水洗涤滤饼,干燥,即得目标化合物h1。
具有HIV-1整合酶抑制活性的化合物h1
实施例2
将实施例1中的步骤(4)中的对三氟甲基苄溴替换为邻氟苄溴,其余步骤相同,最后得到中间体e2。
实施例3
将实施例1中的步骤(5)中的3-氟-4-甲氧基苯胺替换为2,4-二氟苯胺,其余步骤相同,最后得到具有HIV-1整合酶抑制活性的化合物f2。
具有HIV-1整合酶抑制活性的化合物f2
实施例4
将实施例1中的步骤(5)中的化合物e1替换为e2,其余步骤相同,最后得到具有HIV-1整合酶抑制活性的化合物f3。
具有HIV-1整合酶抑制活性的化合物f3
实施例5
将实施例1中的步骤(5)中的化合物e1替换为e2,3-氟-4-甲氧基苯胺替换为2,4-二氟苯胺,其余步骤相同,最后得到具有HIV-1整合酶抑制活性的化合物f4。
具有HIV-1整合酶抑制活性的化合物f4
实施例6
将实施例1中的步骤(6)中的化合物f1替换为化合物f2,其余步骤相同,最后得到具有HIV-1整合酶抑制活性的化合物g2。
具有HIV-1整合酶抑制活性的化合物g2
实施例7
将实施例1中的步骤(6)中的f1替换为f3,其余步骤相同,最后得到具有HIV-1整合酶抑制活性的化合物g3。
具有HIV-1整合酶抑制活性的化合物g3
实施例8
将实施例1中的步骤(6)中的f1替换为f4,其余步骤相同,最后得到具有HIV-1整合酶抑制活性的化合物g4。
具有HIV-1整合酶抑制活性的化合物g4
实施例9
将实施例1中的步骤(7)中的g1替换为g2,其余步骤相同,最后得到具有HIV-1整合酶抑制活性的化合物h2。
具有HIV-1整合酶抑制活性的化合物h2
实施例10
将实施例1中的步骤(7)中的g1替换为g3,其余步骤相同,最后得到具有HIV-1整合酶抑制活性的化合物h3。
具有HIV-1整合酶抑制活性的化合物h3
实施例11
将实施例1中的步骤(7)中的g1替换为g4,其余步骤相同,最后得到具有HIV-1整合酶抑制活性的化合物h4。
具有HIV-1整合酶抑制活性的化合物h4
实施例12
将实施例1中的步骤(5)中的3-氟-4-甲氧基苯胺替换为2-甲基苯胺,其余步骤相同,最后得到具有HIV-1整合酶抑制活性的化合物f5。
具有HIV-1整合酶抑制活性的化合物f5
实施例13
将实施例1中的步骤(6)中的f1替换为f5,其余步骤相同,最后得到具有HIV-1整合酶抑制活性的化合物g5。
具有HIV-1整合酶抑制活性的化合物g5
实施例14
将实施例1中的步骤(7)中的g1替换为g5,其余步骤相同,最后得到具有HIV-1整合酶抑制活性的化合物h5。
具有HIV-1整合酶抑制活性的化合物h5
实施例15
将实施例1中的步骤(4)中的3-三氟甲基苄溴替换为4-三氟甲基-3-溴甲基吡啶,其余步骤相同,最后得到中间体e3。
实施例16
将实施例1中的步骤(5)中的e1替换为e3,3-氟-4甲氧基苯胺替换为2-甲基-5氨基吡啶,其余步骤相同,最后得到具有HIV-1整合酶抑制活性的化合物f6。
具有HIV-1整合酶抑制活性的化合物f6
实施例17
将实施例1中的步骤(6)中的f1替换为f6,其余步骤相同,最后得到具有HIV-1整合酶抑制活性的化合物g6。
具有HIV-1整合酶抑制活性的化合物g6
实施例18
将实施例1中的步骤(7)中的g1替换为g6,其余步骤相同,最后得到具有HIV-1整合酶抑制活性的化合物h6。
具有HIV-1整合酶抑制活性的化合物h6
实施例19
将实施例1中的步骤(5)中的3-氟-4-甲氧基苯胺替换为对氟苄胺,其余步骤相同,最后得到具有HIV-1整合酶抑制活性的化合物f7。
具有HIV-1整合酶抑制活性的化合物f7
实施例20
将实施例1中的步骤(6)中的f1替换为f7,其余步骤相同,最后得到具有HIV-1整合酶抑制活性的化合物g7。
具有HIV-1整合酶抑制活性的化合物g7
实施例21
将实施例1中的步骤(7)中的g1替换为g7,其余步骤相同,最后得到具有HIV-1整合酶抑制活性的化合物h7。
具有HIV-1整合酶抑制活性的化合物h7
对实施例1-21所得的具有HIV-1整合酶抑制活性的化合物进行如下测定:
1.整合酶链转移活性测定
实施例1-21所得的具有HIV-1整合酶抑制活性的化合物的活性如下:
HIV-1整合酶抑制剂活性检测方法主要采用整合酶试剂盒检测。
(1)包被DS DNA:从链霉亲和素涂层的96孔板上去除任何不使用的条形孔,并将它们重新密封在含有干燥剂的铝箔袋中。将所需DS DNA 100×溶液(10μL DSDNA 100×溶液和990μL反应缓冲液)在反应缓冲液中稀释100倍。每孔加入100μL 1×DS DNA溶液,37℃培养箱孵育30min。将反应缓冲液返回37℃水浴。从板孔中抽液,用300μL洗涤缓冲液洗涤5次。每孔加入阻断缓冲液200μL,37℃培养箱孵育30min。
(2)将酶1:300稀释成反应缓冲液(2μL HIV-1整合酶和598μL反应缓冲液)。从板孔中吸出液体,用200μL反应缓冲液洗涤3次。每孔加入反应缓冲液(阴性对照)或整合酶溶液(阳性对照)100μL,37℃培养箱孵育30min。
(3)用200μL反应缓冲液洗涤三次。每个被试品每孔加入反应缓冲液50μL(阳性对照和阴性对照均为反应缓冲液),室温孵育5min。
(4)将所需TS DNA 100×溶液在反应缓冲液中稀释100倍(每mL TS DNA 100×溶液10μL,反应缓冲液990μL)。每孔直接加入50μL的1×TS DNA溶液到50μL缓冲液/测试品中。将盘子在固定的手上轻轻敲打3-5次,混合各种反应。37℃孵育30分钟。
(5)从板孔中抽液,用300μL洗涤液洗涤5次。每孔加入100μL HRP抗体溶液,37℃孵育30min。
(6)从板孔中抽液,用300μL洗涤液洗涤5次。每孔加入100μL TMB过氧化物酶底物溶液,室温孵育10分钟。
(7)在含有TMB底物的孔中直接加入100μL TMB停止液。用移液管尖端戳破任何大气泡。使用设置在450nm的平板阅读器读取孔的吸光度至少0.1秒。在加入TMB停止液后10分钟内读取板。
(8)使用酶标仪测定吸光度A值,通过公式100×[1-(加药组A值-空白对照A值)/(整合酶对照组A值-空白对照A值)]来计算抑制率。
2.细胞毒性实验
细胞毒性实验使用MTT法检测。将待测样品用DMSO溶解,再用磷酸盐缓冲液将其稀释为不同的所需浓度,取出100μL不同浓度的待测化合物加入到接种MT-4细胞(5×10
/>
以上活性测试结果说明,本发明的化合物对T细胞(MT-4)的毒性非常低,只有化合物f3和f4观察到较低的毒性,其他化合物均为发现明显毒性。另一方面,所发明的化合物对HIV-1整合酶链转移具有十分显著的抑制活性,活性最佳的化合物1n IC
- 一种3-(苄氧基)-4-氧代-4H-吡喃-2-羧酸的合成方法
- 一种制备手性1-苄氧羰基六氢哒嗪-3-羧酸和手性1,2-二苄氧羰基六氢哒嗪-3-羧酸的方法
- 一种制备手性1-苄氧羰基六氢哒嗪-3-羧酸和手性1,2-二苄氧羰基六氢哒嗪-3-羧酸的方法