2-氯噻吩的合成及精制方法
文献发布时间:2024-04-18 19:59:31
技术领域
本发明属于化工技术领域,特别涉及一种2-氯噻吩的合成及精制方法,尤其涉及一种2-氯噻吩的工业化合成方法。
背景技术
2-氯噻吩是一种重要的医药中间体,通常是用噻吩经氯化工艺后制得的。目前工业上生产方式如下:噻吩和一定量的盐酸在0-10℃混合,滴加双氧水,获得2-氯噻吩,同时有少量的3-氯噻吩和2,5-二氯噻吩。经过萃取、脱溶、精馏后获得2-氯噻吩产品。2-氯噻吩的产品规格为:2-氯噻吩>98%,3-氯噻吩<1%。
反应方程式如下:
。
如申请号为CN201310479585.5的专利公开了一种2-氯噻吩的合成方法,其步骤如下:在带有机械搅拌的反应容器中加入质量浓度为25-35%的盐酸,噻吩和三乙胺,然后降低温度至-10-0℃,在此温度下缓慢滴加质量浓度为25-35%的双氧水,盐酸,噻吩,三乙胺和双氧水的比例为600ml:95-105g:1-3ml:130-150g;滴毕,在此温度下保温8-12h,保温结束后静置分层,水层用适量乙酸乙酯萃取1-3次,合并有机层,再用饱和食盐水洗涤,有机层减压浓缩至干得到2-氯噻吩。
药典中,对于2-氯噻吩的质量控制主要是两种杂质的控制:3-氯噻吩和2,5-二氯噻吩;申请人在生产过程中发现,2-氯噻吩(沸点为128-129℃)和3-氯噻吩(沸点为137-139℃)的沸点比较接近,纯化难度高,通常需要经过多次精馏(如4-5次)才能得到高纯度的2-氯噻吩。
发明内容
申请人发现在2-氯噻吩的合成过程中,2-氯噻吩的生成是一个较快的反应,3-氯噻吩的生成是一个较慢的反应,加快反应进程可减少3-氯噻吩的生成。因此,在本专利中,提高了反应温度和减少了盐酸的用量,并且让噻吩仅转化30-70%就停止反应。噻吩相对于2-氯噻吩很容易分离,而反应后的3-氯噻吩很少,则一次精馏即可得到高纯度(99.4%以上)的2-氯噻吩。所述技术方案如下:
本发明实施例提供了一种2-氯噻吩的合成及精制方法,包括:噻吩、盐酸和双氧水进行反应,反应完成后经萃取、脱溶和精馏得到产品。盐酸与噻吩的摩尔比为0.5-1.5:1,盐酸与双氧水的摩尔比与现有技术一致,反应温度为30-60℃,噻吩的转化率至30-70%时停止反应。脱溶过程控制(通过控制温度和压力)回收溶剂中噻吩含量为小于5.0wt%(优选小于2.0wt%),让噻吩基本都残留在脱溶残液中以便于回收。脱溶残液送精馏塔的中上部进行精馏,精馏温度可以与现有技术精馏2-氯噻吩的温度差不多(根据需要可稍低);精馏塔的顶部输出噻吩并作为原料,其中下部输出2-氯噻吩(纯度大于99.4%,优选可达99.7%以上)。
在本专利中,如果仅增加温度(不减少盐酸和减少反应时间),会产生较多杂质,无法得到高纯度的2-氯噻吩;如果仅减少盐酸,会导致收率低;如果仅减少反应时间,也会导致收率低。在本专利中,通过多种手段结合,在保证收率的同时,在一次精料下即可得到高纯度的2-氯噻吩。
进一步低,精馏完成后,对精馏塔的釜底液进行二次精馏,二次精馏的温度可以与现有技术精馏2-氯噻吩的温度差不多;二次精馏时,精馏塔的下部输出2,5-二氯噻吩,其中部输出3-氯噻吩,其塔顶输出2-氯噻吩(纯度大于99%)。在本专利中,二次精馏的量非常少,可合并多次反应的釜底液或多个精馏塔的釜底液进行二次精馏,节约能源。
优选地,盐酸与噻吩的摩尔比为0.7-1.0:1。
其中,反应时间为8-10小时。
其中,为了在脱溶过程中保证噻吩少残留在萃取剂中,在保证萃取效果的同时还需要采用低沸点的萃取剂;则萃取溶剂选自二氯甲烷。
优选地,噻吩、盐酸和双氧水的摩尔比为1:0.7:1,反应温度为55℃,反应时间为8h。
具体地,本发明提供的2-氯噻吩的合成及精制方法包括:
(1)合成:噻吩、盐酸和双氧水进行反应,反应温度为40-60℃, 盐酸与噻吩的摩尔比为0.7-1.0:1, 反应时间为8-10小时,噻吩的转化率至30-70%时停止反应。
(2)萃取:采用二氯甲烷对步骤(1)得到的反应液进行萃取,有机相送步骤(3)。
(3)脱溶:有机相通过减压蒸馏进行脱溶,控制回收溶剂中噻吩含量小于5wt%,脱溶残液送步骤(4)。
(4)一次精馏:脱溶残液送第一精馏塔的中上部;第一精馏塔的顶部输出噻吩并作为原料送步骤(1),其中下部输出2-氯噻吩,其釜底液送步骤(5)。
(5)二次精馏:釜底液送第二精馏塔;第二精馏塔的下部输出2,5-二氯噻吩,其中部输出3氯噻吩,其塔顶输出2-氯噻吩。
本发明实施例提供的技术方案带来的有益效果是:
(1)经一次精馏即可得到高纯度的2-氯噻吩,纯度大于99.4%,3-氯噻吩的含量小于0.3%;
(2)精馏次数少,能耗低,设备投入低,更易生产;
(3)在本专利中,精馏的同时实现噻吩(在本专利中,非常容易分离,纯度高)的回收;
(4)由于噻吩再次利用,收率(一次精馏和二次精馏的总收率)与现有技术差不多。
附图说明
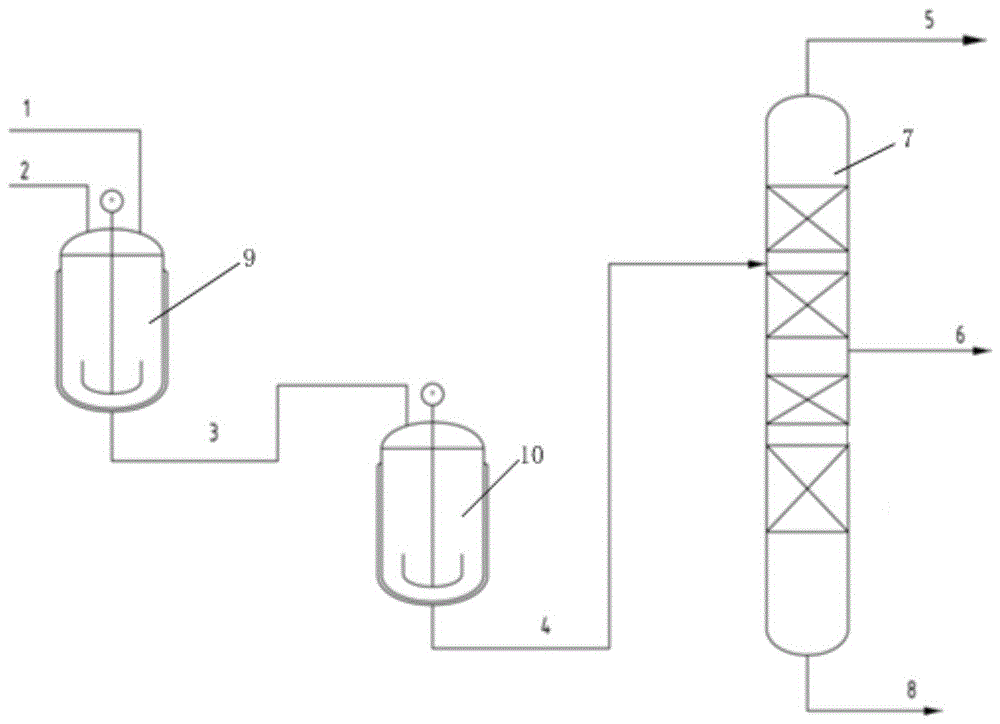
图1是本发明实施例提供的2-氯噻吩的合成及精制系统的原理框图。
图中:1噻吩和盐酸、2双氧水、3反应液、4有机相、5 噻吩、6 2-氯噻吩、7精馏塔、8釜底液、9反应釜、10萃取脱溶釜。
具体实施方式
为使本发明的目的、技术方案和优点更加清楚,下面将结合附图对本发明作进一步地详细描述。
实施例1:
实施例1提供了一种2-氯噻吩的合成及精制方法,包括:将1.0mol噻吩、1mol20wt%盐酸(以HCl计)加入烧瓶中,水浴控温在10℃,滴加双氧水1.5mol,逐渐升高水浴温度至50℃,保温10小时,反应完成后进行萃取(二氯甲烷)和脱溶,脱溶残液取样气相色谱分析,得产物组成如表1所示:
表1
从表1可以看出,反应完成后,产物中含有大量的噻吩,少量的2,5-二氯噻吩,微量的3-氯噻吩。
脱溶残液送精馏塔精馏得到2-氯噻吩,产品组成如表2所示:
表2
实施例2:
实施例2提供了一种2-氯噻吩的合成及精制方法,包括:将1.0mol噻吩、0.7 mol20wt%盐酸(以HCl计)加入烧瓶中,水浴控温在15℃,滴加双氧水1.0mol,逐渐升高水浴温度至55℃,保温8小时,反应完成后进行萃取(二氯甲烷)和脱溶,脱溶残液取样气相色谱分析,得产物组成如表3所示:
表3
从表3可以看出,反应完成后,产物中含有大量的噻吩,少量的2,5-二氯噻吩,微量的3-氯噻吩。
脱溶残液送精馏塔精馏得到2-氯噻吩,产品组成如表4所示:
表4
从表4可以看出,经精馏后的产品中,含有微量的噻吩(由于反应液中噻吩的含量较高,精馏后也会残留少许,但是噻吩非常容易去除(如精馏))和3-氯噻吩,2-氯噻吩的纯度非常高,达到99.7%以上。
精馏塔的釜底液进行二次精馏,产品组成如表5所示:
表5
从表5可以看出,二次精馏也能得到纯度99%的2-氯噻吩,相当于现有技术精馏三次的纯度。
对比例1
将1.0mol噻吩、2mol 20wt%盐酸(以HCl计)加入烧瓶中,水浴控温在15℃,滴加双氧水1.0mol,在15℃下保温反应,保温24小时,反应完成后进行萃取(乙酸乙酯)和脱溶,脱溶残液取样气相色谱分析,得产物组成如表6所示:
表6
从表6可以看出,反应完成后,产物中噻吩基本无残留,含有大量的2,5-二氯噻吩,较多的3-氯噻吩。
脱溶残液送精馏塔进行一次精馏得到2-氯噻吩,产品组成如表7所示:
表7
从表7可以看出,一次精馏后,噻吩基本去除,含有较多的3-氯噻吩,2-氯噻吩的纯度为97%。
一次精馏得到的2-氯噻吩进行二次精馏,产品组成如表8所示:
表8
二次精馏得到的2-氯噻吩进行三次精馏,产品组成如表9所示:
表9
三次精馏得到的2-氯噻吩进行四次精馏,产品组成如表10所示:
表10
从表10可以看出,即使精馏四次,2-氯噻吩的纯度仍然较本专利低;申请人发现,即使再增加三次精馏,2-氯噻吩的纯度也不能达到99.7%。
对比例2
将1.0mol噻吩、2mol 20wt%盐酸(以HCl计)加入烧瓶中,水浴控温在15℃,滴加双氧水1.0mol,升温至55℃,保温反应24小时,反应完成后进行萃取(乙酸乙酯)和脱溶,脱溶残液取样气相色谱分析,得产物组成如表11所示:
表11
从表11可以看出,反应完成后,产物中杂质的含量非常高,2-氯噻吩仅50%左右。即在现有技术的基础上,增加反应温度会大大增加杂质的产生。
对比例3:
将1.0mol噻吩、0.7 mol 20wt%盐酸(以HCl计)加入烧瓶中,水浴控温在15℃,滴加双氧水1.0mol,逐渐升高水浴温度至55℃,保温24小时,反应完成后进行萃取(二氯甲烷)和脱溶,脱溶残液取样气相色谱分析,得产物组成如表12所示:
表12
从表12可以看出,反应完成后,产物中含有较多的3-氯噻吩;相对于本专利来说,延长反应时间,会提升2-氯噻吩的含量(变为1.3倍),但是3-氯噻吩的含量(变为9.4倍)增加更加明显。
对比例4:
将1.0mol噻吩、1.8 mol 20wt%盐酸(以HCl计)加入烧瓶中,水浴控温在15℃,滴加双氧水1.0mol,逐渐升高水浴温度至55℃,保温8小时,反应完成后进行萃取(二氯甲烷)和脱溶,脱溶残液取样气相色谱分析,得产物组成如表13所示:
表13
从表13可以看出,反应完成后,产物中含有大量的噻吩和2,5-二氯噻吩,少量的3-氯噻吩。增加盐酸的量,可以促进反应进行(同时得到更多的2,5-二氯噻吩),增加2-氯噻吩的含量(变为1.4倍),但是3-氯噻吩的含量(变为5.5倍)增加更加明显。
以上所述仅为本发明的较佳实施例,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 一种适用于珠光颜料的大粒径片状α-氧化铝粉末的合成方法
- 一种以片状α-氧化铝为基质的珠光颜料制备方法