液相色谱电化学法高通量检测儿茶酚胺及其代谢物的方法
文献发布时间:2024-04-18 20:01:55
技术领域
本发明涉及分析检测技术领域,尤其涉及液相色谱电化学法高通量检测儿茶酚胺及其代谢物的方法。
背景技术
儿茶酚胺(Catecholamine,CA)类物质主要包括肾上腺素(Epinephrine,E)、去甲肾上腺素(Norepinephrine,NE)和多巴胺(Dopamine,DA)。CA是由肾上腺髓质、肾上腺神经元以及肾上腺外嗜铬体合成与分泌的单胺类神经递质,是具有较强的生理学活性的内源性物质,在大脑和神经信号传导中起着重要的作用。血浆和尿液中儿茶酚胺及其代谢物水平与人体多种生理、病理现象有密切关系,不仅对人体的心血管系统、神经系统、内分泌系统、肾脏、平滑肌组织等的生理活动起着广泛的调节作用,还同时影响人体的代谢。检测生物样本中的儿茶酚胺及其代谢物水平不仅对于嗜铬细胞瘤、副神经节瘤、神经母细胞瘤、高血压、心肌梗塞、肾上腺髓质增生等疾病的诊断和治疗具有重要的临床意义,并且有助于甲状腺功能异常、充血性心力衰竭、糖尿病、肾脏功能不全等疾病的诊断。同时,对于神经电生理等基础医学研究也具有重要的意义。
儿茶酚胺的代谢物主要包含变肾上腺素(Metanphrine,MN)、去甲变肾上腺素(Normetanephrine, NMN)、3-甲氧基酪胺(3-methoxytyramine,3-MT)、香草扁桃酸(Vanillylmandelic acid,VMA)和高香草酸(Homovanillic acid,HVA)。NMN及MN分别是NE和E的中间代谢产物(合称MNs),其仅在肾上腺髓质嗜铬细胞或PPGL肿瘤体内代谢生成,并且以高浓度水平持续存在;MNs的半衰期较CA长,也更加稳定。VMA是E、NE的主要终末代谢产物。嗜铬细胞瘤患者能够分泌大量的肾上腺素和去甲肾上腺素,其中大约60%将最终转化为 VMA,随尿液排出体外。HVA与VMA结构相似,是体内单胺类神经递质多巴胺的终末代谢产物。
嗜铬细胞瘤( pheochromocytoma, PCC) 起源于肾上腺髓质嗜铬细胞,分泌产生儿茶酚胺;副神经节瘤( paraganglioma, PGL)起源于肾上腺外嗜铬细胞,位于胸、腹和盆腔脊柱旁交感神经链,也可来源于沿颈部和颅底分布的舌咽、迷走神经的副交感神经节,后者常不产生CA。PCC 占 80% ~ 85%,PGL 占 15% ~ 20% ,二者总称PPGLs(pheochromocytomas or paragangliomas),是继发性高血压的少见类型,在普通门诊高血压患者中的患病率为0.2%~0.6%。神经母细胞瘤患者体内可同时分泌过量的肾上腺素、去甲肾上腺素及多巴胺,其代谢产物VMA和HVA通过肾脏排泄,在疾病早期即可升高。同时检测尿VMA、HVA不仅可以间接反映体内儿茶酚胺的排泌情况,而且对嗜铬细胞瘤、神经母细胞瘤等中枢神经系统疾病的早期辅助诊断及鉴别具有重要的意义。
目前,临床上关于儿茶酚胺及其代谢物的检测方法主要包括液相色谱-串联质谱法、高效液相色谱法、毛细管电泳法、荧光光度法等。由中华医学会内分泌学分会肾上腺学组在中华内分泌代谢杂志发表的《嗜铬细胞瘤和副神经节瘤诊断治疗的专家共识》中提出,首先推荐液相色谱-质谱法(LC-MS/MS)测定儿茶酚胺及其代谢物。液相色谱-质谱法具有非常高的灵敏度,特异性和准确度,但是LC-MS/MS法复杂的设备操作对操作人员有更高的要求,昂贵的设备及维护成本需要更大的资金投入。除此之外,共识中还推荐液相色谱电化学法(HPLC-ECD)测定儿茶酚胺及其代谢物,相比质谱仪,电化学检测器成本更低,操作简单。
现有技术CN 106610405 A公开了一种先固相萃取然后采用高效液相色谱电化学法检测人尿液中儿茶酚胺类物质和其甲氧基代谢物的方法,但是其仅能检测甲肾上腺素、肾上腺素、去甲苄肾上腺素、间甲肾上腺素四种物质,且色谱检测时间较长。李炳源等人在《高效液相色谱电化学检测生物样品中儿茶酚胺的研究》中也提供了一种检测方法,其也仅能检测少量的几种儿茶酚胺物质,不适用儿茶酚胺代谢物的同时检测,且色谱检测时间较长。
因此,如何在较短的时间内同时检测多种儿茶酚胺及其代谢物,降低检测成本,简化操作,同时又能保证检测方法的灵敏度、特异性和准确度,是目前亟需解决的技术难题。
发明内容
为解决上述技术难题,特提出本发明。
首先,本发明提供了一种液相色谱电化学法高通量检测儿茶酚胺及其代谢物的方法,包括:
将待测样品经WCX固相萃取后,采用液相色谱联合电化学检测器进行检测;
所述液相色谱的条件包括:
C18色谱柱;
流动相A:柠檬酸、磷酸二氢钾、正己烷磺酸钠和乙二胺四乙酸二钾盐的水溶液;
流动相B:甲醇;
洗脱过程中流动相A和流动相B的体积百分比之和为100%;
洗脱程序采用等度洗脱,流动相A的体积百分比为80%~85%;
所述儿茶酚胺及其代谢物为多巴胺(DA)、肾上腺素(E)、去甲肾上腺素(NE)、去甲变肾上腺素(NMN)、变肾上腺素(MN)、3-甲氧基酪胺(3-MT)中的至少一种。
本发明通过对各项检测条件的组合大量摸索优化后发现,将待测样品经WCX固相萃取后,采用上述液相色谱检测条件联合电化学检测器检测后,能够实现快速同时对多种儿茶酚胺及其代谢物的高灵敏度、高特异性和高准确度的检测,大幅降低检测成本,简化操作。
在上述检测方案中,所述液相色谱的检测时间为15min以内。
在一些实施方案中,所述待测样品为尿液。
优选地,流动相A的体积百分比为82%~83%。
本发明进一步经过实验探究后发现,控制流动相A的体积百分比为82%~83%进行等度洗脱,能够使得液相色谱的检测时间在12min以内实现对多巴胺、肾上腺素、去甲肾上腺素、去甲变肾上腺素、变肾上腺素、3-甲氧基酪胺的同时检测。
优选地,所述液相色谱的检测时间为10min以内。
优选地,电化学检测器的检测电压为800V~1200V。
优选地,电化学检测器的检测电压为1000V~1200V。
优选地,电化学检测器的检测电压为1050V~1150V。
本发明进一步经过实验探究后发现,在1050V~1150V检测电压时,多巴胺、肾上腺素、去甲肾上腺素、去甲变肾上腺素、变肾上腺素、3-甲氧基酪胺这6种儿茶酚胺及其代谢物的电化学信号均较强。
优选地,所述C18色谱柱的规格为3μm,4.6
优选地,流动相A为10~20mM柠檬酸、30~40mM磷酸二氢钾、3~5mM正己烷磺酸钠以及0.05~0.2mg/mL乙二胺四乙酸二钾盐二水合物的水溶液。
更优选地,流动相A为12~18mM柠檬酸、32~38mM磷酸二氢钾、3~4mM正己烷磺酸钠以及0.05~0.15mg/mL乙二胺四乙酸二钾盐二水合物的水溶液。
优选地,所述液相色谱的条件还包括:流速为0.5~1.5 mL/min;和/或,柱温为35~45℃;和/或,进样量为20~40μL。
优选地,所述固相萃取的步骤包括:
将待测样品与乙酸铵溶液混合,然后经WCX固相萃取,采用乙酸铵溶液和甲醇分别淋洗,而后采用乙腈和甲酸的水溶液洗脱。
优选地,所述固相萃取的步骤还包括:洗脱后的洗脱液经氮吹吹干,用盐酸和甲醇的水溶液复溶后,采用液相色谱联合电化学检测器进行检测。
采用上述固相萃取的方式制备的待检液,联合上述液相色谱条件分离之后,采用电化学检测器检测时,6种儿茶酚胺及其代谢物的灵敏度、特异性和准确度更佳。
优选地,所述检测方法还包括:采用外标法进行定量检测。
优选地,所述液相色谱为高效液相色谱。
与现有技术相比,本发明的有益效果在于:
本发明提供了一种利用高效液相色谱电化学法高通量同时检测多种儿茶酚胺及其代谢物的方法,该检测方法能够满足6种儿茶酚胺及其代谢物的线性、准确度、精密度、残留和回收率的检测要求,相比质谱检测方法,大幅降低了检测成本,简化操作,方便,快速,同时又能保证检测方法的灵敏度、特异性和准确度,对监测儿茶酚胺及其代谢物具有重要意义。
附图说明
图1是实施例1儿茶酚胺及其代谢物的色谱图。
图2是实施例3儿茶酚胺及其代谢物的色谱图。
图3是对比例1儿茶酚胺及其代谢物的色谱图。
图4是对比例2儿茶酚胺及其代谢物的色谱图。
图5是对比例3儿茶酚胺及其代谢物的色谱图。
图6是对比例4儿茶酚胺及其代谢物的色谱图。
具体实施方式
为使本发明的目的、技术方案和优点更加清楚,下面将对本发明中的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
实施例中未注明具体技术或条件者,均为常规方法或者按照本领域的文献所描述的技术或条件进行,或者按照产品说明书进行。所用试剂和仪器等未注明生产厂商者,均为可通过正规渠道商购买得到的常规产品。
实施例1
本实施例提供了一种高效液相色谱电化学法高通量检测儿茶酚胺及其代谢物的方法,具体步骤如下:
1、主要化合物和试剂如表1所示。
表1 标准品和试剂
2、实验所需仪器设备如表2所示。
表2 仪器设备
3、标准品及质控品配制:
3.1儿茶酚胺及其代谢物储备液配制
多巴胺、肾上腺素、去甲肾上腺素、去甲变肾上腺素、变肾上腺素、3-甲氧基酪胺储备液配制:使用十万分之一电子天平,分别称取多巴胺、肾上腺素、去甲肾上腺素、去甲变肾上腺素、变肾上腺素、3-甲氧基酪适量,分别置于10 mL容量瓶,用5%1M HCl甲醇溶解后并定容至刻度线,配制成1 mg/mL浓度的储备液,转移至15 mL棕色中,贴标签,-80 ℃保存。
3.2儿茶酚胺及其代谢物次级储备液配制
使用移液枪,分别移取各储备液100μL和900μL 5%1M HCl甲醇溶液,配制成100μg/mL的单个次级储备液。
使用移液枪,移取240μL多巴胺次级储备液,120μL肾上腺素次级储备液,120μL去甲肾上腺素次级储备液,120μL去甲变肾上腺素次级储备液,120μL变肾上腺素次级储备液,240μL 3-甲氧基酪胺次级储备液,再加入40μL 5%1M HCl甲醇溶液,混合均匀,配制成儿茶酚胺混合次级储备液1,浓度为:多巴胺:24 μg/mL、肾上腺素:12 μg/mL、去甲肾上腺素:12μg/mL、去甲变肾上腺素:12 μg/mL、变肾上腺素:12 μg/mL、3-甲氧基酪胺:24 μg/mL,贴标签,-80 ℃保存。
移取6μL儿茶酚胺混合次级储备液1,加入1434μL 5%1M HCl甲醇溶液,混合均匀,配制成儿茶酚胺混合次级储备液2,浓度为:多巴胺:0.1μg/mL、肾上腺素:0.05 μg/mL、去甲肾上腺素:0.05μg/mL、去甲变肾上腺素:0.05 μg/mL、变肾上腺素:0.05 μg/mL、3-甲氧基酪胺:0.1 μg/mL,贴标签,-80 ℃保存。
3.3校准品S1至S6配制
标准品配制根据高低配制模式进行,即配制好S1与S6后,通过比例混合配制成S2至S5及LQC,MQC和HQC。
校准品S6配制:选用合适规格的移液枪移取儿茶酚胺混合次级储备液1 0.2 mL,置于合适的HDPE管内,加入超纯水1.8 mL,配制浓度多巴胺:2.4μg/mL、肾上腺素:1.2 μg/mL、去甲肾上腺素:1.2μg/mL、去甲变肾上腺素:1.2 μg/mL、变肾上腺素:1.2 μg/mL、3-甲氧基酪胺:2.4 μg/mL。充分混合后,贴标签,-20 ℃保存。
校准品S1配制:选用合适规格的移液枪移取儿茶酚胺混合次级储备液2 0.2 mL,置于合适的HDPE管内,加入超纯水1.8 mL,配制浓度多巴胺:0.01μg/mL、肾上腺素:0.005μg/mL、去甲肾上腺素:0.005μg/mL、去甲变肾上腺素:0.005μg/mL、变肾上腺素:0.005 μg/mL、3-甲氧基酪胺:0.01 μg/mL。充分混合后,贴标签,-20 ℃保存。
校准品S2配制:准确移取标准品S6 3 μL,移取标准品S1 714 μL,充分混合,配制浓度多巴胺:0.02μg/mL、肾上腺素:0.01μg/mL、去甲肾上腺素:0.01μg/mL、去甲变肾上腺素:0.01 μg/mL、变肾上腺素:0.01 μg/mL、3-甲氧基酪胺:0.02μg/mL。充分混合后,贴标签,-20 ℃保存。
校准品S3配制:准确移取标准品S6 10 μL,移取标准品S1 468 μL,充分混合,配制浓度多巴胺:0.06μg/mL、肾上腺素:0.03μg/mL、去甲肾上腺素:0.03μg/mL、去甲变肾上腺素:0.03 μg/mL、变肾上腺素:0.03 μg/mL、3-甲氧基酪胺:0.06μg/mL。充分混合后,贴标签,-20 ℃保存。
校准品S4配制:准确移取标准品S6 50 μL,移取标准品S1 450 μL,充分混合,配制浓度多巴胺:0.249μg/mL、肾上腺素:0.1245μg/mL、去甲肾上腺素:0.1245μg/mL、去甲变肾上腺素:0.1245 μg/mL、变肾上腺素:0.1245 μg/mL、3-甲氧基酪胺:0.249μg/mL。充分混合后,贴标签,-20 ℃保存。
校准品S5配制:准确移取标准品S6 250 μL,移取标准品S1 250 μL,充分混合,配制浓度多巴胺:1.205μg/mL、肾上腺素:0.6025μg/mL、去甲肾上腺素:0.6025μg/mL、去甲变肾上腺素:0.6025 μg/mL、变肾上腺素:0.6025 μg/mL、3-甲氧基酪胺:1.205μg/mL。充分混合后,贴标签,-20 ℃保存。
3.4质控品配制
低浓度质控品LQC配制:准确移取标准品S6 4μL,移取标准品S1 496 μL,充分混合,配制浓度多巴胺:0.02912μg/mL、肾上腺素:0.01456μg/mL、去甲肾上腺素:0.01456μg/mL、去甲变肾上腺素:0.01456 μg/mL、变肾上腺素:0.01456μg/mL、3-甲氧基酪胺:0.02912μg/mL。充分混合后,贴标签,-20 ℃保存。
中浓度质控品MQC配制:准确移取标准品S6 180μL,移取标准品S1 320 μL,充分混合,配制浓度多巴胺:0.8704μg/mL、肾上腺素:0.4352μg/mL、去甲肾上腺素:0.4352μg/mL、去甲变肾上腺素:0.4352μg/mL、变肾上腺素:0.4352μg/mL、3-甲氧基酪胺:0.8704μg/mL。充分混合后,贴标签,-20 ℃保存。
高浓度质控品HQC配制:准确移取标准品S6 380μL,移取标准品S1 120μL,充分混合,配制浓度多巴胺:1.8264μg/mL、肾上腺素:0.9132μg/mL、去甲肾上腺素:0.9132μg/mL、去甲变肾上腺素:0.9132μg/mL、变肾上腺素:0.9132μg/mL、3-甲氧基酪胺:1.8264μg/mL。充分混合后,贴标签,-20 ℃保存。
4、前处理试剂配制
样本稀释液:精密称取0.7704g乙酸铵,加入200mL纯水,超声混合均匀即可。
20mM乙酸铵:精密称取0.30816g乙酸铵,加入200mL纯水,超声混合均匀即可。
洗脱液:量取196mL乙腈,4mL水和4mL甲酸,共204mL液体,混合均匀即可。
复溶液:量取45mL水,移取5mL 1M HCl甲醇溶液,共50mL液体,混合均匀即可。
5、流动相配制
流动相A液配制:在500 mL纯水中加入终浓度16mM柠檬酸,36mM磷酸二氢钾,3.45mM正己烷磺酸钠和50mg乙二胺四乙酸二钾盐二水合物,超声,混合均匀后使用。
流动性B液配制:甲醇,过滤后超声脱气,待用。
6、前处理方法
(1)加校准品、质控品溶液:移取300 μL标准品溶液/质控品溶液,分别加至新的2ml离心管中;
(2)加样本稀释液:移取300μL样本稀释液依次加至上述离心管,混匀;
(3)活化:移取200μL甲醇至WCX板中,控制液滴速度,加压滴干,再移取200μL 20mM乙酸铵至WCX板中,控制液滴速度,加压滴干;
(4)上样:将步骤(2)中的样品移取至WCX板中,控制液滴速度,加压滴干;
(5)淋洗:移取300μL 20mM乙酸铵至WCX板中,控制液滴速度,加压滴干,再移取300μL甲醇至WCX板中,控制液滴速度,加压滴干;
(6)洗脱:更换样本接收板,移取200μL洗脱液至WCX板中,控制液滴速度,加压滴干;
(7)氮吹:将样本接收板于25℃下氮吹至干;
(8)复溶:加入200μL复溶液至样本接收板中,封膜,混匀3 min;
(9)检测:将样本接收板置于仪器中,采用液相LC20AD检测。
7、高效液相色谱联合电化学检测器检测
1)色谱柱:C18(3μm,4.6
2)流动相:A相:水(含16mM柠檬酸,36mM磷酸二氢钾,3.45mM正己烷磺酸钠,0.1mg/mL乙二胺四乙酸二钾盐二水合物);
B相:甲醇;
3)洗脱程序如表3所示;
表3 洗脱程序条件
4)流速:0.8 mL/min;
5)柱温:40℃;
6)自动进样器温度:10℃;
7)洗针液:甲醇:水(1:1v/v);
8)进样量:30μL。
9)电化学检测器的检测电压为1100V。
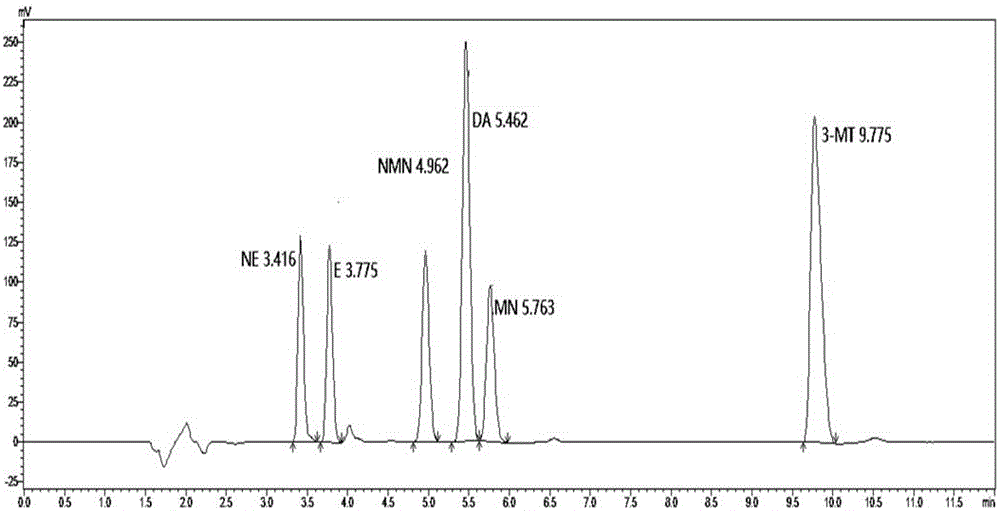
儿茶酚胺及其代谢物的色谱图如图1所示。
实施例2
本实施例对实施例1检测方法的线性范围、精密度和准确度、残留以及回收率进行测试。
1、线性范围
配制校准品和质控品,按照实施例1的前处理方法对样品进行处理,由加入标准品的量(x)与仪器计算出峰面积(y)进行线性回归,要求线性回归的相关系数r≥0.990。
根据实验数据计算得到以下回归方程:
DA:y=667.462x-760.984(r=0.9996),
E:y=511.931x-105.030(r=0.9998),
NE:y=527.938x-396.232(r=0.9995),
NMN:y=608.897x-168.955(r=0.9999),
MN:y=558.733x-283.918(r=0.9999),
3-MT:y=767.284x-988.730(r=0.9997),
结果表明,DA和3-MT在10-2400 ng/mL范围内呈良好的线性关系,E、NE、NMN和MN在5-1200 ng/mL范围内呈良好的线性关系。
2、精密度和准确度
配制定量下限(LLOQ)和质控品三个浓度的样品,定量下限浓度是校准品S1的浓度,按照校准品S1的配制方法进行配制。按照前处理方法对样品进行处理,处理5个平行样本,计算精密度和准确度。计算样品的变异系数和相对偏差,要求变异系数(CV)≤15%,准确度的相对偏差(B)≤ ±15%。
实验结果如表4所示。
表4
结论:各化合物定量下限、低值质控品、中值质控品和高值质控品变异系数(CV)≤15%,准确度的相对偏差(B)≤ ±15%。
3、残留
准备定量上限样本和空白样本,定量上限浓度是校准品S6的浓度,按照校准品S6的配制方法进行配制,空白样本是10%1M HCl甲醇即复溶液。按实施例1进行前处理,通过先进定量上限样本,随后连续进三个空白样本,来评价是否有残留。要求空白样本的峰面积<20%LLOQ。
实验结果如表5所示。
表5
结论:各化合物空白样本的峰面积<20%LLOQ,没有残留。
4、回收率
取30μL低值和高值质控品样本,加入270μL尿液,配制两个加标回收样本,DA和3-MT的低值加标浓度是29.12ng/mL,高值加标浓度是1826.4ng/mL,NE、E、NMN和MN的低值加标浓度是14.56ng/mL,高值加标浓度是913.2ng/mL,按照实施例1的前处理方法对样本进行处理。要求加标回收率为80%~120%。
实验结果如表6所示。
表6
/>
结论:低值和高值加标样本加标回收率均为80%~120%。
综上,实施例1的检测方法可同时检测多巴胺、肾上腺素、去甲肾上腺素、去甲变肾上腺素、变肾上腺素和3-甲氧基酪胺,具有灵敏度高,重复性好,准确性高,特异性好等特点。该方法中多巴胺(DA)和3-甲氧基酪胺(3-MT)的线性范围为10-2400 ng/mL,肾上腺素(E)、去甲肾上腺素(NE)、去甲变肾上腺素(NMN)和变肾上腺素(MN)的线性范围为5-1200ng/mL,相关系数均r ≥ 0.990;定量下线、低值、中值、高值质控品精密度的变异系数(CV)均≤15%,准确度的相对偏差(B)均≤ ±15%,加标回收率为80%~120%。
实施例3
本实施例提供了一种高效液相色谱电化学法高通量检测儿茶酚胺及其代谢物的方法,具体步骤仅与实施例1不同的是:
洗脱程序中流动相A与流动相B的体积比调整为84:16。
儿茶酚胺及其代谢物的色谱图如图2所示,在15min内6个化合物全部出峰。
对比例1
本对比例提供了一种高效液相色谱电化学法高通量检测儿茶酚胺及其代谢物的方法,具体步骤仅与实施例1不同的是:
洗脱程序中流动相A与流动相B的体积比调整为95:5。
儿茶酚胺及其代谢物的色谱图如图3所示,15min只有前三个化合物出峰,全部出峰需要60min。
对比例2
本对比例提供了一种高效液相色谱电化学法高通量检测儿茶酚胺及其代谢物的方法,具体步骤仅与实施例1不同的是:
流动相:A相:水(含50mM磷酸二氢钾,5mM辛烷磺酸钠,pH为3.0)。
儿茶酚胺及其代谢物的色谱图如图4所示,30min内只有4个化合物出峰,6个化合物全部出峰需要1h。
对比例3
本对比例提供了一种高效液相色谱电化学法高通量检测儿茶酚胺及其代谢物的方法,具体步骤仅与实施例1不同的是:
流动相:A相:水(含16mM柠檬酸,18mM磷酸二氢钾,5mM辛烷磺酸钠,0.1mg/mL乙二胺四乙酸二钾盐二水合物)。
儿茶酚胺及其代谢物的色谱图如图5所示,在15min内6个化合物全部出峰,但是多巴胺和变肾上腺素出峰时间重合(7.243min)。
对比例4
本对比例提供了一种高效液相色谱电化学法高通量检测儿茶酚胺及其代谢物的方法,具体步骤仅与实施例1不同的是:
流动相:A相:水(含20mM柠檬酸,20mM磷酸二氢钾,8mM辛烷磺酸钠,0.1mg/mL乙二胺四乙酸二钾盐二水合物);
电化学检测器的检测电压为800V。
儿茶酚胺及其代谢物的色谱图如图6所示,在对比例3的基础上调整添加剂量,在20min内6个化合物全部出峰,但是多巴胺和变肾上腺素出峰时间重合(10.039min)。
最后应说明的是:以上实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的精神和范围。