一种内切酶活性的测定方法
文献发布时间:2024-07-23 01:35:21
技术领域
本发明涉及酶活测定技术领域,具体涉及一种内切酶活性的测定方法。
背景技术
酶类几乎都是蛋白质,各类蛋白的纯化方式、不同的人操作手法不同,就会导致等质量的蛋白纯度不同,酶类蛋白的活性不同。因此,蛋白类酶的活性按照质量投入时,就会出现使用不同纯化方式和/或不同人纯化的酶在相同的反应体系中结果不同,影响实验研究的准确性。
因此,如何提供一种新的内切酶活性的测定方法,保证可以对每个批次纯化后的蛋白进行酶活测定,按照酶活性在反应体系中投入后,保证投入的酶活性一致具有重要意义。
发明内容
基于此,本发明提供了一种内切酶活性的测定方法。
本发明采用如下技术方案:一种内切酶活性的测定方法,所述内切酶为pAG-MNase酶,其反应体系中梯度酶量反应呈“S”形,所述方法具体包括以下步骤:
S1,将至少两批次的标准品进行梯度稀释后分别确定其EC50值M
S2,按照公式E
S3,将酶的比活力M
作为一种优选的实施方式,反应体系为:λDNA 5μL,10×反应缓冲液2μL、酶2μL、10mg/mL BSA 0.2μL、ddH
进一步,所述10×反应缓冲液为500mMTris-HCl、50mMCaCl
作为一种优选的实施方式,步骤S1中,37℃反应10min后,65℃10min反应,反应结束后置于冰上,向各管反应液中加入100mM EGTA,pH 8.0,2μL/管,涡旋振荡混匀,瞬时离心,置于25℃反应10min,涡旋振荡混匀,瞬时离心,取20μL溶液至新的离心管中,加入20μL体积比为10%的TCA溶液,涡旋振荡混匀,瞬时离心,置于冰上静置后离心,取上清18μL加入62μL无核酸水进行检测,每个样品做至少2个平行;检测后,将各反应液的平均A260吸光度强度值A减去空白对照的平均A260吸光度强度均值A0,即得校正A260吸光度强度均值ΔA。
作为一种优选的实施方式,EC50确定时,横坐标为待测酶投入质量ng或标品投入活力U的lg值,纵坐标的反应后校正A260吸光度强度均值ΔA。
作为一种优选的实施方式,EC50确定时的公式为:ΔA=Bottom+(Top-Bottom)/(1+((X/EC50)^(-HillSlope)),其中,EC50为酶的半最大效应浓度。
作为一种优选的实施方式,所述标准品的确定需要满足以下条件:将至少两批次的标准品进行梯度稀释后分别测定并做出其梯度酶量反应曲线,每轮重复实验内变异系数≤15%,两轮独立重复实验结果间变异系数≤15%。
作为一种优选的实施方式,所述标准品的确定还需要满足以下条件:将至少两批次的标准品以及竞品进行梯度稀释后测定并做出其梯度酶量反应曲线,每轮实验计算数据可靠性,及回归值P值,p值<0.01。
作为一种优选的实施方式,标准品的稀释方法如下:首先将标准品稀释成1U/μL的酶液b,将酶液b以1/2稀释的方式梯度稀释11次,反复吸吹,轻弹混匀,瞬时离心,再轻柔振荡,瞬时离心。
作为一种优选的实施方式,待测酶的稀释方法如下:当待测酶为质量浓度时:将待测酶稀释至62.5ng/μL的酶液a
作为一种优选的实施方式,酶活C
与现有技术相比,本发明的核心效果在于:可以稳定的对内切酶进行酶活测定,减少酶活批次之间的差异,具有更稳定的反应体系。
附图说明
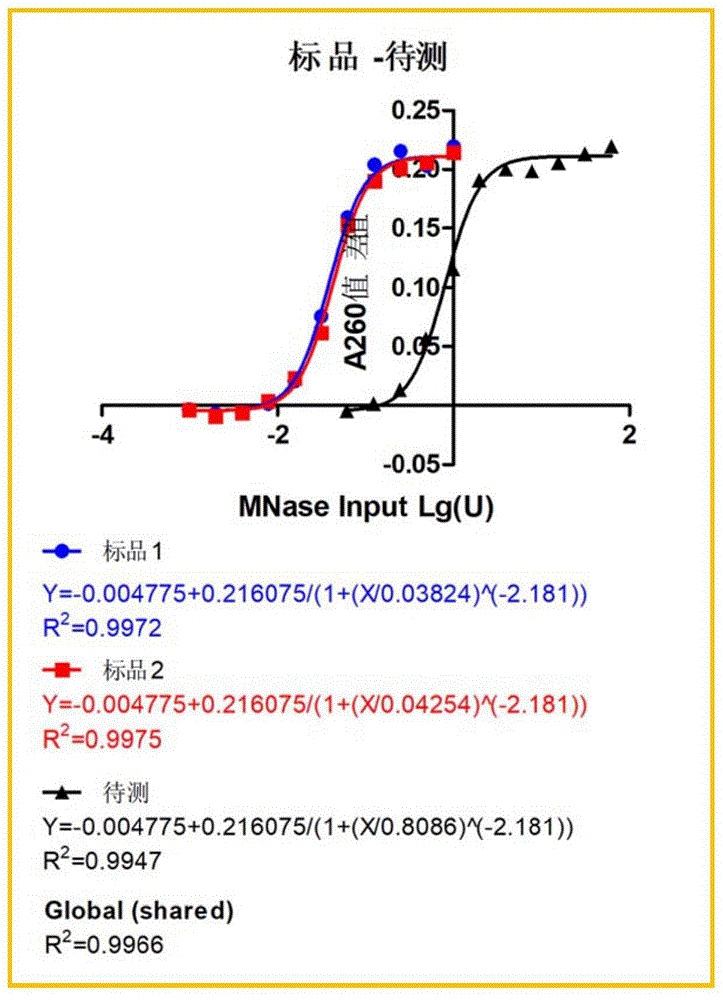
图1为本发明实施例1中标品1、标品2和待测品所得拟合曲线;
图2为本发明实施例1中标品1、竞品NEB和待测品所得拟合曲线;
图3为本发明实施例1中标品2、竞品NEB和待测品所得拟合曲线;
图4为本发明实施例2中不同温度对片段化反应的影响;
图5为本发明实施例2中不同pH条件对片段化反应的影响。
具体实施方式
一种内切酶活性的测定方法,该方法适用于反应体系中梯度酶量反应呈“S”形的酶,尤其适用于pAG-MNase内切酶。该方法具体包括以下步骤:
S1,将至少两批次的标准品进行梯度稀释后分别确定其EC50值M
S2,按照公式E
S3,将酶的比活力Mo乘以蛋白浓度,即得酶活C
在该方法中,EC50值的确定优选的为通过酶标仪比色法进行检测,其检测原理为,使用TCA(三氯乙酸)沉淀大片段核酸后,使用酶标仪在260nm处进行检测,使用EC50值进行酶活计算。其中,反应体系为:λDNA 5μL(1μg),10×反应缓冲液2μL、酶2μL、10mg/mL牛血清白蛋白(BSA)0.2μL、ddH
详细的方法可按如下步骤进行操作:37℃反应10min后,65℃10min反应,反应结束后置于冰上,向各管反应液中加入100mM乙二醇双(2-氨基乙基醚)四乙酸(EGTA),pH 8.0,2μL/管,涡旋振荡混匀,瞬时离心,置于25℃反应10min,涡旋振荡混匀,瞬时离心,取20μL溶液至新的离心管中,加入20μL体积比为10%的TCA溶液,涡旋振荡混匀,瞬时离心,置于冰上静置后离心,取上清18μL加入62μL无核酸水进行检测,每个样品做至少2个平行;检测后,将各反应液的平均A260吸光度强度值A减去空白对照的平均A260吸光度强度均值A
在该方法中,EC50确定时,横坐标为待测酶投入质量ng或标品投入活力U的lg值,纵坐标的反应后校正A260吸光度强度均值ΔA。EC50确定时的公式为:ΔA=Bottom+(Top-Bottom)/(1+((X/EC50)^(-HillSlope)),其中,EC50为酶的半最大效应浓度。
另外,在该方法中,标准品的确定需要满足以下条件:将至少两批次的标准品(如下述实施例中的标品1、标品2)进行梯度稀释后分别测定并做出其梯度酶量反应曲线,每轮重复实验内变异系数≤15%,两轮独立重复实验结果间变异系数≤15%。以及将至少两批次的标准品以及竞品进行梯度稀释后测定并做出其梯度酶量反应曲线,每轮实验计算数据可靠性,及回归值P值,p值<0.01。
该方法中,标准品的稀释方法如下:首先将标准品稀释成1U/μL的酶液b,将酶液b以1/2稀释的方式梯度稀释11次,反复吸吹,轻弹混匀,瞬时离心,再轻柔振荡,瞬时离心。
待测酶的稀释方法如下:当待测酶为质量浓度时:将待测酶稀释至62.5ng/μL的酶液a
按照上述稀释方法进行时,酶活C
下面结合具体实施例对本发明作进一步的详细说明,以使本领域的技术人员更加清楚地理解本发明。以下各实施例,仅用于说明本发明,但不止用来限制本发明的范围。基于本发明中的具体实施例,本领域普通技术人员在没有做出创造性劳动的情况下,所获得的其他所有实施例,都属于本发明的保护范围。
在本发明实施例中,若无特殊说明,所有原料组分均为本领域技术人员熟知的市售产品;在本发明实施例中,若未具体指明,所用的技术手段均为本领域技术人员所熟知的常规手段。在本发明实施例中,所使用的原料均为常规市售产品。
部分试剂说明:
酶稀释液(1×稀释液)为爱博泰克(ABclonal)商业化试剂,货号为:RM20820。
实施例1
一种pAG-MNase酶活力的测试方法,测试方法具体包括以下步骤:
步骤1,待测酶稀释:
(1)待测酶为质量浓度时
按下列公式计算第一步稀释所需1×稀释液体积:1×稀释液体积=20×(pAG-MNase质量浓度mg/mL-1)μL,取计算所得体积的1×稀释液加入200μL离心管中,再加入20μL待测pAG-MNase酶液,反复吹吸(20次以上),轻弹混匀,瞬时离心,再轻柔振荡,瞬时离心,将酶液稀释至1mg/mL。如果质量浓度过高使得稀释倍数超过10倍,则分次稀释,使稀释倍数不超过10倍。将1mg/mL酶液以1/4稀释的方式稀释2次,每次将10μL酶液加入30μL 1×稀释液,换用200μL枪头反复吸吹(20次以上),轻弹混匀,瞬时离心,再轻柔振荡,瞬时离心,稀释至62.5ng/μL的酶液a
将待测酶梯度稀释:将酶液a
(2)待测酶单位为U/μL时
基于本公司产品为例,产品浓度为10U/μL,首先进行5倍预稀释,将10μL酶液加入30μL 1×稀释液,换用200μL枪头反复吹吸(20次以上),轻弹混匀,瞬时离心,再轻柔振荡,瞬时离心,稀释至5U/μL,再进行2倍预稀释,将20μL 5倍稀释后的酶液加入20μL 1×稀释液,换用200μL枪头反复吹吸(20次以上),轻弹混匀,瞬时离心,再轻柔振荡,瞬时离心,稀释至1U/μL的酶液a
待测酶液梯度稀释:将酶液a
步骤2,标准品稀释:
基于本公司产品为例,产品浓度为10U/μL,首先进行5倍预稀释,将10μL酶液加入40μL 1×稀释液,换用200μL枪头反复吹吸(20次以上),轻弹混匀,瞬时离心,再轻柔振荡,瞬时离心,稀释至5U/μL,再进行2倍稀释,将20μL 5倍稀释后的酶液加入20μL 1×稀释液,换用200μL枪头反复吹吸(20次以上),轻弹混匀,瞬时离心,再轻柔振荡,瞬时离心,稀释至1U/μL的酶液b。
标准品梯度稀释:将酶液b以1/2稀释的方式梯度稀释11次,每次将20μL酶液加入20μL1×稀释液中,反复吹吸(20次以上),轻弹混匀,瞬时离心,再轻柔振荡,瞬时离心。
步骤3,竞品NEB产品稀释(Micrococcal Nuclease,Cat:M0247S,2X10
将200U/μL酶液以1/10稀释的方式稀释2次,首先将2μL酶液加入18μL1×稀释液,换用200μL枪头反复吸吹(20次以上),轻弹混匀,瞬时离心,再轻柔振荡,瞬时离心,稀释至20U/μL,再次将3μL 20U/μL的酶液加入27μL 1×稀释液,换用200μL枪头反复吹吸(20次以上),轻弹混匀,瞬时离心,再轻柔振荡,瞬时离心,稀释至2U/μL的酶液c。
竞品梯度稀释:将酶液c以1/2稀释的方式梯度稀释11次,每次将20μL酶液加入20μL 1×稀释液中,反复吹吸(20次以上),轻弹混匀,瞬时离心,再轻柔振荡,瞬时离心。
步骤4,将PCR仪预热到37℃,设置如下表1所示程序:
表1
步骤5,按照片段化体系添加试剂,将酶单独添加,酶之外的试剂混为Mix,再分装到各个离心管中,置于冰上,将pAG-MNase分别加于200μL管盖,设置一个空白对照,将1×稀释液加于200μL管盖,离心混匀后立即置于预热的PCR仪,进行37℃10min,65℃10min反应,热盖温度75℃,反应结束后立即置于冰上。其中,片段化体系如下表2所示:
表2
其中,10X Reaction Buffer:500mM Tris-HCl、50mM CaCl
步骤6,向各管反应液中加入200mM EGTA,pH 8.0,2μL/管,涡旋振荡混匀,瞬时离心,将联排管置于25℃反应10min。
步骤7,将联排管涡旋振荡混匀,瞬时离心,取20μL溶液至新的1.5mL离心管,再向各1.5mL离心管中加入20μL 10wt%的TCA溶液,涡旋振荡混匀,瞬时离心,置于冰上静置30min。
步骤8,将静置后的1.5mL离心管置于台式离心机离心,室温12000rpm,离心5min。
步骤9,用移液器向酶标板各孔加入无核酸水62μL,从1.5mL离心管中吸取样本上清加18μL至96孔酶标板上(80μL/孔),每个样做2个平行检测。
步骤10,将酶标板置于酶标仪上,注意板放置方向。振板1min,核酸检测方案-不预读空板程序进行检测,按照下表3设置孔板类型,选取260nm处实验数据进行记录;
表3
步骤11,将酶标仪数据导出后进行数据处理。
步骤12,选取待测p-MNase样本和空白对照的A260吸光度强度值,2批次标品和空白对照的A260吸光度强度值,求出2个复孔的均值,将各反应液的平均A260吸光度强度值(A)减去空白对照的平均A260吸光度强度均值(A
步骤13,以待测pAG-MNase投入质量(单位:ng)取Lg值和各批次标品投入活力(单位:U)取Lg值为横坐标,各梯度的矫正A260吸光度强度均值(ΔA)为纵坐标,使用GraphPad软件做散点图,拟合四参数方程曲线,获得2条矫正A260吸光度强度均值于MNase投入活力Lg值关系的曲线,以及1条校正A260吸光度强度均值于待测pAG-MNase投入质量Lg值的关系曲线,R2≥0.98。
公式2:ΔA=Bottom+(Top-Bottom)/(1+((X/EC50)^(-HillSlope)),其中,ΔA—校正A260吸光度强度均值,EC50—MNase半最大效应浓度,单位:U或者ng。
步骤14,分别用2批次标品所得EC50值(M1,M2,单位:U)除以待测pAG-MNase所得EC50值(M0,单位:ng),计算待测pAG-MNase的酶投入活力E
公式3:E
步骤15,通过公式3计算所得酶比活力E
公式4:C
步骤16,以每轮重复实验内各梯度酶投入量计算所得待测酶比活力值的平均值,作为待测酶比活力标准值,每轮重复实验内变异系数≤15%视为合格,此外,两轮独立重复实验结果间变异系数≤15%视为合格。
变异系数=STDEV值/平均值。
步骤17,每轮实验要计算数据可靠性,根据数据可靠性分析表计算,回归项p值<0.01,偏离平行项和模型失拟项不做要求。
(1)数据拟合结果如下表1~3及图1~3所示:
表1
表2
表3
由上述表1~3和图1~3所示,标品、待测品及竞品趋势相同,且拟合后R2≥0.98,说明该方法可行。
(2)数据重复性结果见下表4:
表4
(3)数据可靠性结果如下表5~7所示,其中表5、6、7为表1中标品1与待测品、标品2与待测品、标品1与标品2的可靠性结果,表8、9、10为表2中标品1与待测品、NEB竞品与待测品、标品1与竞品的可靠性结果,表11、12、13为表3中标品2与待测品、竞品NEB与待测品、标品2与竞品的可靠性结果。
表5第一组数据可靠性结果(标品1-待测品)
表6第一组数据可靠性结果(标品2-待测)
表7第一组数据可靠性结果(标品1-标品2)
表8第二组数据可靠性结果(标品1-待测)
表9第二组数据可靠性结果(NEB-待测)
表10第二组数据可靠性结果(标品1-NEB)
表11第三组数据可靠性结果(MEB-待测)
表12第三组数据可靠性结果(标品2-待测)
表13第三组数据可靠性结果(NEB-标品2)
由上述结果可知,回归值p值<0.01,符合要求。
实施例2温度的影响
参考市面上的产品信息,改变反应温度,比较不同温度条件下的片段化结果,具体执行步骤如下:
(1)基于本公司产品为例,产品浓度为10U/μL,首先进行5倍预稀释,将10μL酶液加入40μL 1×稀释液,换用200μL枪头反复吹吸(20次以上),轻弹混匀,瞬时离心,再轻柔振荡,瞬时离心,稀释至5U/μL,再进行2倍预稀释,将20μL 5倍稀释后的酶液加入20μL 1×稀释液,换用200μL枪头反复吹吸(20次以上),轻弹混匀,瞬时离心,再轻柔振荡,瞬时离心,稀释至1U/μL的酶液。
待测酶液梯度稀释:将酶液以1/3稀释的方式梯度稀释6次,每次将10μL酶液加入20μL 1×稀释液中,反复吹吸(20次以上),轻弹混匀,瞬时离心,再轻柔振荡,瞬时离心。
(2)将PCR仪预热到12℃、16℃、20℃、25℃、37℃,即设置如下程序:
(3)按照片段化体系添加试剂,将酶单独添加,酶之外的试剂混为Mix,再分装到各个离心管中,置于冰上,将pAG-MNase分别加于200μL管盖,离心混匀后立即置于预热的PCR仪,分别进行12℃/16℃/20℃/25℃/37℃15min反应,热盖温度75℃,反应结束后立即置于冰上;另外,0℃15min反应置于冰上反应,4℃15min反应置于4℃冰箱。
片段化体系:
其中,10X Reaction Buffer:500mM Tris-HCl、50mM CaCl
(4)向各管反应液中加入200mM EGTA,pH 8.0,2μL/管,涡旋振荡混匀,瞬时离心,将联排管置于25℃反应10min,终止片段化反应。
(5)使用2%琼脂糖凝胶进行检测,每个样上样量200ng,即取反应后的样本4.4μL,加入Nuclease-free Water 15.6μL,再用排枪吸取4μL 6X Loading Buffer加入到新的八联排中,振荡混匀并瞬时离心;加入2%琼脂糖凝胶,200V 30min,并取5μL DNALadder加入适当的胶孔中。
结果如图4所示,图中各泳道为:M:D2000 plus;1:酶投入量1U;2:酶投入量3
由结果可知,仅在预热的PCR仪,进行37℃15min反应时,随着酶投入量的增加,各体系中呈现出梯度变化,由此表明酶量的多少是影响模板切割效率的唯一因素,酶投入量大,酶活高时,切割DAN片段的能力强,酶的投入量较少,酶活较低时,切割DNA片段的能力若或者不切割,证明了本发明酶活测定的准确性及可行性。
实施例3酶反应缓冲液的影响
改变Reaction Buffe的pH值,比较不同pH值条件下的片段化结果。具体执行步骤如下:
(1)分别配制pH值为6.5、6.8、7.5、8.8的10×Reaction Buffer(由500mM Tris-HCl、50mM CaCl
(2)基于本公司产品为例,产品浓度为10U/μL,首先进行5倍预稀释,将10μL酶液加入40μL 1×稀释液,换用200μL枪头反复吹吸(20次以上),轻弹混匀,瞬时离心,再轻柔振荡,瞬时离心,稀释至5U/μL,再进行2倍预稀释,将20μL 5倍稀释后的酶液加入20μL 1×稀释液,换用200μL枪头反复吹吸(20次以上),轻弹混匀,瞬时离心,再轻柔振荡,瞬时离心,稀释至1U/μL的酶液。
待测酶液梯度稀释:将酶液以1/3稀释的方式梯度稀释6次,每次将10μL酶液加入20μL 1×稀释液中,反复吹吸(20次以上),轻弹混匀,瞬时离心,再轻柔振荡,瞬时离心。
(3)将PCR仪预热到37℃,即设置如下程序:
(4)按照片段化体系添加试剂,将酶单独添加,酶之外的试剂混为Mix,不同pH值的10×Reaction Buffer以及分别配制Mix,再分装到各个离心管中,置于冰上,将pAG-MNase分别加于200μL管盖,离心混匀后立即置于预热的PCR仪,进行37℃15min,反应,热盖温度75℃,反应结束后立即置于冰上。
片段化体系:
(5)向各管反应液中加入200mM EGTA,pH 8.0,2μL/管,涡旋振荡混匀,瞬时离心,将联排管置于25℃反应10min,终止片段化反应。
(6)使用2%琼脂糖凝胶进行检测,每个样上样量200ng,即取反应后的样本4.4μL,加入Nuclease-free Water 15.6μL,再用排枪吸取4μL 6X Loading Buffer加入到新的八联排中,振荡混匀并瞬时离心;加入2%琼脂糖凝胶,200V 30min,并取5μL DNALadder加入适当的胶孔中。
结果如图5所示,图中各泳道为:M:D2000 plus;1:酶投入量1U;2:酶投入量3
由上述结果可知,反应体系pH在6.5~8.8时,随着酶投入量的增加,各体系中呈现出梯度变化,说明该方法具有较高的pH普适性。
在此有必要指出的是,以上实施例仅限于对本发明的技术方案做进一步的阐述和说明,并不是对本发明的技术方案的进一步的限制,本发明的方法仅为较佳的实施方案,并非用于限定本发明的保护范围。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 一种魁蚶来源的具有免疫增强活性的β-1,3葡聚糖内切酶及其编码多核苷酸
- 一种产单宁酶假丝酵母菌的筛选及其酶活性测定方法
- 一种平板培养基酶活性测定方法
- 一种腺苷脱氨酶活性测定方法及腺苷脱氨酶检测试剂盒
- 一种核酸内切酶Ⅷ活性测定方法
- 一种核酸内切酶Ⅷ活性测定方法