一种血清中前列腺素A2的检测方法
文献发布时间:2024-07-23 01:35:21
技术领域
本发明涉及一种血清中前列腺素A2的检测方法,属于分析检测技术领域。
背景技术
液相色谱和质谱联用技术的研究开始于20世纪70年代。之后,随着接口技术和电离技术的不断进步,液相色谱串联质谱技术已广泛应用于药物分析、毒物筛查、临床检验等领域。液相色谱串联质谱技术具有灵敏度高、特异性强、同时准确定量多种化合物的优势,大大弥补了常规生化和免疫检测方法学上存在的不足,可更好地助力临床精准诊断和治疗。随着现代医学的发展,传统的疾病诊断标志物已无法满足临床需求,越来越多新的疾病诊断标志物被探索出来。液相色谱串联质谱技术可用于检测激素、氨基酸、长链脂肪酸、有机酸、维生素、神经酰胺及其体内代谢产物等多种小分子物质,具有极其重要的临床应用价值。
前列腺素A2(Prostaglandin A2,PGA2)是前列腺素E2(Prostaglandin E2,PGE2)的内源性代谢产物,在体内是通过PGE2脱水而形成。PGA2能够诱导p53依赖性细胞凋亡,进而发挥抗肿瘤活性。此外,PGA2也具有抗病毒活性,在无毒性剂量下能够抑制病毒复制。目前,可供用于临床诊疗的肿瘤标志物数量较少,且现有的肿瘤标志物往往存在敏感性和特异性低的缺点,不能满足临床精准诊疗的需要。而对于病毒感染来说,除了核酸检测之外,临床也缺乏有效监测病毒感染的特异性指标。因此,肿瘤和感染患者中血清PGA2水平的精准定量具有极其重要的潜在临床意义。
PGA2在体内含量极低(pg级),且稳定性较差,因此其检测难度较大。之前,人们已经开发出基于气相色谱质谱联用技术的PGA2检测方法,但气相色谱质谱联用技术仅限于应用于300℃左右及以下可以汽化、并且能离子化的待测物或样本,而在加热过程中易分解的、极性太强的化合物,如有机酸类等,则需进行衍生化处理之后才可进行气相色谱质谱分析。因此,气相色谱质谱联用技术难度大、要求高,且PGA2易降解的特性,使其在高温加热过程中不稳定,进而可能导致检测结果不准确。而随着分析技术的发展,液相色谱串联质谱技术则提供了另一种精准检测PGA2的可能。
发明内容
本发明的目的是提供一种血清中前列腺素A2的检测方法,可以快速、准确地检测出血清中PGA2的含量。
本发明提供的血清中前列腺素A2的检测方法,包括如下步骤:
S1、待测样本经前处理,得到上清液;
所述待测样本为血清;
S2、将所述上清液进行液相色谱串联质谱检测;
所述液相色谱串联质谱检测的液相条件如下:
0~6min,流动相A的体积含量由70%调节至50%,流动相B的体积含量由30%调节至50%;
6~6.5min,流动相A的体积含量由50%调节至3%,流动相B的体积含量由50%调节至97%;
6.5~7.9min,流动相A的体积含量保持为3%,流动相B的体积含量保持为97%;
7.9~8min,流动相A的体积含量由3%调节至70%,流动相B的体积含量由97%调节至30%;
8~10min,流动相A的体积含量保持为70%,流动相B的体积含量保持为30%;
所述流动相A为体积含量为0.05%的甲酸溶液,所述流动相B为体积含量为0.05%的甲酸乙腈溶液;
所述液相色谱串联质谱检测的质谱条件如下:
喷雾电压为-4500V,温度为550℃,雾化气为55psi,辅助加热气为55psi,气帘气为35psi。
本发明检测方法中,所述前处理的步骤如下:
将所述待测样本与水混合后涡旋(加入PGA2-D4内标),所得上清液转移至SPE板中,依次采用水和正己烷进行淋洗,最后采用洗脱剂进行洗脱,收集洗脱液,经氮气吹干、复溶后离心;
所述洗脱剂为甲醇与乙腈体积比为1:1的混合液;
所述SPE板为Cleanert 2mg PEP((Agela&Phenomenex)微孔板。
本发明检测方法中,所述液相色谱串联质谱检测的液相条件如下:
色谱柱为C18色谱柱;
柱温为40℃;
进样体积为25uL;
进样器温度为2~8℃;
流速为0.3ml/min。
本发明检测方法中,在检测之前,所述检测方法还包括如下步骤:
采用甲醇冲洗色谱柱以进行活化。
本发明检测方法中,在所述淋洗之前,依次采用甲醇和水活化和平衡所述SPE板。
本发明利用液相色谱的高分离性能,以及质谱的高选择性和高灵敏度,能够精准地检测血清中PGA2的水平,为临床肿瘤和感染的诊断提供指导和帮助。
附图说明
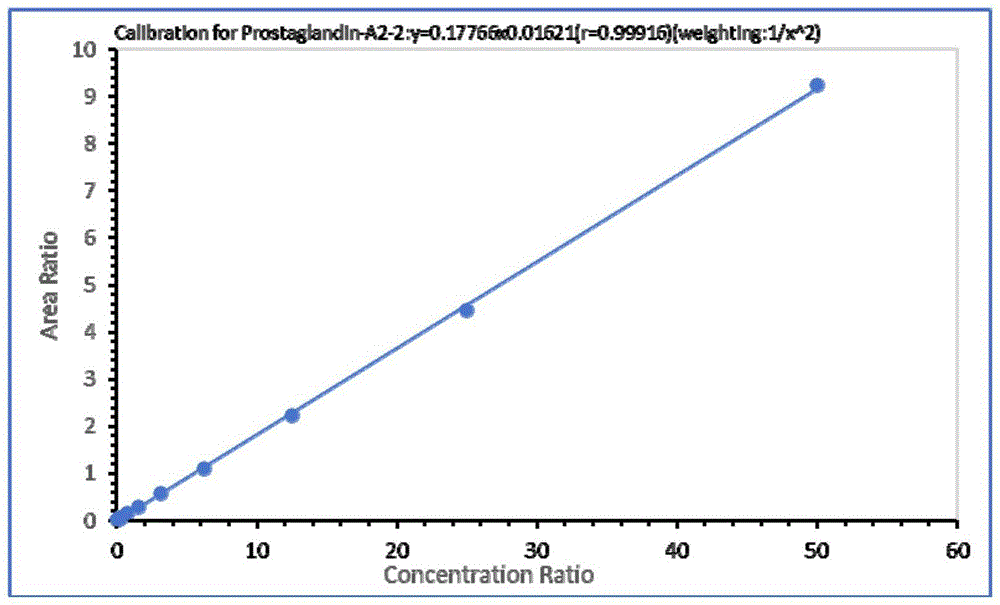
图1为PGA2的校准曲线。
图2为PGA2的线性最低点的物质峰。
图3为内标PGA2-D4的色谱峰。
图4-图7分别为校准曲线最高点C12待测物PGA2的色谱图、校准曲线最高点C12内标色谱图、携带污染空白管待测物的色谱图、携带污染空白管内标色谱图。
图8-图9为采用不同微孔板处理后的色谱图。
图10-图11为不同分离时间时的色谱图。
具体实施方式
下述实施例中所使用的实验方法如无特殊说明,均为常规方法。
下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
一、流动相的配制
1、流动相A的配制(以配制1L流动相A为例)
在1000mL超纯水中加入0.5mL甲酸(减量配制时按比例缩小各组分添加量),采用超声震荡10分钟排出气体后即可使用,在20~25℃保存,有效期为一周。
2、流动相B的配制(以配制1L流动相B为例)
在1000mL乙腈中加入0.5mL甲酸(减量配制时按比例缩小各组分添加量),采用超声震荡10分钟排出气体后即可使用,在20~25℃保存,有效期为一个月。
3、新色谱柱活化方法:甲醇以0.2ml/min冲洗8小时以上即达到活化效果,可以使用,将启用日期作为色谱柱编号标记在色谱柱上。色谱柱参数:C18,2.6um,2.1×100mm。
二、检测样本处理
1、预处理
SPE板的活化与平衡:取96孔SPE板加入200μL甲醇活化,静置流干;加入200μL水平衡,静置流干。SPE板型号:Cleanert 2mg PEP微孔板。
2、样本前处理
1)取2mL 96孔板,依次加入200μL标曲/质控/待测样本、200μL含PGA2-D4的沉淀剂(甲醇:0.1M硫酸锌=7:3),400μL水,涡旋混匀,4000rpm离心10min;
2)取650μL上述离心后上清转移到预处理后的SPE板中,静置2min,正压仪控制流速(2~3秒/滴)压干;
3)加入200μL水淋洗,静置5min,正压仪控制流速(2~3秒/滴)逐步压干;
4)加入200μL正己烷淋洗,静置5min,正压仪控制流速(2~3秒/滴)逐步压干,完全压干并保持2min;
5)加入200μL洗脱液(甲醇:乙腈=1:1)洗脱,收集洗脱液至1.2mL 96孔板中,静置5min,正压仪压干;
6)将收集的洗脱液用氮气(如氮吹仪有加热功能,温度设为40℃)吹干,残渣中加入100μL复溶液(100mM氨水30%乙腈水)复溶,4000rpm离心5min后,取上清转移至350μL 96孔板后待测。
在表1和表2所示条件下进行液相色谱串联质谱检测,得到的色谱图如图8-图9所示,其中,图8是采用Oasis HLB(Waters)微孔板处理后的色谱图,图9图是采用Cleanert2mg PEP(Agela&Phenomenex)微孔板处理后的色谱图,对比两个图可以看出,采用OasisHLB微孔板时的基线较高,会导致定量不准确,而采用Cleanert2mg PEP微孔板时的基线较低,更加符合检测需求。
Cleanert PEP是吡咯烷酮化聚苯乙烯/二乙烯基苯固相萃取柱,极性极强;WatersOasis HLB吸附剂是由亲脂性二乙烯苯和亲水性N-乙烯基吡咯烷酮两种单体按一定比例聚合成的大孔共聚物,其保留机理为反相,通过一个“特殊的极性捕获基团”来增加对极性物质的保留提供很好的水浸润性。在大部分情况下,两者的萃取效果相当,但是在本发明中发现Qasis HLB基线较高,而Cleanert PEP基线较低,更适宜PGA2的检测。
三、色谱条件
色谱条件如表1所示。
表1色谱条件
流动相及梯度是反复摸索之后确定的,请见图10和图11,初始的液相时间设置为7min/样本,发现有些样本中特征峰附近会存在干扰峰,而且位置接近,影响定量,后来便把液相时间设置为10min/样本。
表2原7min流动相梯度(分离效果不好)
四、质谱条件
色谱条件如表3所示。
表3质谱条件
离子对参数(可适当做优化调整)如表4所示:
表4离子对参数
*离子对-1为定量离子对,-2为定性离子对
五、仪器分析
1、实验前检查仪器状态是否良好,然后打开计算机电源,进入Windows操作系统后登录Analyst软件,进入工作站主界面。
2、双击Hardware configuration,单击选LC-MS后再单击右侧Activate profile激活液相色谱质谱联用仪,LC-MS模块联机后会变显示为绿色对号,表示联机正常。
3、点击菜单栏Equilibrate,选中要运行的液质联用方法,设置5~10min的平衡时间。
4、双击Acquire,在序列编辑菜栏中,新建或另存为方式,编好相应的样品序列表(样品编号,样品盘位置,仪器采集方法等内容),然后选中要分析的样品序列并点击Submit按钮提交。
5、待平衡时间结束后,主界面右下角的仪器状态转变为Ready,点击菜单栏的Start sample,开始进行样品分析和数据采集。
6、样品分析采集完毕后在Quantitate模块或者使用Multiquant建立定量列表,进行定量分析。
六、待机
1、双击Acquire,在菜单栏中点击Standby按钮,缓慢降低流速至零,待系统压力降为0后,双击Hardware configuration,单击选LC-MS后再单击右侧Deactivate profile去激活液相色谱质谱联用仪,断开联机。
2、退出Analyst软件,关闭计算机。
七、方法的性能验证结果
1、线性
将已知浓度的标准品做一系列倍比稀释(50、25、12.5、6.25、3.125、1.5625、0.7813、0.3906、0.1953、0.0977、0.0488、0.0244ng/mL)加入等量的内标品,采用最小二乘法对校准点进行拟合形成校准曲线,其线性要求r>0.99,各浓度点的值与理论值的偏差小于15%,将检测值与理论值偏差小于20%的最低值作为定量限Limit of Quantification(LOQ),试验得到线性范围为0.024~50ng/mL,如图1、图2所示,标准曲线r=0.99916,线性良好,LOQ=0.024ng/mL,足以表明本发明检测方法灵敏度高,能满足临床检测和科学研究的需要。
2、精密度和携带污染
将制备好的高、中、低质控每天检测3次,连续3天,计算得到日内精密度变异系数分别为2.01%、0.57%、0.08%,日间精密度分别为5.65%、6.53%、5.38%,均小于10%,证明精密度良好(表5)。在校准曲线最高点后紧接着测定空白样本,未见明显信号响应,见图4-图7,证明本发明检测方法无携带污染。
表5PGA2的检测精密度
3、准确度和基质效应
(1)提取回收率
制备基质加入内标和低、中、高值标准品后进行前处理流程,然后上机进样检测,得到样本峰面积A;基质进行前处理后加入内标和低、中、高值标准品,上机进样检测,得到样本峰面积B,按照前处理标准操作流程操作,分别配置高低两个浓度的样本,每个浓度平行制备3份,得到表5中的数据,提取回收率分别为95.85%、92.97%和97.00%,满足临床检验需要。
Extraction Recovery(%)=A/B×100%
(2)加标回收率:取6份不同来源基质,在其中3份中分别入已知浓度的低、中、高值标准品,进行相同的前处理和进样分析流程,平行测定三次,利用公式加标回收率%=(实测加标后的样本浓度-实测基质样本浓度)/理论加标浓度×100%计算,得到表6数据,加标回收率为98.07%~103.60%,说明本发明检测方法的准确度良好。
表6PGA2的回收率和基质效应
(3)基质效应
取3份不同来源的血清样本,分别配制成以下三种溶液:A:血清样本+内标和标准品;B:血清样本+内标;C:纯溶液标准品,上机检测,通过计算公式基质效应=(A-B)/C×100%得到基质效应为85%、89.9%、95.43%,见表6,在80%~120%范围内,满足实验要求。
- 一种检测生物制品中牛血清白蛋白的试纸及制备方法
- 一种基于液相色谱串联质谱法检测血清中25-羟基维生素D的方法及试剂盒
- 一种血清中C-肽含量的化学发光法测定试剂盒及检测方法
- 一种利用氮化硼量子点-金纳米粒子纳米复合物检测人血清中乙酰胆碱酯酶含量的方法
- 一种用于检测牛乳品中A2β-酪蛋白含量的特征肽及方法
- 一种同时测定血浆中脂蛋白磷脂酶A2与C反应蛋白含量的检测方法及试剂盒