一种检测农产品复杂基质中盐酸吗啉胍的方法
文献发布时间:2024-07-23 01:35:21
技术领域
本发明涉及分析化学领域,特别涉及一种检测农产品复杂基质中盐酸吗啉胍的方法。
背景技术
盐酸吗啉胍(Moroxydine hydrochloride)是一种吗啉类广谱病毒防治剂,具有较强的碱性和水溶性。稀释后的药液喷施到植物表面后,通过气孔进入植物体内,抑制或破坏核酸和脂蛋白的形成,阻止病毒的复制过程,起到防治病毒的作用。且该药物价格低廉,对蔬菜、瓜类、果树、大田作物等的花叶病、蕨叶病、条斑病、叶枯病等病毒性疾病有一定的治疗、钝化和保护作用,目前作为广谱病毒防治剂广泛应用于农业生产当中。其化学结构式如下:
盐酸吗啉胍由一个吗啉环和一个胍基团构成,含有氨基,属于有机碱,其为水溶性化合物,提取时需采用极性的水溶液,但是由于农产品中包括高含水、高含油以及含水量低等不同的复杂基质类型,常规的水溶液很难满足提取和净化的需求。目前对农产品中盐酸吗啉胍的检测分析研究较少,赵琳(赵琳和张晓波等2013)等人将10g糙米和5g植株加入40mL 5%三氯乙酸,振荡提取60min,上清液中加入0.5mol/L庚烷磺酸钠溶液0.5mL混合,用Oasis HLB或C18固相萃取小柱净化,UPLC-MS/MS分析。在糙米和植株中的检出限为0.005mg/kg。0.5mg/kg的平均回收率为105.4%,相对标准偏差为7.2%。郑海香(郑海香等2012)等人将10g烟草样品经三氯乙酸提取,庚烷磺酸钠中和,液液萃取净化后,高效液相色谱(DAD检测器)进行测定,反相高效液相色谱分析。在0.05~1.0mg/kg时,方法的平均回收率为79.88~90.98%,相对标准偏差为1.27~4.05%。邵辉(邵辉,等.2011)等人将10g番茄样品采用三氯乙酸提取,HLB固相萃取柱净化,UPLC-MS/MS分析。定量限为0.005mg/kg,0.5mg/kg的平均回收率为86.7%,相对标准偏差为6.3%。以上技术均需要使用较长的提取时间,且无法满足对多种复杂基质提取的需求。
盐酸吗啉胍为水溶性物质,出峰时间较早,容易受到一定的杂质干扰。Hilic色谱柱对极性及极性很强的分析物进行保留的同时,对碱性溶质的峰形分离表现出色,可提供更良好的峰形。如果采用C18色谱柱可使用0.1%甲酸水溶液+乙腈溶液作为流动相,具有较好的稳定性;使用Hilic色谱柱时,则需配制甲酸+甲酸铵的缓冲盐体系作为流动相,提升本方法的适用性,该流动相也适用于C18色谱柱。盐酸吗啉胍化学结构中含有很多氨基,所以易形成一个缓冲溶液体系,有助于农药在色谱柱中的平衡,延长农药在色谱柱中的保留时间。
发明内容
鉴于此,本发明提出一种检测农产品复杂基质中盐酸吗啉胍的方法,在农药残留分析中,提取溶剂必须适用于不同水分、油脂、糖分和物质含量的复杂基质中,才能提取出具有各种极性的化合物。在谷物、茶叶这类水分低的样品中,极性溶剂也不能完全提取,必须采用含水20~40%的溶剂或预先向试样中加入等量的水之后再行提取,因此本方法加入乙腈+水作为提取溶剂,并加入酸从待测样品中直接提取待测成分,提取溶剂采用乙腈+酸+水的形式。盐酸吗啉胍易溶于水,本方法中不加入NaCl使提取液中有机相和水相分层,通过重复三次提取的方式,保证方法的回收率。采用超高相液相色谱三重四级杆质谱联用技术成功检测出农产品复杂基质中盐酸吗啉胍,提取方法快速简便,使用常规的提取溶剂,按照含水量和含油量不同选择不同的提取和净化条件。盐酸吗啉胍在茶叶中定量限0.1mg/kg,其余基质中定量限0.05mg/kg,适用于常规分析,可满足对农产品复杂基质中盐酸吗啉胍的定量检测要求。
本发明的技术方案为:
一种检测农产品复杂基质中盐酸吗啉胍的方法,包括以下步骤:
a)、提取处理:称取待测农产品复杂基质试样至离心管中,加入乙腈+酸+水溶液进行涡旋振荡并离心,倾出提取液,向装剩余残渣的离心管中再次加入乙腈+酸+水溶液再次进行涡旋振荡并离心,合并提取液,进行定容,待净化;
b)、净化处理:吸取少量提取液,加入净化剂,涡旋振荡并离心,取上层清液经微孔滤膜过滤后制得待测溶液;
c)、将制得的待测溶液通过色谱柱进行高效液相色谱分离,然后通过质谱进行分析测定。
优选的,蔬菜、水果类产品提取处理:称取10g试样于50mL离心管中,加入20mL乙腈-三氟乙酸-水溶液,涡旋振荡10min后,以8000r/min离心5min,倾出提取液,向装剩余残渣的离心管中加入10mL乙腈-三氟乙酸-水溶液再次涡旋振荡10min后,以8000r/min离心5min,合并提取液,用提取液定容至50mL,待净化。
优选的,所述乙腈-三氟乙酸-水溶液的体积比为49.5:1:49.5。
优选的,谷物、油料、坚果、食用菌、植物油、香辛料和糖料类产品提取处理:称取5g于50mL离心管中,除植物油外,其他产品补水至样品完全润湿,静置30min;加入10mL乙腈-甲酸-水溶液,涡旋振荡10min后,以8000r/min离心5min,取出提取液,向装剩余残渣的离心管中再加入5mL乙腈-甲酸-水溶液,再次涡旋振荡10min后,以8000r/min离心5min,合并提取液,用提取液定容至25mL,待净化。
优选的,茶叶类产品提取处理:称取2g试样于50mL离心管中,补水至样品完全润湿,静置30min,加入10mL乙腈-甲酸-水溶液,涡旋振荡10min后,以8000r/min离心5min,倾出提取液,向装剩余残渣的离心管中加入5mL乙腈-甲酸-水溶液,再次涡旋振荡10min后,以8000r/min离心5min,合并提取液,用提取液定容至20mL,待净化。
优选的,所述乙腈-甲酸-水溶液的体积比为10:1:89。
优选的,步骤b)净化处理:吸取5mL提取液,油料、坚果和植物油等高含油量样品须使用氨水调节pH至8.5,其它低含油量样品可不调节pH值,并加入150mg PSA,涡旋振荡2min后,以8000r/min离心5min,取上层清液经0.22μm微孔滤膜过滤后,供液相色谱-串联质谱测定。
优选的,步骤c)中的高效液相色谱:色谱柱为C18或Hilic,50mm×2.1mm;流动相:A相为0.1%甲酸+5mmol甲酸胺溶液,B相为乙腈,其梯度洗脱条件为:0~1.5min,流动相B保持5%;1.5~3.5min,流动相B从5%变为95%;3.5~5min,流动相B从95%变为5%;柱温:30℃,流速:0.25mL/min,进样量:2μL。
优选的,步骤c)中的质谱工作条件:扫描方式:电喷雾正离子扫描;检测方式:多反应监测(MRM);雾化气流量:600L/h;喷雾电压:5.50kV;毛细管温度:500℃;碰撞气类型:N
优选的,所述农产品复杂基质包括苹果、桃、葡萄和柑橘的水果类产品,甘蓝、芹菜、番茄、茄子、马铃薯、胡萝卜、菜豆和韭菜的蔬菜类产品,糙米、小麦和玉米的谷物类产品,油料作物花生、坚果花生、食用菌平菇、菜籽油、绿茶、香辛料胡椒以及糖料作物甘蔗。
与现有技术相比,本发明的有益效果有:
(1)本发明根据农药本身的极性以及不同农产品基质在提取溶剂中的溶解度,对农产品复杂基质采用乙腈+酸+水溶液的方式进行重复三次提取,提取方法快速简便。
(2)本发明提取溶剂采用乙腈+酸+水溶液,根据样品中含水量不同选择不同酸,高含水量样品使用三氟乙酸、低含水量样品使用甲酸,避免三氟乙酸在含水量低且含油量高的样品分析时出现凝胶状,不能分离得到提取液。
(3)本发明净化中根据样品含油量不同调节pH值,高含油量样品须使用氨水调节pH至8.5,低含油量样品可不调节pH值。避免乙腈+甲酸+水溶液提取高含油量样品时提取液浑浊,不能过膜,且容易导致仪器污染。
(4)采用超高效液相色谱-串联质谱法检测盐酸吗啉胍的含量,在0.01-0.2μg/mL浓度范围内,盐酸吗啉胍的线性相关系数均大于0.999,所得结果误差在合理范围内,符合食品安全国家标准《食品中农药最大残留限量》中的规定,而且测定盐酸吗啉胍在茶叶中定量限0.1mg/kg,其余农产品基质定量限为0.05mg/kg,适用于盐酸吗啉胍残留量的准确、快速分析。
(5)本发明优选PSA作为净化剂,净化效果更好、回收率高且成本更低。
附图说明
图1为本发明的不同提取溶剂对盐酸吗啉胍提取效果对比图。
图2为本发明的不同净化剂对盐酸吗啉胍的净化效果对比图。
图3为本发明的高含油量样品在三氟乙酸体系提取时呈现凝胶状现象图。
图4为含油量高的样品使用甲酸体系提取时呈现浑浊现象图。
图5为本发明的盐酸吗啉胍在0.005mg/kg芹菜基质中C18和Hilic色谱柱分离典型色谱图(A为C18柱;B为Hilic柱)。
图6为本发明盐酸吗啉胍的裂解途径。
具体实施方式
为了更好理解本发明技术内容,下面提供具体实施例,对本发明做进一步的说明。
本发明实施例所用的实验方法如无特殊说明,均为常规方法。
本发明实施例所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
实施例1
一种检测农产品复杂基质中盐酸吗啉胍的方法,其包括以下步骤:
a)、蔬菜、水果类提取处理:称取葡萄10g(精确至0.01g)试样于50mL离心管中,加入20mL乙腈-三氟乙酸-水溶液,涡旋振荡10min后,以8000r/min离心5min,倾出提取液,向装剩余残渣的离心管中加入10mL乙腈-三氟乙酸-水溶液,重复上述提取处理,合并3次提取液,用提取液定容至50mL,待净化。
b)、从提取液中吸取5mL,加入150mg PSA,涡旋振荡2min后,以8000r/min离心5min,取上层清液经0.22μm微孔滤膜过滤后,供液相色谱-串联质谱测定;
c)、将制得的待测溶液通过色谱柱进行高效液相色谱分离,然后通过质谱进行分析测定,
其中,色谱工作条件:UPLC色谱柱为Hilic,50mm×2.1mm;流动相:A相为0.1%甲酸+5mmol甲酸胺溶液,B相为乙腈,其梯度洗脱条件为:0~1.5min,流动相B保持5%;1.5~3.5min,流动相B从5%变为95%;3.5~5min,流动相B从95%变为5%;柱温:30℃,流速:0.25mL/min,进样量:2μL;
质谱工作条件:扫描方式:电喷雾正离子扫描;检测方式:多反应监测(MRM);雾化气流量:600L/h;喷雾电压:5.50kV;毛细管温度:500℃;碰撞气类型:N
实施例2
一种检测农产品复杂基质中盐酸吗啉胍的方法,其包括以下步骤:
a)、蔬菜、水果类产品提取处理:称取柑橘10g(精确至0.01g)试样于50mL离心管中,加入20mL乙腈-三氟乙酸-水溶液,涡旋振荡10min后,以8000r/min离心5min,倾出提取液,向装剩余残渣的离心管中加入10mL乙腈-三氟乙酸-水溶液,重复上述提取处理,合并3次提取液,用提取液定容至50mL,待净化。
b)、从提取液中吸取5mL,加入150mg PSA,涡旋振荡2min后,以8000r/min离心5min,取上层清液经0.22μm微孔滤膜过滤后,供液相色谱-串联质谱测定;
c)、将制得的待测溶液通过色谱柱进行高效液相色谱分离,然后通过质谱进行分析测定,
其中,色谱工作条件:UPLC色谱柱为C18,50mm×2.1mm;流动相:A相为0.1%甲酸+5mmol甲酸胺溶液,B相为乙腈,其梯度洗脱条件为:0~1.5min,流动相B保持5%;1.5~3.5min,流动相B从5%变为95%;3.5~5min,流动相B从95%变为5%;柱温:30℃,流速:0.25mL/min,进样量:2μL;
质谱工作条件:扫描方式:电喷雾正离子扫描;检测方式:多反应监测(MRM);雾化气流量:600L/h;喷雾电压:5.50kV;毛细管温度:500℃;碰撞气类型:N
实施例3
一种检测农产品复杂基质中盐酸吗啉胍的方法,其包括以下步骤:
a)、蔬菜、水果类产品提取处理:称取芹菜10g(精确至0.01g)试样于50mL离心管中,加入20mL乙腈-三氟乙酸-水溶液,涡旋振荡10min后,以8000r/min离心5min,倾出提取液,向装剩余残渣的离心管中加入10mL乙腈-三氟乙酸-水溶液,重复上述提取处理,合并3次提取液,用提取液定容至50mL,待净化。
b)、从提取液中吸取5mL,加入150mg PSA,涡旋振荡2min后,以8000r/min离心5min,取上层清液经0.22μm微孔滤膜过滤后,供液相色谱-串联质谱测定;
c)、将制得的待测溶液通过色谱柱进行高效液相色谱分离,然后通过质谱进行分析测定,
其中,色谱工作条件:UPLC色谱柱为C18或Hilic,50mm×2.1mm;流动相:A相为0.1%甲酸+5mmol甲酸胺溶液,B相为乙腈,其梯度洗脱条件为:0~1.5min,流动相B保持5%;1.5~3.5min,流动相B从5%变为95%;3.5~5min,流动相B从95%变为5%;柱温:30℃,流速:0.25mL/min,进样量:2μL;
质谱工作条件:扫描方式:电喷雾正离子扫描;检测方式:多反应监测(MRM);雾化气流量:600L/h;喷雾电压:5.50kV;毛细管温度:500℃;碰撞气类型:N
本方法分别采用C18色谱柱和Hilic色谱柱分别对盐酸吗啉胍进行进样,其色谱图见图5,两根色谱柱均能有较好峰形,但是由于盐酸吗啉胍为水溶性农药,在C18柱上保留性较差,故出峰靠前;而Hilic色谱柱可对极性及极性很强的分析物进行保留的同时,对碱性溶质的峰形分离表现出色,可提供更良好的峰形。盐酸吗啉胍化学结构中含有很多氨基,所以易形成一个缓冲溶液体系,有助于农药在色谱柱中的平衡,延长农药在色谱柱中的保留时间。本方法优选Hilic色谱柱进行该农药的实验,盐酸吗啉胍在Hilic色谱柱条件下的保留时间为1.64min。本方法测定盐酸吗啉胍也可使用C18色谱柱进行分析,其保留时间为0.63min。
实施例4
一种检测农产品复杂基质中盐酸吗啉胍的方法,其包括以下步骤:
a)、蔬菜、水果类产品提取处理:称取胡萝卜10g(精确至0.01g)试样于50mL离心管中,加入20mL乙腈-三氟乙酸-水溶液,涡旋振荡10min后,以8000r/min离心5min,倾出提取液,向装剩余残渣的离心管中加入10mL乙腈-三氟乙酸-水溶液,重复上述提取处理,合并3次提取液,用提取液定容至50mL,待净化。
b)、从提取液中吸取5mL,加入150mg PSA,涡旋振荡2min后,以8000r/min离心5min,取上层清液经0.22μm微孔滤膜过滤后,供液相色谱-串联质谱测定;
c)、将制得的待测溶液通过色谱柱进行高效液相色谱分离,然后通过质谱进行分析测定,
其中,色谱工作条件:UPLC色谱柱为C18,50mm×2.1mm;流动相:A相为0.1%甲酸+5mmol甲酸胺溶液,B相为乙腈,其梯度洗脱条件为:0~1.5min,流动相B保持5%;1.5~3.5min,流动相B从5%变为95%;3.5~5min,流动相B从95%变为5%;柱温:30℃,流速:0.25mL/min,进样量:2μL;
质谱工作条件:扫描方式:电喷雾正离子扫描;检测方式:多反应监测(MRM);雾化气流量:600L/h;喷雾电压:5.50kV;毛细管温度:500℃;碰撞气类型:N
实施例5
a)、提取处理:称取小麦5g(精确至0.01g)于50mL离心管中,补水至样品完全润湿,静置30min,。加入10mL乙腈-甲酸-水溶液,涡旋振荡10min后,以8000r/min离心5min,取出全部提取液,向装剩余残渣的离心管中加入5mL乙腈-甲酸-水溶液,重复上述提取处理,合并3次提取液,用提取液定容至25mL,待净化。
b)、从提取液中吸取5mL,加入150mg PSA,涡旋振荡2min后,以8000r/min离心5min,取上层清液经0.22μm微孔滤膜过滤后,供液相色谱-串联质谱测定;
c)、将制得的待测溶液通过色谱柱进行高效液相色谱分离,然后通过质谱进行分析测定,
色谱工作条件:UPLC色谱柱为Hilic,50mm×2.1mm;流动相:A相为0.1%甲酸+5mmol甲酸胺溶液,B相为乙腈,其梯度洗脱条件为:0~1.5min,流动相B保持5%;1.5~3.5min,流动相B从5%变为95%;3.5~5min,流动相B从95%变为5%;柱温:30℃,流速:0.25mL/min,进样量:2μL;
质谱工作条件:扫描方式:电喷雾正离子扫描;检测方式:多反应监测(MRM);雾化气流量:600L/h;喷雾电压:5.50kV;毛细管温度:500℃;碰撞气类型:N
实施例6
a)、提取处理:称取花生5g(精确至0.01g)于50mL离心管中,补水至样品完全润湿,静置30min。加入10mL乙腈-甲酸-水溶液,涡旋振荡10min后,以8000r/min离心5min,取出全部提取液,向装剩余残渣的离心管中加入5mL乙腈-甲酸-水溶液,重复上述提取处理,合并3次提取液,用提取液定容至25mL,待净化。
b)、从提取液中吸取5mL,使用氨水调节pH至8.5后加入150mg PSA,涡旋振荡2min后,以8000r/min离心5min,取上层清液经0.22μm微孔滤膜过滤后,供液相色谱-串联质谱测定;
c)、将制得的待测溶液通过色谱柱进行高效液相色谱分离,然后通过质谱进行分析测定,
色谱工作条件:UPLC色谱柱为Hilic,50mm×2.1mm;流动相:A相为0.1%甲酸+5mmol甲酸胺溶液,B相为乙腈,其梯度洗脱条件为:0~1.5min,流动相B保持5%;1.5~3.5min,流动相B从5%变为95%;3.5~5min,流动相B从95%变为5%;柱温:30℃,流速:0.25mL/min,进样量:2μL;
质谱工作条件:扫描方式:电喷雾正离子扫描;检测方式:多反应监测(MRM);雾化气流量:600L/h;喷雾电压:5.50kV;毛细管温度:500℃;碰撞气类型:N
实施例7
a)、提取处理:称取核桃5g(精确至0.01g)于50mL离心管中,补水至样品完全润湿,静置30min。加入10mL乙腈-甲酸-水溶液,涡旋振荡10min后,以8000r/min离心5min,取出全部提取液,向装剩余残渣的离心管中加入5mL乙腈-甲酸-水溶液,重复上述提取处理,合并3次提取液,用提取液定容至25mL,待净化。
b)、从提取液中吸取5mL,使用氨水调节pH至8.5后加入150mg PSA,涡旋振荡2min后,以8000r/min离心5min,取上层清液经0.22μm微孔滤膜过滤后,供液相色谱-串联质谱测定;
c)、将制得的待测溶液通过色谱柱进行高效液相色谱分离,然后通过质谱进行分析测定,
色谱工作条件:UPLC色谱柱为Hilic,50mm×2.1mm;流动相:A相为0.1%甲酸+5mmol甲酸胺溶液,B相为乙腈,其梯度洗脱条件为:0~1.5min,流动相B保持5%;1.5~3.5min,流动相B从5%变为95%;3.5~5min,流动相B从95%变为5%;柱温:30℃,流速:0.25mL/min,进样量:2μL;
质谱工作条件:扫描方式:电喷雾正离子扫描;检测方式:多反应监测(MRM);雾化气流量:600L/h;喷雾电压:5.50kV;毛细管温度:500℃;碰撞气类型:N
实施例8
a)、提取处理:称取平菇5g(精确至0.01g)于50mL离心管中,补水至样品完全润湿,静置30min。加入10mL乙腈-甲酸-水溶液,涡旋振荡10min后,以8000r/min离心5min,取出全部提取液,向装剩余残渣的离心管中加入5mL乙腈-甲酸-水溶液,重复上述提取处理,合并3次提取液,用提取液定容至25mL,待净化。
b)、从提取液中吸取5mL,加入150mg PSA,涡旋振荡2min后,以8000r/min离心5min,取上层清液经0.22μm微孔滤膜过滤后,供液相色谱-串联质谱测定;
c)、将制得的待测溶液通过色谱柱进行高效液相色谱分离,然后通过质谱进行分析测定,
色谱工作条件:UPLC色谱柱为Hilic,50mm×2.1mm;流动相:A相为0.1%甲酸+5mmol甲酸胺溶液,B相为乙腈,其梯度洗脱条件为:0~1.5min,流动相B保持5%;1.5~3.5min,流动相B从5%变为95%;3.5~5min,流动相B从95%变为5%;柱温:30℃,流速:0.25mL/min,进样量:2μL;
质谱工作条件:扫描方式:电喷雾正离子扫描;检测方式:多反应监测(MRM);雾化气流量:600L/h;喷雾电压:5.50kV;毛细管温度:500℃;碰撞气类型:N
实施例9
a)、提取处理:称取菜籽油5g(精确至0.01g)于50mL离心管中,加入10mL乙腈-甲酸-水溶液,涡旋振荡10min后,以8000r/min离心5min,取出全部提取液,向装剩余残渣的离心管中加入5mL乙腈-甲酸-水溶液,重复上述提取处理,合并3次提取液,用提取液定容至25mL,待净化。
b)、从提取液中吸取5mL,使用氨水调节pH至8.5后加入150mg PSA,涡旋振荡2min后,以8000r/min离心5min,取上层清液经0.22μm微孔滤膜过滤后,供液相色谱-串联质谱测定;
c)、将制得的待测溶液通过色谱柱进行高效液相色谱分离,然后通过质谱进行分析测定,
色谱工作条件:UPLC色谱柱为Hilic,50mm×2.1mm;流动相:A相为0.1%甲酸+5mmol甲酸胺溶液,B相为乙腈,其梯度洗脱条件为:0~1.5min,流动相B保持5%;1.5~3.5min,流动相B从5%变为95%;3.5~5min,流动相B从95%变为5%;柱温:30℃,流速:0.25mL/min,进样量:2μL;
质谱工作条件:扫描方式:电喷雾正离子扫描;检测方式:多反应监测(MRM);雾化气流量:600L/h;喷雾电压:5.50kV;毛细管温度:500℃;碰撞气类型:N
实施例10
a)、提取处理:称取胡椒5g(精确至0.01g)于50mL离心管中,补水至样品完全润湿,静置30min。加入10mL乙腈-甲酸-水溶液,涡旋振荡10min后,以8000r/min离心5min,取出全部提取液,向装剩余残渣的离心管中加入5mL乙腈-甲酸-水溶液,重复上述提取处理,合并3次提取液,用提取液定容至25mL,待净化。
b)、从提取液中吸取5mL,加入150mg PSA,涡旋振荡2min后,以8000r/min离心5min,取上层清液经0.22μm微孔滤膜过滤后,供液相色谱-串联质谱测定;
c)、将制得的待测溶液通过色谱柱进行高效液相色谱分离,然后通过质谱进行分析测定,
色谱工作条件:UPLC色谱柱为Hilic,50mm×2.1mm;流动相:A相为0.1%甲酸+5mmol甲酸胺溶液,B相为乙腈,其梯度洗脱条件为:0~1.5min,流动相B保持5%;1.5~3.5min,流动相B从5%变为95%;3.5~5min,流动相B从95%变为5%;柱温:30℃,流速:0.25mL/min,进样量:2μL;
质谱工作条件:扫描方式:电喷雾正离子扫描;检测方式:多反应监测(MRM);雾化气流量:600L/h;喷雾电压:5.50kV;毛细管温度:500℃;碰撞气类型:N
实施例11
a)、提取处理:称取甘蔗5g(精确至0.01g)于50mL离心管中,补水至样品完全润湿,静置30min。加入10mL乙腈-甲酸-水溶液,涡旋振荡10min后,以8000r/min离心5min,取出全部提取液,向装剩余残渣的离心管中加入5mL乙腈-甲酸-水溶液,重复上述提取处理,合并3次提取液,用提取液定容至25mL,待净化。
b)、从提取液中吸取5mL,加入150mg PSA,涡旋振荡2min后,以8000r/min离心5min,取上层清液经0.22μm微孔滤膜过滤后,供液相色谱-串联质谱测定;
c)、将制得的待测溶液通过色谱柱进行高效液相色谱分离,然后通过质谱进行分析测定,
色谱工作条件:UPLC色谱柱为Hilic,50mm×2.1mm;流动相:A相为0.1%甲酸+5mmol甲酸胺溶液,B相为乙腈,其梯度洗脱条件为:0~1.5min,流动相B保持5%;1.5~3.5min,流动相B从5%变为95%;3.5~5min,流动相B从95%变为5%;柱温:30℃,流速:0.25mL/min,进样量:2μL;
质谱工作条件:扫描方式:电喷雾正离子扫描;检测方式:多反应监测(MRM);雾化气流量:600L/h;喷雾电压:5.50kV;毛细管温度:500℃;碰撞气类型:N
其中,植物油提取液为下层,其它基质提取液均为上层,且注:测定谷物、食用菌、香辛料和糖料中盐酸吗啉胍时,可不调节pH;测定油料作物和坚果时,需要用氨水调节pH至8.5±0.1。在测试中发现,油料、坚果和植物油由于含油量较大,在加入三氟乙酸体系后,样品呈现凝胶状,其可能的原因是样品中含有大量油脂,在酸性条件下凝固,即使在10000rpm的高速离心后,样品和提取液仍然不能很好分离,很难收集到提取液,使得回收率降低。在改用甲酸体系提取后,样品和提取液能较好分离,但是提取液浑浊(图4),在加入各类净化剂后仍然净化效果较差,因此使用调节pH进行处理。
实施例12
a)、茶叶类产品提取处理:称取2g绿茶(精确至0.01g)于50mL离心管中,补水至样品完全润湿,静置30min。加入10mL乙腈-甲酸-水溶液,涡旋振荡10min后,以8000r/min离心5min,倾出提取液,向装剩余残渣的离心管中加入5mL乙腈-甲酸-水溶液,重复上述提取处理,合并3次提取液,用提取液定容至20mL,待净化;
b)、从提取液中吸取5mL,加入150mg PSA,涡旋振荡2min后,以8000r/min离心5min,取上层清液经0.22μm微孔滤膜过滤后,供液相色谱-串联质谱测定;
c)、将制得的待测溶液通过色谱柱进行高效液相色谱分离,然后通过质谱进行分析测定;
其中,色谱工作条件:UPLC色谱柱为C18或Hilic,50mm×2.1mm;流动相:A相为0.1%甲酸+5mmol甲酸胺溶液,B相为乙腈,其梯度洗脱条件为:0~1.5min,流动相B保持5%;1.5~3.5min,流动相B从5%变为95%;3.5~5min,流动相B从95%变为5%;柱温:30℃,流速:0.25mL/min,进样量:2μL;
质谱工作条件:扫描方式:电喷雾正离子扫描;检测方式:多反应监测(MRM);雾化气流量:600L/h;喷雾电压:5.50kV;毛细管温度:500℃;碰撞气类型:N
对比实施例1
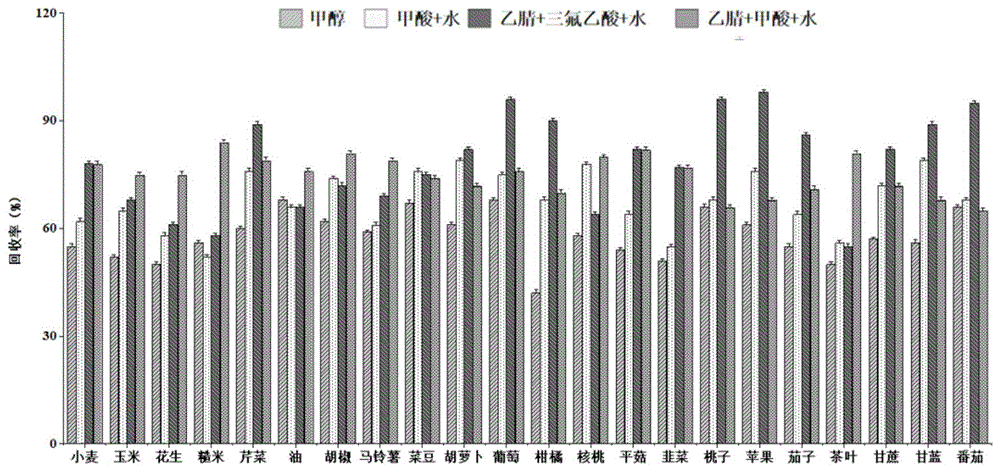
采用不同提取剂对盐酸吗啉胍的提取效果对比:通过采用甲醇、甲酸+水、乙腈+三氟乙酸+水(49.5+1+49.5,体积比)、乙腈+甲酸+水(10+1+89,体积比)4种提取剂对盐酸吗啉胍的提取效果,通过对样品的回收率进行分析,结果如图1所示。乙腈+三氟乙酸+水、乙腈+甲酸+水两种混合溶液的提取效率优于其它两种溶剂,其中,蔬菜水果类样品使用三氟乙酸体系的提取效果优于甲酸体系,但是对于其它种类样品甲酸提取效果优于三氟乙酸体系;这是由于对于含水量高的样品用强酸提取更有效,但是浓度过低会导致酸性能力不足达不到回收效果,而浓度过高又不稳定,对仪器也容易造成损害。因此本方法优选乙腈+三氟乙酸+水对水果、蔬类样品进行提取,而其他样品优选乙腈+甲酸+水提取。
实施例13
采用不同净化剂对盐酸吗啉胍的净化效果对比;盐酸吗啉胍属水溶性,因此本方法中不加入NaCl使提取液中有机相和水相分层,提取液中含有杂质较多。通过C18、GCB(石墨碳)、PSA三种常用净化剂对样品净化,对比3种净化剂的净化效果,净化的效果见图2。在相同条件下,回收率均能满足标准,与其他净化剂相比,PSA净化效果更好、回收率高且成本更低,综合考虑,最终优选PSA作为本方法的净化剂。
实施例14
在测试中发现,油料和坚果由于含油量较大,在加入三氟乙酸体系后,样品呈现凝胶状,如图3所示,其可能的原因是样品中含有大量油脂,在酸性条件下凝固,即使在10000rpm的高速离心后,样品和提取液仍然不能很好分离,很难收集到提取液,使得回收率降低。在改用甲酸体系提取后,样品和提取液能较好分离,但是提取液浑浊见图4,在加入各类净化剂后仍然净化效果较差,因此尝试使用调节pH进行处理。经使用氨水将pH调节为8.5±0.1,可以保证样品分离,并且回收率达到要求。因此,本方法在后续实验时,对于含油量高的样品,前处理过程中须调节pH,保证样品与提取液良好分离。
实施例15
盐酸吗啉胍由吗啉和盐酸组成,其在质谱中离子化的裂解规律是其准确定量的关键。本方法首先在正离子检测模式下进行激发,使用直接进样的方式优化质谱分析盐酸吗啉胍的条件。盐酸吗啉胍在质谱中的裂解碎片如图6所示,在一级质谱中,盐酸吗啉胍产生稳定的[M+H]
为考察本方法对于盐酸吗啉胍的回收率,用不含待测农药及其代谢物的10大类共计22种植物源性食品的样品做添加回收率和精密度实验,样品添加农药混合标准溶液后,放置30min,使农药被样品完全吸收,然后各类样品分别按照本方法阐述的进行提取、净化和上机测试,共测定了3种农药及其代谢物在22种植物源性食品中3个添加水平(其它为0.05、0.1和1mg/kg,茶叶为0.1mg/kg、0.5mg/kg、1mg/kg)和5次平行实验。《农药残留检测方法国家标准编制指南》中对不同添加水平的回收率和相对标准偏差要求见表1。
表1不同添加水平对回收率的要求
数据分析后发现,各基质中不同添加水平的回收率绝大部分能够满足上表中的要求。现对不同类型基质中的代表性基质(糙米、小麦、玉米、花生、甘蓝、芹菜、番茄、茄子、马铃薯、萝卜、菜豆、韭菜、苹果、桃、葡萄、柑橘、核桃、平菇、菜籽油、绿茶、花椒、甘蔗)的回收率结果见表2。
表2盐酸吗啉胍中22种基质的添加回收率
(其中绿茶的添加水平为0.1mg/kg、0.5mg/kg、1mg/kg)
由以上各个基质的回收率数据可以看出,本方法所测定的3种农药及其代谢物中,平均回收率绝大部分分布在70%-110%之间,少部分分布在110%-120%之间,RSD绝大部分在10%以下。说明本方法的回收率很好,能够满足残留检测的要求。
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 混合制剂中盐酸吗啉胍含量和乙酸铜含量的检测方法
- 混合制剂中盐酸吗啉胍含量和乙酸铜含量的检测方法