一种靶向药物的高通量筛选方法
文献发布时间:2023-06-19 12:04:09
技术领域
本发明属于药物筛选技术领域,具体涉及一种靶向药物的高通量筛选方法。
背景技术
高通量药物筛选是指以分子水平和细胞水平的实验方法为基础,以微板等形式作为实验工具载体,以自动化操作系统执行实验过程,以灵敏快速的检测仪器采集实验结果数据,以计算机对实验数据进行分析处理,同一时间对大量样品进行检测,并以相应的数据库支持整个体系运转的技术体系。随着越来越多新靶点的出现、合成化学的发展以及新检测技术的应用,也出现了许多高通量药物筛选手段,如差式扫描荧光法(differentialscanning fluorimetry),表面等离子共振法(surface plasmon resonance),生物膜干涉法(bio-layer interferometry),放大化学发光亲和均相检测(Alpha Screen)及荧光偏正法(fluorescence polarization)等。
在基于靶点的药物筛选中,放大化学发光亲和均相检测(Amplified LuminescentProximity Homogeneous Assay Screen,ALPHA Screen)技术,由于其灵敏、实时、快速、方便等众多优点,是高通量筛选的常用手段之一。该技术是基于微珠的均相亲和检测方法。但是,ALPHA技术所得结果可能与胞内真实状况不同,很难直接关注蛋白质的相互作用过程。而且,由于其串联式的放大信号反应,产生了高度灵敏的信号,只要在200nm范围内存在受体微珠(Acceptor beads),就可以在520-620nm产生光信号,所以容易出现因微珠发生异常靠近而产生的假阳性信号。
表面等离子共振法(surface plasmon resonance)在金属表面固定一层受体分子(receptor),加在缓冲液中的配体(ligand)与固定的受体的反应将导致谱峰发生可以观测到的位移。通过分析共振角的改变,就可以得到分子间相互作用的信息。这些信息数据传输到计算机上,通过分析可以实时、定量、灵敏地监测到生物大分子的相互作用。但该方法需要使用专用的芯片,检测成本较高,与细胞实验和动物药效学实验结果有时会存在差异。
表面增强拉曼光谱分析技术:光的散射是指光通过不均匀介质时一部分光偏离原方向传播的现象,偏离原方向的光称为散射光。拉曼散射则是波长发生改变的散射光,即入射光与介质的分子运动相互作用引起的频率发生改变的散射。每种物质都有自己特有的拉曼光谱,拉曼峰的位置和强度、拉曼谱线的数目直接与样品分子振动或转动能级有关。因此,与红外吸收光谱类似,解析拉曼光谱也可以得到有关分子振动或转动的信息进而用于物质定性、定量分析。但是拉曼散射是个非常弱的过程,散射光强仅约为入射光强的10
直到二十世纪七十年代科研工作者发现当一些分子被吸附到某些粗糙的金属(如银、铜、金等)表面上时,它们的拉曼散射强度会增加10
发明内容
为了解决上述技术问题,本发明公开了一种抑制剂的高通量筛选和捕获方法,所述方法具有信号灵敏,并且极近距离触发,可以排除大部分因微珠发生异常靠近而产生的假阳性信号等优势。
本发明提供了一种靶向药物的高通量筛选方法,包括以下步骤:
步骤S1,分别制备金属纳米粒子探针和磁珠探针,
步骤S2,将所述金属纳米粒子探针或磁珠探针与拉曼信号分子偶联;
步骤S3,将所述步骤S2制得的金属纳米粒子探针、磁珠探针分别与靶蛋白或靶蛋白结合受体蛋白偶联;
步骤S4,将所述步骤S3制得的金属纳米粒子探针和磁珠探针同时加入待筛选化合物中,孵育;
步骤S5,检测拉曼信号,判断所述待筛选化合物是否为靶向药物,所述靶向药物具有抑制靶蛋白或核酸与其结合受体结合的活性。
在某些实施例中,所述金属纳米粒子探针为金纳米粒子探针或银纳米粒子探针。
在某些实施例中,所述拉曼信号分子为4-MBA或4-ABP。
在某些实施例中,所述靶蛋白为PDEδ蛋白、PD-L1蛋白或PARP1蛋白。
在某些实施例中,所述结合受体为具有能够与靶蛋白或核酸结合活性的受体蛋白。
在某些实施例中,所述步骤S5中的判断具体为:
若检测到SERS信号明显由强变弱或由弱变强,则所述待筛选化合物为针对特定靶点的靶向药物;
若SERS信号没有明显改变,则所述待筛选化合物非针对特定靶点的靶向药物。
在某些实施例中,所述方法还包括设置阳性对照,所述阳性对照为已知具有抑制靶蛋白与靶蛋白结合受体结合活性的抑制剂。
在某些实施例中,所述抑制剂可以为小分子化合物、蛋白质或核酸。
在某些实施例中,所述方法还包括对靶向药物的回收。
本发明还提供了本发明上述的高通量筛选方法在在混合物中进行分子垂钓得到靶向药物的应用。
本发明相对与现有技术具有的效果:
1)本发明首次将拉曼信号检测应用到抑制剂筛选技术领域,并结合金属纳米粒子及磁珠,形成简单灵敏的抑制剂筛选方法。
2)本发明的方法测量干扰小,所需样品量少,信号强且灵敏度高,并可以实现高通量分子垂钓,同时操作简便,整个检测过程无需进行清洗,为抑制剂的筛选途径提供了新策略。
附图说明
为了更清楚地说明本发明具体实施方式或现有技术中的技术方案,下面将对具体实施方式描述中所需要使用的附图作简单地介绍。
图1为实施例3中两种拉曼探针制备过程示意图。
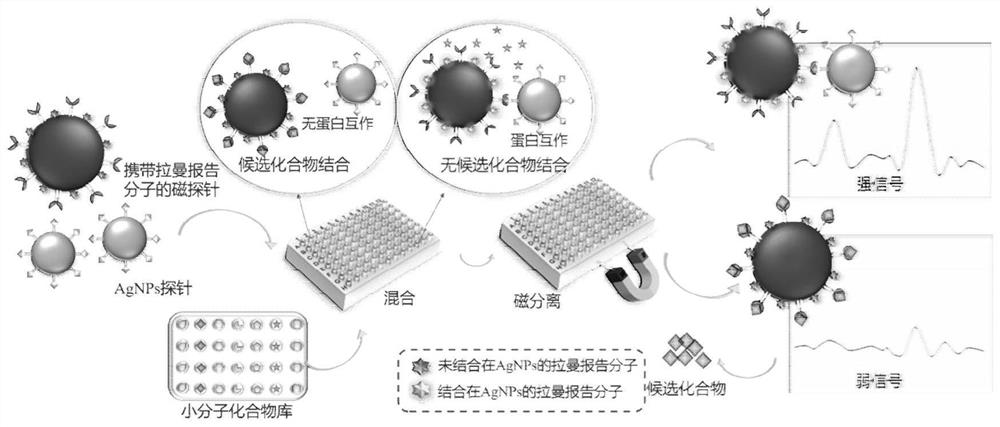
图2为实施例4中抑制剂筛选过程示意图。
图3为实施例4中加入不同浓度抑制剂时拉曼信号检测结果。
图4为实施例5中抑制剂的筛选及回收过程示意图。
图5为实施例5中抑制剂回收后质谱检测结果。
图6为实施例6两种拉曼探针制备及小分子药物筛选过程的示意图。
图7为实施例6实验的拉曼信号检测结果。
图8为实施例6加入不同浓度阳性药后拉曼信号变化情况。
图9为实施例7中两种拉曼探针制备及小分子抑制剂筛选过程示意图。
图10为A)AgNPs,AgNPs@PD-1的紫外可见光谱。B)AgNP,AgNPs@PD-1,MNs-COOH,Mns-COOH@PD-L1的平均大小和zeta电位。C)加入BMS-202,洗涤后,电子显微镜观察收集到的磁珠,没有探针结合。D)不加阳性药,银和磁性探针结合洗涤后的电镜图像。
图11为实施例8拉曼信号检测结果。
图12为实施例9拉曼信号检测结果。
图13为实施例10中两种探针相互作用及抑制剂筛选过程示意图。
图14为实施例10拉曼信号检测结果。
具体实施方式
根据下述实施例,可以更好地理解本发明。然而,本领域的技术人员容易理解,实施案例所描述的内容仅用于说明和解释本发明,并不用于限制权利要求书中所详细描述的本发明。除非特别说明,本发明采用的试剂、方法和设备如无特别说明,均为常规方法,所使用的试验材料如无特别说明,均可从商业公司获取。
实施例1制备金(银)纳米粒子
1.制备金纳米粒子
金纳米粒子(AuNPs):取一只250mL三口烧瓶,用王水(1份浓硝酸加入3份浓盐酸)清洗,第二天取出冲洗干净后再用注射用水洗涤三次,保证壁内纯净无杂质。放入烘箱烘干,冷却至室温。烧瓶中加入2mL 1%的HAuCl
2.制备银纳米粒子
银纳米粒子(AgNPs):取一只250mL三口烧瓶在酸缸浸泡过夜,第二天取出冲洗干净后再用注射用水洗涤三次,保证壁内纯净无杂质。放入烘箱烘干,冷却至室温。期间精确称量0.018g硝酸银粉末,小心倒入三口烧瓶中。加入100mL超纯水,摇匀,接上冷凝管并设定电热套温度为100℃,将溶液搅拌加热至微沸后立刻快速加入2mL 1%的柠檬酸钠溶液。观察溶液颜色变化:浅黄,到深黄,再到灰绿,观察到颜色不再发生变化,稍稍降温到90℃,继续磁力搅拌,维持40min得到银纳米粒子分散体系。用紫外分光光度计测定银纳米粒子的吸光度曲线:将银纳米粒子5倍稀释后检测,观察最大吸收波长范围(420-480nm)正确,并没有其他杂峰,即可初步判断合成粒径均一。利用公式:A=kbc(A=Amax,k=3×10
实施例2磁纳米粒子的制备
百迈格生产的商品化羧基磁珠,配置浓度为0.5mg/mL,用磁珠保存液冲洗并磁分离重复三次,重悬在保存液中以防止磁珠聚集,于4℃保存备用。
实施例3合成PDEδ蛋白抑制剂筛选相关的两种拉曼探针(拉曼信号分子偶联在磁珠上时)
具体过程如图1中所示:
(1)AgNPs@Fc融合KRAS银探针的制备
取1mL 0.29nM银纳米粒子(AgNPs),加入10μL吐温20,反应30min后加入5.0μL 1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(EDC)(2.5mM)和5.0μL N-羟基琥珀酰亚胺(NHS)(2.5mM),反应1h,离心去除多余的活化剂,重悬浮。加入4μL 0.05mg/mL ProteinA(金黄色葡萄球菌表面的一种蛋白)的B结构域(Domain B),反应2h,离心弃上清后重悬。加入5μL乙醇胺(15.0mM)封闭,反应30min,离心弃上清后重悬。加入5μL 0.5mg/mL融合表达Fc段的KRAS蛋白,反应2h,离心弃上清后重悬。加入5μL IgG Fc封闭,反应30min,离心弃上清后重悬备用。
(2)MNs@Fc融合PDEδ磁探针的制备
取400μL 0.5mg/mL羧基磁珠,加入5μL吐温20,反应30min后加入10μL 100mM EDC/NHS,反应1h,离心去除多余的活化剂,重悬浮。加入5μL 0.5mg/mL Domain B,和20μL 5mM拉曼信号分子,4-氨基联苯(4-aminobiphenyl,4-ABP)反应2h,离心弃上清后重悬。加入5μL乙醇胺(15.0mM)封闭,反应30min,离心弃上清后重悬。加入5μL 0.5mg/mL融合表达Fc段的PDEδ蛋白,反应2h,离心弃上清后重悬。加入5μL IgG Fc封闭,反应30min,离心弃上清后重悬备用。
实施例4表面增强拉曼光谱仪检测探针孵育后的拉曼信号及抑制剂的高通量筛选
具体过程如图2中所示,
(1)阴性对照:免疫亲和结构的评价
拉曼探针现用现配,按3:1的体积比例混合金属探针和磁探针,分别于孵育20分钟、30分钟,40分钟、50分钟、60分钟、过夜后进行取样,立即进行拉曼信号检测并做图,选择时间较短斜率较小而信号强的时间为拟定的孵育时间。
(2)实验组:免疫亲和抑制效力评价
准确吸取金属纳米粒子探针150μL和磁纳米粒子探针50μL,同时加入用于筛选的1μg/mL化合物1μL,按得到的孵育时间孵育后分别在毛细玻璃管中或标准PCR管中进行拉曼信号检测,设定曝光时间为5s,曝光次数为10次取平均值,判断该化合物是否有抑制结合作用。如果拉曼信号变得很弱,则有作为抑制剂的可能。从化合物库中,最后筛选得到新的抑制剂。
(3)定量和阳性对照
分别对筛选得到的抑制剂进行定量实验。用PBS缓冲液稀释抑制剂,设置浓度梯度。以PDEδ已知抑制剂Deltarasin为例,得到相浓度梯度,在384孔板中,用高通量进样装置取样,平行进行拉曼检测,绘制对应的浓度梯度曲线,得到对应的检测限。并对此进行评价,挑选出更有潜力的抑制剂,检测结果如图3所示。
实施例5对混合物中抑制剂的筛选、垂钓及回收
从中药提取物等混合物及已知化合物库等,可以用同样的方法检测到其中可以开发成新药的抑制剂,样品经过简单前期处理,去除脂类、蛋白等杂质,如混合样品不易溶于水,可先用少量DMSO溶解样品,并离心去除杂质。
具体过程如图2,4所示,金属球和磁珠通过亲和反应彼此靠近,当距离达到10nm内拉曼报告分子的信号会得到指数级的增强。在针对特定靶点的新药筛选过程中,当有药物结合在靶点上时,金属球和磁珠不能通过亲和反应彼此靠近,距离不能达到10nm内,拉曼报告分子仅有极弱的拉曼信号。之后用磁铁回收偶联磁珠的蛋白和结合的抑制剂,先用PBS清洗,再通过酸性溶液清洗,使蛋白结构改变,抑制剂脱落,离心取上清,回收小分子抑制剂。进一步用核磁共振、质谱等方法进行结构鉴定,并用于后续的活性检测等。
以Deltarasin为例,与MNs@Fc融合PDEδ磁探针混合后,进行磁分离,用注射用水洗涤磁探针3次后,加入1%乙酸清洗磁珠,离心取上清,上清液用冻干机冻干,去除水分和乙酸,收集样品进行质谱检测。结果如图5显示,得到样品分子量604.2与Deltarasin一致,且纯度较高。
实施例6合成PDEδ蛋白抑制剂筛选相关的两种拉曼探针(拉曼信号分子偶联在银纳米粒子上)
具体过程如图6所示:
偶联蛋白时为了减少与非位点特异性结合,提高蛋白的靶向效率,我们在PDEδ蛋白的C端添加了一段Fc标签并使用Protein A结构域B高效特异性的识别PDEδ的Fc段。
1mL AgNPs(0.25nM)加入20μL吐温20混匀20min防止AgNPs聚沉。然后,加入10μL2.5mM NHS和10μL 2.5mM EDC混匀活化1小时,离心(3500g,6min),吸去上清后用等体积的注射用水将AgNPs重悬。迅速加入4μL Domain B(0.05mg/mL)偶联1.5小时,离心(3500g,6min),吸去上清后用等体积的注射用水将AgNPs重悬。用拉曼信号分子4-巯基苯甲酸(4-Mercaptobenzoic acid,4-MBA)组装到AgNPs表面,加入2μL PDEδ(0.05mg/mL)和25μl 4-MBA(1.0mM)偶联1.5小时,离心(3500g,6min),吸去上清后用等体积的注射用水将AgNPs重悬得到AgNPs4-MBA@Domain B@PDEδ探针。
加入1μL乙醇胺(15mM)和1μLBSA(0.5mgmL-1)封闭20min,离心(3500g,6mins),吸去上清后用等体积的注射用水将AgNPs重悬。
为了完成KRAS4B和磁珠的偶联,合成一段生物素化C端K-Ras4B多肽,与链霉亲和素修饰的磁珠(300nm,0.5mg/mL)偶联。
Deltarasin(一种小分子KRAS-PDEδ抑制剂)作为阳性药。在有Deltarasin(2.0μL,5μg/mL)和没有Deltarasin的情况下混合两种探针(银探针:磁探针=500μL:150μL),分别对两组混合物进行磁分离,用注射用水洗涤磁探针3次后用等体积注射用水重悬。
磁富集后,在633nm的激发光下进行SERS检测,检测结果如图7所示,可以看到明显的拉曼信号变化,加入阳性药后,拉曼信号明显降低。如图8所示,阳性药浓度越高,信号峰越低。
实施例7合成PD-L1蛋白抑制剂筛选相关的两种拉曼探针(拉曼信号分子偶联在银纳米粒子上)
具体过程如图9所示:
在PD-1/PD-L1方案中,银纳米粒子的合成方法同上。探针的制备,AgNPs(1.0mL,0.30nM)加入5.0μL 1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(EDC)(2.5mM)和5.0μLN-羟基琥珀酰亚胺(NHS)(2.5mM)反应1h来活化AgNPs表面的羧基,离心除去多余的活化剂(4000g,7min)。之后加入3.0μL 1mg/mL PD-1和20μL拉曼信号分子4-氨基联苯(4-aminobiphenyl,4-ABP)(5.0mM)偶联2小时,离心除去未偶联的PD-1(4000g,7min)得到AgNPs4-ABP@PD-1探针。
同理,使用5.0μL 100mM EDC和100mM NHS活化表面修饰羧基的磁珠(400μL,300nm,0.5mg/mL),反应1h后,磁分离除去多余活化剂。加入3.0μL PD-L1(1mg/mL)偶联2h,磁分离去除未偶联的PD-L1,用PBS缓冲液重悬后得到MNs@PD-L1。
检测结果如图10A,B所示,通过将PD-1与AgNPs偶联,在紫外-可见光谱中观察到9nm的红移。与AgNPs相比,AgNPs@PD-1的平均流体动力学尺寸增加了14.82nm(从24.46到39.28nm),Zeta电位从-37.70改变到-13.2mV。另外,通过共轭PD-L1,MNs-COOH的Zeta电位相对于MNs-COOH从-25.93变到-19.90mV,证明了PD-L1在其表面上修饰成功。
实施例8
小分子抑制剂BMS-202可诱导PD-L1的二聚化,并使人的PD-1/PD-L1复合物解离,作为阳性药对该体系进行验证。BMS-202(1.8μM,18μM,180μM)与银探针和磁探针混合,对照组加入相应量的溶剂DMSO(AgNPs4-ABP@PD-1:MNs@PD-L1=3:1)分别对两组混合物进行磁分离,洗涤磁探针3次后用等体积注射用水重悬。磁富集后,在633nm的激发光下进行SERS检测,如图11所示,加入阳性药后,拉曼信号明显降低。如图10C,D所示,电子显微镜下,有阳性药时,银探针和磁珠没有结合,没加阳性药时,磁珠和银探针发生结合。
实施例9
Durvalumab是一种与PD-L1受体有高亲和力结合的IgG1单克隆抗体,在本实验中也用于验证该方法的可行性。按照上述方法成功合成SERS探针后,按照3:1的比例(AgNPs@PD-1@4-ABP:MNs@PD-L1)配置成筛选系统,并向系统中加入一定体积的Durvalumab,使Durvalumab的最终浓度为(0.01μM,0.1μM,1μM,10μM),对照组加入等体积的PBS,以4-ABP在1143cm-1处的特征峰为主要观察目标,判断峰高是否发生变化,结果如图12所示,可观察到随着阳性药浓度增高,拉曼信号降低。
实施例10
紫外线辐射、DNA分子自身的不稳定性及细胞分裂时DNA复制产生的错误均可引起DNA损伤。聚(ADP-核糖)聚合酶-1(Poly(ADP-ribose)polymerase–1,PARP-1),通过与单链损伤DNA(single-strand breaks,SSB)损伤位点结合并产生聚(ADP-核糖)翻译后修饰,继而来调控基因组修复。PARP1已成为越来越多癌症的有效临床靶点。临床PARP1抑制剂与PARP1酶催化中心相结合,进而阻断底物烟酰胺腺嘌呤二核苷酸(NAD+)的结合;阻止聚(ADP-核糖)的产生,引起PARP1捕获,导致DNA-PARP复合物长期存在,无法进行后续的修复。
如图13所示,基于上述原理,联合SERS技术设计两种拉曼探针:修饰了PARP1蛋白的磁探针和修饰了SSB的银探针。PARP1能够识别结合SSB,同时牵引着两种拉曼探针的靠近。拉曼检测结果如图14所示,当体系中只存在NAD+时,PARP1催化NAD+分解,并以分解产物ADP核糖为底物,使PARP1自身发生“PAR化”,驱使PARP1和SSB的分离,磁探针上的拉曼报告分子距离具有拉曼增强效应的银探针较远,不引发SERS效应,拉曼检测强度低;当体系中存在NAD+和PARP1抑制剂(如加入5nM Olaparib)时,PARP1抑制剂能够竞争性结合NAD+结合位点,抑制PARP1-SSB复合物的分离,两种拉曼探针距离较近,引发SERS效应得到较高的拉曼信号,因此可以通过拉曼信号的高低筛选有效的PARP1抑制剂。
以上显示和描述了本发明的基本原理、主要特征和本发明的优点。本行业的技术人员应该了解,本发明不受上述实施例的限制,上述实施例和说明书中描述的只是说明本发明的原理,在不脱离本发明精神和范围的前提下本发明还会有各种变化和改进,这些变化和改进都落入要求保护的本发明范围内。
- 一种靶向药物的高通量筛选方法
- 一种电动转盘系统和微藻高通量筛选装置以及微藻的高通量筛选方法