制备治疗性T淋巴细胞的方法
文献发布时间:2023-06-19 12:14:58
本申请要求2018年12月4日提交的序列号为62/775,323的我们共同未决的美国临时专利申请的优先权,并且将其通过引用并入本文中。
名为102402.0082PCT_ST25、大小为2kb的序列表的ASCII文本文件的内容创建于2019年11月27日,通过EFS-Web与本申请一起以电子方式提交,将其内容通过引用以其整体并入。
技术领域
本发明的领域是组学数据的计算分析,特别地它涉及乳腺癌患者中可以通过肿瘤浸润淋巴细胞靶向的肿瘤相关抗原的鉴定。
背景技术
背景描述包括可用于理解本发明的信息。并不承认本文提供的任何信息是现有技术或与当前要求保护的发明相关,也不承认具体地或隐含地引用的任何出版物是现有技术。
本文中的所有出版物和专利申请都通过引用并入,其程度如同每个单独的出版物或专利申请被具体地且单独地指明通过引用并入一样。在并入的参考文献中的术语的定义或用法与本文提供的该术语的定义不一致或相反时,适用本文提供的该术语的定义,而不适用该术语在该参考文献中的定义。
最近的研究证明,肿瘤浸润淋巴细胞(TIL)的数目与患有Her2+和三阴性乳腺癌(TNBC)的患者的结局和对化疗的应答正向相关。此外,对免疫检查点抑制剂的首次研究显示了这些患者中的有希望的结果。然而,那些TIL的靶标仍然是未知的。因此,分离和克隆繁殖是有问题的,并且许多不同的抗原的可能性进一步加重与分离肿瘤反应性T细胞相关的困难。
因此,仍然需要为基于T细胞的癌症免疫疗法提供改进的方法和组合物。
发明内容
本发明的主题涉及用于鉴定和/或扩增从肿瘤浸润白细胞(TIL)群体中获得的新抗原反应性T细胞的各种组合物和方法。最优选地,因而所获得的T细胞对患者和肿瘤特异性新抗原有反应性,并且可以用作治疗患者癌症的治疗性淋巴细胞。
在本发明主题的一个方面,诸位发明人设想了产生治疗性T细胞的方法,该方法包括从患者的肿瘤组织获得患者特异性组学数据的步骤,从同一患者的匹配正常组织获得患者特异性组学数据的进一步步骤,以及比较来自该肿瘤组织和该匹配正常组织的组学数据以鉴定肿瘤特异性新抗原的又另一步骤。在又另一步骤中,产生重组抗原呈递细胞,其包含编码肿瘤特异性新抗原的重组核酸。然后使从肿瘤浸润白细胞获得的多个T细胞与重组抗原呈递细胞接触,并且从该多个T细胞中分离与重组抗原呈递细胞接触时表达活化标记物(例如,细胞因子或趋化因子如IFN-γ)的一个或多个T细胞,从而获得治疗性T细胞。在先验施用给患者之前可以进一步扩增这些细胞(可以补充向患者施用免疫刺激细胞因子和/或检查点抑制剂)。
如将理解的,可以在使这些T细胞与重组抗原呈递细胞接触之前扩增该多个T细胞,和/或分离的T细胞在克隆上可能是不同的。在需要的情况下,可以对分离的T细胞针对肿瘤特异性新抗原的特异性进行测试。
在一些实施例中,患者特异性组学数据是BAMBAM格式、SAMBAM格式、FASTQ格式或FASTA格式,并且来自肿瘤的患者特异性组学数据包括突变信息、拷贝数信息、插入信息、缺失信息、取向信息和/或断点信息。典型地,但非必需地,通过确定新抗原的表达、新抗原与患者的MHC复合体的等于或小于200nM的结合常数(解离常数)和/或基于SNP的新抗原的排除来进一步鉴定肿瘤特异性新抗原。
在另外的实施例中,重组抗原呈递细胞衍生自自体或HLA匹配的抗原呈递细胞,并且优选是患者的树突细胞。在需要的情况下,编码肿瘤特异性新抗原的重组核酸可以进一步编码至少第二肿瘤特异性新抗原。可替代地或另外地,编码肿瘤特异性新抗原的重组核酸也可以编码共刺激分子、免疫刺激细胞因子或细胞因子类似物、OX40配体或包含OX40配体的融合蛋白、和/或CD40配体或包含CD40配体的融合蛋白。
因此,从不同的角度来看,诸位发明人还设想了如本文所呈现的产生的分离的T细胞以及包含药学上可接受的载体组合如本文所呈现的产生的多个分离的T细胞的药物组合物。最典型地,该组合物将配制用于输液并包含至少10
本发明主题的多个对象、特征、方面和优点将根据以下关于优选实施例的详细描述以及附图而变得更清楚。
附图说明
图1显示了示例性抗原分类的图表。
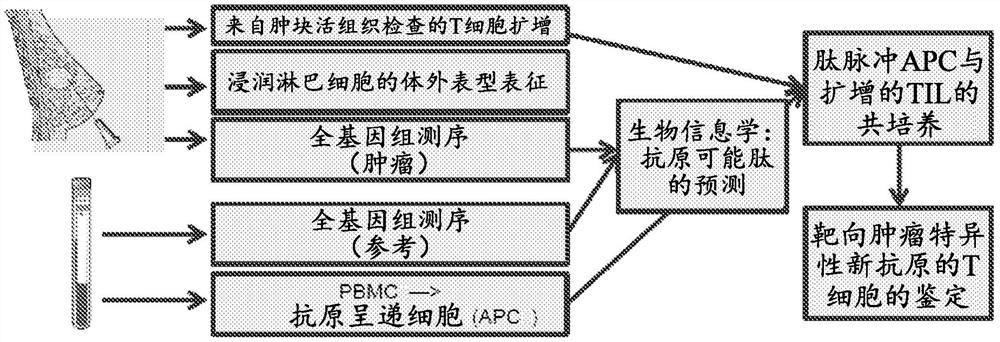
图2显示了用于产生靶向肿瘤特异性新抗原的T细胞的示例性工作流程图。
图3显示了肿瘤浸润淋巴细胞的示例性FACS。
图4显示了示出通过流式细胞术测量的不同类型乳腺癌中的T细胞百分比的示例性结果。
图5显示了用于扩增靶向肿瘤特异性新抗原的T细胞的示例性工作流程图。
图6显示了通过分泌IFN-γ的细胞的流式细胞分选而分离的活化肿瘤浸润白细胞的示例性FACS结果。
图7显示了基于肽刺激后48h的CD137表达分离的活化肿瘤浸润白细胞的示例性FACS结果。
图8示意性地示出了通过T细胞受体和T细胞受体的结构进行的抗原识别。
图9显示了TCR的CDR3部分的示例性序列。
图10A显示了如下的示例性结果,其指示用全长突变型(mut)和野生型(wt)抗原RBMX对自体EBV-LCL进行逆转录病毒转导,以证明内源表达蛋白的加工和呈递(暗色)。作为阳性对照肽,使用肽负载的EBV-LCL(浅色)。
图10B显示了如下的示例性结果,其指示在IFN-γ诱导HLA II类上调后,将突变型RBMX转导入HLA II类阴性乳腺癌细胞系MCF-7细胞系中引起T细胞识别。
图10C显示了如下的示例性结果,其指示MCF-7细胞中的HLA-DP 04:01过表达也导致有效呈递突变型抗原。
图10D显示了如下的示例性结果,其指示自体EBV-LCL上负载的RBMX mut转导的MCF-7细胞的细胞裂解物诱导特异性T细胞应答。
图11显示了在有和没有肽池的情况下针对自体EBV-LCL,重新测试各种T细胞克隆中的IFN-γ表达的示例性结果。
图12显示了针对HLA II类肽池的各种肽测试且对肽28(衍生自RBMX蛋白的肽)发生特异性反应的3E1 T细胞克隆中的IFN-γ表达的示例性条形图。
图13显示了其他两个P28特异性T细胞克隆的IFN-γ表达的示例性条形图。
图14显示了滴定野生型和突变型肽28的示例性结果。
图15显示了在MHC-I类或II类阻断抗体的情况下T细胞克隆中的IFN-γ表达的示例性结果。
图16显示了在MHC II类限制分子的情况下T细胞克隆中的IFN-γ表达的示例性结果。
图17显示了识别肽28的T细胞克隆1A35的IFN-γ表达的示例性结果。
图18显示了识别HLA-II限制分子中的肽28的细胞克隆1A35的IFN-γ表达的示例性结果。
图19显示了TCR的CDR3部分的另一示例性序列。
具体实施方式
现在诸位发明人已经发现了产生可引发针对患者的肿瘤细胞的特异性细胞毒性免疫应答的治疗性T细胞的方法,该方法是通过从患者的肿瘤活组织检查或其他肿瘤样品的TIL分离和任选地扩增对患者真实的一种或多种肿瘤特异性新抗原产生应答的T细胞。最典型地,将在计算机上由患者的肿瘤样品的组学数据鉴定肿瘤特异性新抗原,并且对新抗原的应答性将优选(但不一定)使用自体抗原呈递细胞。从不同的角度来看,可以仅仅使用来自肿瘤样品和匹配正常样品以及先前获得的血液样品的组学数据完全体外/体内进行所设想的方法。
例如,在本发明主题的一个方面,诸位发明人设想了产生治疗性T细胞的方法,该方法包括从患者的肿瘤组织获得患者特异性组学数据的步骤,从同一患者的匹配正常组织获得患者特异性组学数据的进一步步骤,以及比较来自该肿瘤组织和该匹配正常组织的组学数据以鉴定肿瘤特异性新抗原的又另一步骤。在又另一步骤中,产生重组抗原呈递细胞,其包含编码肿瘤特异性新抗原的重组核酸。然后使从肿瘤浸润白细胞获得的多个T细胞与重组抗原呈递细胞接触,并且从该多个T细胞中分离与重组抗原呈递细胞接触时表达活化标记物(例如,细胞因子或趋化因子如IFN-γ)的一个或多个T细胞,从而获得治疗性T细胞,可以将其进一步扩增。
如本文所用,术语“肿瘤”是指以下项并且可与以下项互换使用:一种或多种癌细胞、癌组织(包括转移)、恶性肿瘤细胞或恶性肿瘤组织,它们可以位于或者发现于人体的一个或多个解剖位置中。应当注意,如本文所用的术语“患者”包括经诊断患有病症(例如,癌症)的个体以及出于检测或鉴定病症的目的经历检查和/或测试的个体二者。因此,患有肿瘤的患者是指诊断为患有癌症的个体以及被怀疑患有癌症的个体二者。如本文所用,术语“提供(provide)”或“提供(providing)”是指并且包括制造、产生、放置、使得能使用、转移或准备使用中的任何行为。
可以将新抗原表征为在肿瘤细胞中表达的随机突变,这些随机突变产生独特的抗原和肿瘤特异性抗原。因此,从不同的角度来看,可以通过考虑突变的类型(例如,缺失、插入、颠换、转换、易位)和突变的影响(例如,无义、错义、移码等)来鉴定新抗原,因此这些新抗原可以被用作第一内容过滤器,通过该过滤器可以消除沉默突变和其他不相关的(例如,未表达的)突变。还应当理解,新抗原序列可以被定义为具有相对较短长度(例如,7-11mer)的序列延伸物,其中此类延伸物将包括氨基酸序列中的一个或多个变化。最典型地,发生改变的氨基酸将在中心氨基酸位置处或附近。例如,典型的新抗原可以具有A
因此,应当理解,取决于发生改变的氨基酸的位置,单个氨基酸改变可以在包括这些改变的氨基酸的许多新抗原序列中呈现。有利地,这种序列变异性允许新抗原的多种选择,并因此增加了然后可以基于一种或多种所希望的特征(例如,对患者HLA类型的最高亲和力,最高结构稳定性等)而选择的潜在有用的靶标的数目。最典型地,新抗原将被计算为具有长度为在2-50个之间的氨基酸,更典型地为5-30个之间的氨基酸,并且最典型地为9-15个之间的氨基酸,其中发生改变的氨基酸优选以改善其与MHC结合的方式位于中心或以其他方式定位。例如,在由MHC-I复合体呈递表位的情况下,典型的新抗原长度将为约8-11个氨基酸,而经由MHC-II复合体呈递的典型新抗原的长度将具有约13-17个氨基酸。如将容易理解的,由于在新抗原中改变的氨基酸的位置可能不是中心,实际肽序列和新抗原的实际拓扑可能变化甚大。
当然,应当认识到,新抗原的鉴定或发现可以从多种生物材料开始,包括新鲜活组织检查、冷冻的或其他保存的组织或细胞样品、循环的肿瘤细胞、外泌体、无细胞循环DNA和/或RNA、多种体液(尤其是血液)等。因此,适合的组学分析方法包括核酸测序,特别是对DNA进行操作的NGS方法(例如Illumina测序、ion torrent测序、454焦磷酸测序、纳米孔测序等),RNA测序(例如RNAseq、基于逆转录的测序等),以及蛋白质测序或基于质谱的测序(例如SRM、MRM、CRM等)。因此,组学数据可以覆盖整个基因组、外显子组、转录组或其部分。
照此,并且特别是对于基于核酸的测序,应该特别认识到,对肿瘤组织的高通量基因组测序将允许快速鉴定新抗原。然而,必须意识到,在将如此获得的序列信息与标准参考序列比较时,正常发生的患者间变异(例如,由于SNP、短的插入缺失(indel)、重复的不同数目等)以及杂合性将导致相对大量的潜在假阳性新抗原。值得注意的是,在将患者的肿瘤样品与相同患者的匹配的正常(即,非肿瘤)样品进行比较的情况下,可以消除这种不准确性。此外,可以消除包含由于SNP引起的变化的序列。
在本发明主题的一个尤其优选的方面,通过肿瘤和匹配的正常样品的全基因组测序和/或外显子组测序(通常按覆盖深度为至少10x,更典型地至少20x)来进行DNA分析。可替代地,还可以从先前序列确定已建立的序列记录(例如,SAM、BAM、FASTA、FASTQ或VCF文件)提供DNA数据。因此,数据集可以包括未处理的或已处理的数据集,并且示例性数据集包括具有BAMBAM格式、SAMBAM格式、FASTQ格式或FASTA格式的那些。然而,尤其优选的是,以BAMBAM格式或作为BAMBAM diff对象提供数据集(参见例如,US 2012/0059670 A1和US2012/0066001 A1)。此外,应当注意,数据集反映了同一患者的肿瘤和匹配的正常样品,以便如此获得患者和肿瘤特异性信息。因此,可以排除不引起肿瘤的遗传种系改变(例如,沉默突变、SNP等)。当然,应该认识到,肿瘤样品可能来自初始肿瘤、治疗开始时的肿瘤、复发的肿瘤和/或转移部位等。在大多数情况下,患者的匹配的正常样品可能是血液或与肿瘤相同组织类型的未患病组织。
同样,可以按多种方式进行序列数据的计算分析。然而,在最优选的方法中,如例如在US 2012/0059670 A1和US 2012/0066001 A1中所披露,使用BAM文件和BAM服务器通过肿瘤和正常样品的位置引导的同步比对,在计算机中进行分析。这样的分析有利地减少假阳性抗原,并且显著地减少对存储器和计算资源的需求。
应当注意,应该读取针对计算机的任何语言,以包括任何合适的计算装置的组合,这些计算装置包括服务器、接口、系统、数据库、代理、端、引擎、控制器或单独或共同操作的其他类型的计算装置。应当理解,计算装置包括处理器,该处理器被配置为执行存储在有形的非暂时性计算机可读存储介质(例如,硬盘驱动器、固态驱动器、RAM、闪存、ROM等)上的软件指令。软件指令优选地配置计算装置,以提供如下文关于所披露的设备所讨论的角色、职责或其他功能。此外,所披露的技术可以体现为计算机程序产品,其包括存储软件指令的非暂时性计算机可读介质,这些软件指令使处理器执行与基于计算机的算法、过程、方法或其他指令的实现相关联的所披露的步骤。在特别优选的实施例中,多种服务器、系统、数据库或接口交换数据使用标准化协议或算法,可能地基于HTTP、HTTPS、AES、公钥-私钥交换、web服务API、已知的金融交易协议、或其他电子信息交换方法。装置之间的数据交换可以通过以下进行:分组交换网络,即因特网、LAN、WAN、VPN或其他类型的分组交换网络;电路交换网络;信元交换网络;或其他类型的网络。
从不同的角度来看,可以建立患者和癌症特异性计算机模拟序列集合,这些序列集合具有预定长度为5和25个之间的氨基酸的新抗原,并且包括至少一个发生改变的氨基酸。对于每个发生改变的氨基酸,这种集合将典型地包括至少两个、至少三个、至少四个、至少五个或至少六个成员,其中发生改变的氨基酸的位置是不同的。这种集合然后可以用于进一步过滤(例如,通过亚细胞定位、转录/表达水平、MHC-I和/或II亲和力等),如下文详细的描述。例如,使用同步的位置引导分析肿瘤和匹配的正常序列数据,诸位发明人先前从多种癌症和患者中鉴定出多种癌症新抗原,包括以下癌症类型:BLCA、BRCA、CESC、COAD、DLBC、GBM、HNSC、KICH、KIRC、KIRP、LAML、LGG、LIHC、LUAD、LUSC、OV、PRAD、READ、SARC、SKCM、STAD、THCA和UCEC。所有新抗原数据都可以在美国专利公开2018/0141998中找到,将其通过引用并入本文。
根据癌症的类型和阶段,应该注意的是,当将检查点抑制剂给予患者时,并非所有已鉴定的新抗原都必定导致患者的治疗上同样有效的反应。实际上,在本领域中熟知,仅一部分新抗原将产生免疫应答。为了增加治疗上希望的应答的可能性,可以进一步过滤新抗原。当然,应当理解,出于本文提出的方法的目的,下游分析不需要考虑沉默突变。然而,优选的突变分析将除了提供突变的类型(例如,缺失、插入、颠换、转换、易位),还提供突变影响的信息(例如,无义、错义等),并且可以这样充当第一内容过滤器,通过该过滤器消除沉默突变。例如,可以选择新抗原用于进一步考虑,其中突变是移码、无义、和/或错义突变。
在进一步的过滤方法中,新抗原还可以针对亚细胞定位参数进行详细的分析。例如,如果新抗原被鉴定为具有膜相关位置(例如,位于细胞的细胞膜的外部)和/或如果计算机模拟结构计算确认了新抗原可能暴露于溶剂,或呈现出结构稳定的表位(例如,J ExpMed[实验医学杂志]2014)等,则可以选择新抗原序列用于进一步考虑。
关于过滤新抗原,通常设想新抗原尤其适合于在组学(或其他)分析揭示出新抗原实际表达的情况下使用。可以按本领域已知的所有方式对新抗原的表达和表达水平进行鉴定,并且优选的方法包括定量RNA(hnRNA或mRNA)分析和/或定量蛋白质组学分析。最典型地,包含新抗原的阈值水平将是以下表达水平,该表达水平是相应匹配的正常序列的表达水平的至少20%、至少30%、至少40%、或至少50%,因此确保(新)表位对于免疫系统是至少潜在‘可见的’。因此,通常优选的是,组学分析还包括对基因表达的分析(转录组学分析),以此帮助鉴定具有突变的基因的表达水平。
本领域存在已知的许多转录组分析方法,并且所有已知方法均被认为适用于本文。例如,优选的物质包括mRNA和初级转录物(hnRNA),并且RNA序列信息可以从逆转录的多聚腺苷酸
类似地,可以按多种方式进行蛋白质组学分析以确定新抗原的RNA的实际翻译,并且本文设想了蛋白质组学分析的所有已知方式。然而,特别优选的蛋白质组学方法包括基于抗体的方法和质谱方法。此外,应当注意,蛋白质组学分析不仅可以提供有关蛋白质本身的定性或定量信息,而且也可以包括蛋白质具有催化活性或其他功能活性的蛋白质活性数据。用于进行蛋白质组学测定的一种示例性技术描述于通过引用并入本文的US 7473532中。鉴定并甚至定量蛋白质表达的另外适合的方法包括多种质谱分析(例如,选择性反应监测(SRM)、多反应监测(MRM)和连续性反应监测(CRM))。
在过滤的又另一个方面,可以将新抗原与包含已知人序列(例如,患者或患者集合的序列)的数据库进行比较,以此避免使用与人相同的序列。而且,过滤还可以包括去除由于患者中的SNP而导致的新抗原序列,其中这些SNP同时存在于肿瘤和匹配的正常序列中。例如,dbSNP(单核苷酸多态性数据库)是由美国国家生物技术信息中心(NCBI)与美国国家人类基因组研究所(NHGRI)合作开发和托管的关于不同物种内和不同物种之间的遗传变异的免费公共档案。尽管数据库的名称仅暗示了一类多态性(单核苷酸多态性(SNP))的集合,但实际上它包含相对广泛的分子变异:(1)SNP;(2)短缺失和插入多态性(插入缺失/DIP);(3)微卫星标记或短串联重复序列(STR);(4)多核苷酸多态性(MNP);(5)杂合的序列;和(6)命名的变体。dbSNP接受明显中性的多态性,对应于已知表型的多态性和无变异的区域。
使用如上所述的这样的数据库和其他过滤选项,可以过滤患者和肿瘤特异性新抗原以去除那些已知序列,产生具有多个新抗原序列的序列集,该多个新表位序列具有显著减少的假阳性。
然而,尽管进行了过滤,应当认识到,并不是所有新抗原都对免疫系统可见,因为这些新抗原还需要在患者的MHC复合体上呈递。实际上,只有一部分新抗原对呈递具有足够的亲和力,并且大多数MHC复合体将排除最常见(如果不是全部)的新抗原的使用。因此,在免疫疗法的背景下,应当由此显而易见的是,在新抗原被结合并由MHC复合体呈递的情况下,这些新抗原将更可能有效。从另一个角度看,检查点抑制剂治疗成功需要多种新抗原通过MHC复合体呈递,其中新抗原必须对患者的HLA类型具有最小的亲和力。因此,应当理解,有效的结合和呈递是新抗原的序列与患者的特定HLA类型的组合功能。最典型地,HLA类型的确定包括至少三种MHC-I亚型(例如,HLA-A、HLA-B、HLA-C)和至少三种MHC-II亚型(例如,HLA-DP、HLA-DQ、HLA-DR),优选地其中每种亚型被确定为至少4位深度。然而,本文还设想更大的深度(例如,6位、8位)。
一旦确定了患者的HLA类型(使用例如已知的基于抗体的化学或计算机确定),就可以根据数据库计算或获取HLA类型的结构解决方案,然后将其用于计算机中的对接模型中以确定(典型地是过滤的)新抗原与HLA结构解决方案的结合亲和力。如以下将进一步讨论的,用于确定结合亲和力的合适系统包括NetMHC平台(参见例如,Nucleic Acids Res.[核酸研究]2008年7月1日;36(Web服务器卷):W509-W512.)。然后选择对先前确定的HLA类型具有高亲和力(例如,具有小于200nM、小于100nM、小于75nM、小于50nM的K
可以使用本领域熟知的湿化学中的多种方法来进行HLA确定,并且所有这些方法均被认为适合用于本文。然而,在尤其优选的方法中,还可以使用包含大多数或所有已知和/或常见的HLA类型的参考序列,在计算机中从组学数据预测HLA类型,如更详细的下文所显示的。
例如,在根据本发明主题的一个优选的方法中,通过数据库或测序仪提供映射至染色体6p21.3(或在该处附近/该处发现HLA等位基因的任何其他位置)的相对大量的患者序列读段。最典型地,序列读段将具有约100-300个碱基的长度,并且包含元数据,包括读段质量、比对信息、取向、位置等。例如,合适的格式包括SAM、BAM、FASTA、GAR等。虽然不限于本发明的主题,但通常优选的是,患者的序列读段提供至少5x、更典型地至少10x、甚至更典型地至少20x,以及最典型地至少30x的覆盖深度。
除了患者序列读取之外,设想的方法进一步采用一个或多个参考序列,这些参考序列包括多个已知不同的HLA等位基因的序列。例如,典型的参考序列可以是合成的(没有相应的人或其他哺乳动物对应物)序列,该序列包括至少一种HLA类型的序列区段,该HLA类型具有该HLA类型的多个HLA等位基因。例如,合适的参考序列包括针对HLA-A的至少50个不同等位基因的已知基因组序列的集合。可替代地或另外地,参考序列也可以包括针对HLA-A的至少50个不同等位基因的已知RNA序列的集合。当然,并且如下面进一步详细讨论的,参考序列不限于HLA-A的50个等位基因,而是关于HLA类型和等位基因的数目/组成可以具有替代组成。最典型地,参考序列将是计算机可读格式,并且将从数据库或其他数据存储设备提供。例如,合适的参考序列格式包括FASTA、FASTQ、EMBL、GCG或GenBank格式,并且可以直接从公共数据存储库(例如IMGT,国际免疫遗传学(International ImMunoGeneTics)信息系统,或等位基因频率网络数据库(The Allele Frequency Net Database),EUROSTAM,URL:www.allelefrequencies.net)的数据中获取或构建。可替代地,参考序列还可以基于一个或多个预定标准(例如等位基因频率、种族等位基因分布、常见或稀有等位基因类型等)从各个已知的HLA等位基因构建。
使用参考序列,现可以将患者序列读取穿过迪布鲁英(de Bruijn)图,以鉴定具有最佳匹配的等位基因。在该上下文中,应注意,每个个体携带针对每种HLA类型的两个等位基因,并且这些等位基因可能非常类似,或在一些情况下甚至是相同的。如此高的相似度给传统的比对方案带来了重大问题。本发明人现已发现,可以使用以下方法来解析HLA等位基因,并且甚至是非常密切相关的等位基因,在该方法中通过将序列读取分解成相对小的k-mer(典型地具有10-20个之间的碱基长度),并且通过实施加权投票过程(其中每个患者序列读取基于与等位基因的序列匹配的序列读取的k-mer,针对每个等位基因提供投票(“定量读取支持”))来构建迪布鲁英图。然后,等位基因的累积最高投票指示最可能预测的HLA等位基因。另外,通常优选与等位基因匹配的每个片段也用于计算该等位基因的总覆盖率和覆盖深度。
得分可以根据需要被进一步改进或完善,尤其是在许多最高命中(top hit)相似的情况下(例如,其得分的很大一部分来自高度共享的k-mer集)。例如,得分细化可以包括加权方案,其中从未来考虑中去除与当前最高命中基本上相似(例如,>99%,或其他预定值)的等位基因。然后,将当前最高命中所使用的k-mer的计数重新加权一个因子(例如,0.5),并通过将这些加权计数相加来重新计算每个HLA等位基因的得分。重复此选择过程以找到新的最高命中。使用RNA序列数据可甚至进一步改进该方法的准确性,该RNA序列数据允许鉴定由肿瘤表达的等位基因,这些等位基因有时可能只是DNA中存在的2个等位基因中的1个。在设想的系统和方法的进一步有利的方面,可以处理DNA或RNA、或DNA和RNA两者的组合以做出高度准确的HLA预测,并且可以源自肿瘤或血液DNA或RNA。在另外的方面中,用于高准确性计算机模拟的HLA分型的合适方法和考虑描述于美国专利申请20180237949(通过引用并入本文)中。
一旦鉴定了患者和肿瘤特异性新抗原和HLA类型,例如使用NetMHC,可以通过将新抗原与HLA对接并确定最佳结合物(例如,最低K
当然,应该意识到,可以使用除NetMHC之外的系统将患者的HLA类型与患者特异性和癌症特异性新抗原进行匹配,并且合适的系统包括NetMHC II、NetMHCpan、IEDB分析资源(URL immunepitope.org)、RankPep、PREDEP、SVMHC、Epipredict、HLABinding等(例如,参见J Immunol Methods[免疫学方法杂志]2011;374:1-4)。在计算最高亲和力时,应注意,可以使用新抗原序列的集合,其中发生改变的氨基酸的位置被移动(同上)。可替代地或另外地,通过添加N末端和/或C末端修饰可以实施对新抗原的修饰,以进一步增加表达的新抗原与患者的HLA型的结合。因此,如鉴定,新抗原可以是天然的,或可以被进一步修饰以更好地匹配特定的HLA型。而且,在需要时,可以计算相应的野生型序列(即,没有氨基酸变化的新抗原序列)的结合以确保高的差别亲和力。例如,新抗原及其相应的野生型序列之间的MHC结合中尤其优选的高差别亲和力是至少2倍、至少5倍、至少10倍、至少100倍、至少500倍、至少1000倍等。
因而鉴定的一个或多个肿瘤特异性新抗原序列可以作为一个或多个重组DNA序列克隆到重组表达载体中,该重组表达载体可在可以从患者的全血中获得的哺乳动物细胞中表达,优选在人抗原呈递细胞中、甚至更优选在自体抗原呈递细胞(例如,树突细胞)中表达。如将容易理解的,新抗原序列典型地具有至少九个氨基酸的长度,或至少12个氨基酸,或至少15个氨基酸,或至少20个氨基酸,或至少30个氨基酸,并且甚至可以表达为全长蛋白质。可以进一步使这类抗原呈递细胞增殖以增加可用细胞的数目。如将易于理解的,设想了引入具有编码肿瘤特异性新抗原的序列的重组核酸的任何合适的方法,包括转染、病毒递送等。在进一步设想的方面,特别是在使用多种新抗原的情况下,抗原可以布置在单个多肽链中,典型地在两个新抗原之间具有柔性肽接头。这种布置有利地增加了抗原呈递的可能性,该抗原呈递将活化先前在TIL群体中分离的T细胞。
另外,设想的重组核酸还可包括编码增强T细胞刺激的蛋白质的其他序列。例如,合适的蛋白质包括共刺激分子(例如,B7-1、B7-2等)、免疫刺激细胞因子(例如,IL2、IL15等)或细胞因子类似物(例如,ALT-803)、OX40配体或包含OX40配体的融合蛋白(例如,OX40受体/OX40配体融合物)和/或CD40配体或包含CD40配体的融合蛋白(例如,CD40受体/CD40配体融合物)。因此,基于上文,应当理解,可以制备自体或HLA相容性抗原呈递细胞,其表达一种或多种患者和肿瘤特异性新抗原,其能够与来自TIL群体的T细胞富有成效地接合以基于患者和肿瘤特异性新抗原活化一个或多个T细胞。
因此,如此产生的重组抗原呈递细胞(表达新抗原)可以进一步与自体或HLA相容性T细胞接触以鉴定对重组抗原呈递细胞有反应性的T细胞。虽然设想了适于与表达肿瘤特异性新抗原的抗原呈递细胞反应的任何T细胞,但优选的T细胞包括患者的自体T细胞,更优选地为从肿瘤组织获得(例如,通过活组织检查等)的TIL群体中的T细胞。设想通过接触表达肿瘤特异性新抗原的抗原呈递细胞来活化具有识别肿瘤特异性新抗原的T细胞受体的一部分T细胞群体。当然,应该注意,可以在使T细胞与抗原呈递细胞接触之前扩增TIL中的TIL和/或T细胞,并且认为所有已知的体外TIL/T细胞扩增的方式都适用于本文。因此,通过测量在活化时T细胞表达和/或释放的一种或多种活化标记物(例如,细胞因子和/或趋化因子)来检测或定量这种活化。这种细胞因子和/或趋化因子可包括IL-2、IL-12、IL-17、γ干扰素、粒细胞-巨噬细胞菌落刺激因子。
诸位发明人进一步设想了可以对通过重组抗原呈递细胞活化的T细胞进行分离,并且任选地进一步扩增,以增加特异性识别肿瘤特异性新抗原的T细胞的群体。可以设想任何合适的扩增T细胞的方法,包括将分离的T细胞用抗CD3抗体刺激,并用IL-2(例如,10-20U/ml)活化3天、7天、14天等。任选地,通过重新接触表达肿瘤特异性新抗原的抗原呈递细胞,可以进一步测试这类分离的(和任选地扩增的)T细胞对肿瘤特异性新抗原的特异性。
在一些实施例中,这种分离的(和任选地扩增的)T细胞可以进一步配制在药学上可接受的载体(例如,用于注射等)中并施用于患者以治疗肿瘤。不希望受任何特定理论的束缚,诸位发明人设想识别肿瘤上的肿瘤特异性新抗原的此类T细胞可以体内特异性地靶向肿瘤细胞,以引发治疗有效的免疫应答,并增加免疫疗法的效率和特异性。如本领域中容易认识到的,进一步的免疫刺激(例如,用免疫刺激细胞因子和/或检查点抑制剂)可以增强T细胞的治疗效果。
如图1所示,人组织可以携带各种类型的抗原。其中,诸位发明人设想,非自抗原的肿瘤特异性新抗原可以提供高亲合力、特异性T细胞应答。
在示例性工作流程中,如图2所示,通过非特异性刺激扩增在诊断的时间点取得的来自乳腺癌活组织检查的肿瘤浸润白细胞(TIL)。另外,诸位发明人使用Gentle Macs分离器组合流式细胞术,以研究肿瘤组织中的TIL数目。此外,诸位发明人对肿瘤组织和作为参考的自体白细胞进行了全基因组测序,以确定患者和肿瘤特异性突变。这里,位置引导的同步比对使得检测导致非同义氨基酸变化的突变。针对编码基因的RNA表达分析这些突变,以及确定潜在的新抗原。评估新抗原与患者特异性HLA分子的潜在结合,截止Kd等于或小于200nM。然后合成潜在新抗原的肽,负载到自体抗原呈递细胞(APC)上并与TIL共培养。产生IFNγ的所有T细胞被克隆地扩增并重新测试肽特异性以鉴定新抗原特异性T细胞克隆。
图3显示了在GentleMACS解离后乳房块活组织检查中浸润淋巴细胞(例如,CD19+、CD4+、CD8+等)的离体表型表征的实例的FACS数据,图4显示了通过流式细胞术测量的不同类型的乳腺癌中的CD3+T细胞的百分比。可以看出,与激素受体(HR)或Her2阳性类型乳腺癌和健康供体(HD)相比,TNBC中的TIL频率较高。
图5显示了下文所选实验的示例性工作流程,其中表达肿瘤特异性新抗原(通过肽-脉冲方案)的抗原呈递细胞(APC)和T细胞共培养。通过FACS进一步分选T细胞并扩增。更具体地,图6显示了通过分泌IFN-γ的细胞的FACS得到的活化T细胞分离的结果。注意,通过接触抗原呈递细胞(APC)可以增加活化的CD4+T细胞的数目。图7显示了刺激后48小时基于CD137表达的活化T细胞分离。与CD4+T细胞相比,CD8+T细胞的活化增加,但在较小程度上。
从分离的T细胞中,诸位发明人鉴定了CDR3部分的序列(如图8所示)。基于CDR3测序,显示三个分离的T细胞克隆代表了证明T细胞应答的多克隆性的各个克隆。图9显示了鉴定为SEQ ID NO:1、SEQ ID NO:2和SEQ ID NO:3的三个T细胞克隆的氨基酸序列CDR3部分。
通过全长突变型(mut)和野生型(wt)抗原RBMX对自体EBV-LCL(EBV感染淋巴细胞系)细胞进行逆转录病毒转导,并使如此制备的细胞与反应性T细胞接触。图10A证明了内源表达蛋白(蓝色)的加工和呈递。作为阳性对照,使用了肽负载的EBV-LCL(灰色)。图10B显示在IFN-γ诱导HLA II类上调后,将突变型RBMX转导入HLA II类阴性乳腺癌细胞系MCF-7细胞系引起T细胞识别。如图10C所示,MCF-7细胞中的HLA-DP 04:01过表达也导致有效呈递突变型抗原。此外,如图10D所示,自体EBV-LCL上负载的RBMX mut转导的MCF-7细胞的细胞裂解物诱导特异性T细胞应答。
然后在IFNγELISA中将扩增的T细胞克隆在有和没有肽池的情况下针对自体EBV-LCL重新测试。在所有克隆中,如图11所示,克隆3E1显示出潜在的新抗原(PP)特异性。然后,针对HLA II类肽池的各种肽测试CD4+T细胞克隆3E1。图12显示克隆3E1针对肽28特异性反应,该肽是衍生自RBMX蛋白的肽。此外,如图13所示,在TIL中发现了其他两种P28特异性T细胞克隆。所有这些都识别突变型P28,而不是野生型对应物。图14示出了野生型和突变型肽28的滴定。
诸位发明人进一步证实,这种反应是HLA类型特异性的。如图15所示,MHC-II类阻断抗体在IFNγELISA中阻断了T细胞应答,证实表位的MHC-II类限制。进一步如图16所示,患者的HLA II类限制分子被逆转录病毒转录到HeLa细胞中。肽28在患者的HLA-DP限制分子中呈递。其中两个克隆识别HLA-DPB1*0401中的肽,且一个识别HLA-DPB1*0201中的肽。HLA-DPB1*0401限制的T细胞克隆还识别HLA-DPB1*0402中的肽。自体EBV-LCL作为阳性对照。
除了在初始诊断的时间点获得的活组织检查外,诸位发明人还在新抗原疗法后分析了切除的肿瘤组织。图17显示可以鉴定识别肽28的第四T细胞克隆(1A35)。1A35克隆可以识别HLA DP B1*04:01和B1*04:02中的肽28,如图18所示,并且图19显示了1A35克隆的CDR3区(SEQ ID NO:4)的序列,该克隆与在初始诊断下鉴定的克隆相比是不同的。
因此,相比于其他类型的乳腺癌或没有恶性肿瘤的患者,本发明人对多于300名患者的肿瘤活组织检查的流式细胞术分析显示TNBC中的TIL频率较高。筛选一名患者中的新抗原特异性T细胞使得鉴定在诊断的时间点取得的从Her2+乳腺癌组织分离的三个肽特异性CD4+T细胞克隆。所有T细胞克隆特异性识别相同的肿瘤特异性突变,而不是野生型对应物。此外,诸位发明人证明这些T细胞克隆还识别内源表达的突变型抗原。这验证了加工和呈递各种蛋白质的能力。有趣的是,诸位发明人还可以分离识别在新佐剂疗法后切除的肿瘤组织中相同的新抗原的T细胞克隆。基于CDR3测序,诸位发明人可以证明四种T细胞克隆代表各个克隆。这证实了免疫应答的多克隆性质。此外,诸位发明人表明,在患者的两种不同HLA限制分子中呈递相同的新抗原,其中有三个克隆识别HLA-DPB1*0401中的新抗原且一个识别HLA-DPB1*0201中的新抗原。这些结果进一步强调了这种新抗原的免疫原性。
总之,数据证明患有Her2+乳腺癌的患者中的TIL的肿瘤特异性。此外,诸位发明人表明鉴定乳腺癌患者中各个癌症特异性T细胞靶标的可行性。这些结果可能有助于靶向患者特异性免疫疗法的未来发展。
本文的说明书和随后的整个权利要求中,“一个/一种(a/an)”以及“该(the)”的含义包括复数参照物,除非上下文清楚地另外指明。而且,如本文的说明书中所使用,“在……中(in)”的含义包括“在……中(in)”和“在……上(on)”,除非上下文另有明确说明。除非上下文指示相反,否则本文所列出的所有范围应被解释为包括其端点,并且开放式范围应被解释为包括商业实用值。类似地,除非上下文指出相反的情况,否则应将所有值的列表视为包含中间值。
此外,除非本文另外指示或另外与上下文明显矛盾,否则本文所述的所有方法均能够以任何合适的顺序进行。关于本文某些实施例提供的任何和所有实例或示例性语言(例如,“如”)的使用仅旨在更好地说明本发明,而不对原本要求保护的本发明范围构成限制。本说明书中的任何语言都不应当被解释为指示任何未要求保护的要素是实践本发明所必需的。
本文披露的本发明的可替代的要素或实施例的分组不应被解释为限制。每个组成员可以单独或以与组中其他成员或本文发现的其他要素的任何组合被提及和要求保护。出于方便和/或专利性的原因,组中的一个或多个成员可以包括在组中或从组中删除。当发生任何这种包括或删除时,在本文中认为本说明书含有经修改从而满足所附权利要求中使用的所有马库什群组(Markush group)的书面描述的组。
对于本领域技术人员应当清楚的是,在不脱离本文的发明构思的情况下,除了已经描述的那些之外的更多修改是可能的。因此,本发明主题仅受限于所附权利要求的范围。此外,在解释本说明书和权利要求时,所有术语都应当以与上下文一致的尽可能广泛的方式来解释。特别地,术语“包含/包括”(“comprises”和“comprising”)应当被解释为以非排他性方式提及要素、组分或步骤,从而指示所提及的要素、组分或步骤可以与未明确提及的其他要素、组分或步骤一起存在、或使用、或组合。在本说明书的权利要求提及选自由A、B、C……和N组成的组的某物中的至少一种的情况下,该文本应当被解释为仅需要该组中的一种要素,而不是A加N或B加N等。
- 一种活性化T淋巴细胞培养液及其活性化T淋巴细胞的制备方法
- 制备治疗性T淋巴细胞的方法