一种氘标记的依维莫司稳定同位素化合物及其制备方法
文献发布时间:2023-06-19 19:28:50
技术领域
本发明属于稳定同位素标准物的有机合成技术领域,涉及一种稳定同位素标记的依维莫司-D4(Everolimus-D4)及其制备方法,具体涉及一种采用稳定同位素标记原料氘代乙二醇-D4和非同位素的雷帕霉素为起始物合成依维莫司-D4(Everolimus-D4)的方法。
背景技术
依维莫司(Everolimus)是诺华(Novartis)公司研发的新一代的大环内醋类免疫抑制剂及抗肿瘤药物,其由雷帕霉素的42-OH衍生为42-O-(2-羟乙基)而成,故依维莫司又称42-O-(2-羟乙基)-雷帕霉素;其分子式为C
依维莫司临床上主要用来预防肾移植和心脏移植手术后的排斥反应;目前,也可用于治疗已使用过两种抑制血管内皮生长因子受体激酶抑制剂舒尼替尼和索拉非尼的晚期肾癌患者,毒副作用较轻微。相较于雷帕霉素,依维莫司的羟乙基结构增加了水溶性,提高了口服生物利用度,药理学性质明显改善,减少了不良反应,具有更高的治疗指数和人体耐受性。
稳定同位素示踪技术在临床药代动力学研究中应用十分广泛;采用气相色谱-质谱或液相色谱-质谱联机技术检测生物样本中标记药物和未标记药物,具有较高的测试专属性和灵敏度。
稳定同位素标记的依维莫司(如依维莫司-D4),对调查非氘代依维莫司在体内的代谢途径及评估其作用机制的研究不可缺少,是依维莫司的临床检测和药物研究的重要工具。
现有技术中,已有的稳定同位素标记的依维莫司以依维莫司-D4(Everolimus-D4)为主。目前该产品的生产工艺也很少见有公开发表;然而,国外产品的主要问题是产品中一般都含有高达约10%的杂质或产品中含有少量的非同位素的依维莫司,有些生产商都对其产品特意注明了这一点,如:加拿大Toronto Research Chemicals公司对其产品就注明如下:Everolimus-d4(~90%),美国Cayman Chemicals公司也对其产品就注明如下:≥99%deuterated forms(d1-d4),≤1%d0,这就很大范围地限制了该产品作为稳定同位素标准品的作用,对很多高标准、高精度的研究甚至是不合格的;此外,稳定同位素标记的依维莫司的具体生产工艺也很少见有公开报道。
鉴于此,特提出本发明。
发明内容
针对现有技术所存在的不足,本发明提供了一种氘标记的依维莫司稳定同位素化合物及其制备方法。
为实现上述目的,本发明的技术方案如下:
一种氘标记的依维莫司稳定同位素化合物的制备方法,包括以下步骤:
S1、在氮气保护下,将雷帕霉素的甲苯溶液加热至40-60℃并保温,再将含2-(叔丁基二甲基甲硅烷氧基)乙基三氟甲磺酸酯-d4和有机碱的甲苯溶液逐滴加入,搅拌并加热,保温反应16-32h后自然冷却至室温,随后对反应液进行后处理,得到叔丁基二甲基甲硅烷氧基-依维莫司-d4;
S2、在0℃冰水浴和氮气保护下,将盐酸溶液加入到叔丁基二甲基甲硅烷氧基-依维莫司-d4的甲醇溶液中,搅拌反应0.5-3h,随后对反应液进行再处理,得到氘标记的依维莫司稳定同位素化合物;
步骤S1中,所述有机碱为N,N-二异丙基乙基胺、三乙胺、吡啶和2,6-二甲基吡啶中的至少一种。
在上述技术方案中,步骤S1中,对应于790mg的雷帕霉素,所述2-(叔丁基二甲基甲硅烷氧基)乙基三氟甲磺酸酯-d4和有机碱的加入量分别为275-350mg和0.2-0.5ml。
进一步地,在上述技术方案中,步骤S1中,所述含2-(叔丁基二甲基甲硅烷氧基)乙基三氟甲磺酸酯-d4和有机碱的甲苯溶液的加入次数为2-6次,且每次加入的间隔时间为45-75min。
在上述技术方案中,步骤S2中,对应于600mg的叔丁基二甲基甲硅烷氧基-依维莫司-d4,所述盐酸溶液的摩尔浓度和加入体积分别为0.75-1.2mol/L和0.8-1.25ml。
再进一步地,在上述技术方案中,在步骤S1之前,还包括:
S0:采用氘代乙二醇-D4为原料,制得2-(叔丁基二甲基甲硅烷氧基)乙基三氟甲磺酸酯-d4。
详细地,在上述技术方案中,步骤S0具体包括:
S01:在0℃冰水浴和氮气保护下,依次将叔丁醇钾的四氢呋喃溶液和叔丁基二甲基氯硅烷的四氢呋喃溶液逐滴加入到氘代乙二醇-D4的四氢呋喃溶液中,随后移除冰水浴,将反应液搅拌2-4h并自然升温至室温,加水稀释后用乙醚萃取,取有机相并用饱和NaCl水溶液洗涤,减压去除有机溶剂后,再用硅胶柱层析分离并用体积比为2:1的正己烷-乙醚洗脱,得到2-(叔丁基二甲基甲硅烷基氧基)乙醇-d4;
S02:在0℃冰水浴和氮气保护下,依次将N,N-二异丙基乙基胺和三氟磺酸酐逐滴加入到2-(叔丁基二甲基甲硅烷基氧基)乙醇-d4的正己烷溶液中,搅拌1-3h,减压去除有机溶剂后,再用硅胶柱层析分离并用体积比为4:1的正己烷-乙醚洗脱,得到2-(叔丁基二甲基甲硅烷氧基)乙基三氟甲磺酸酯-d4。
具体地,在上述技术方案中,步骤S01中,对应于2.22g的氘代乙二醇-D4,所述叔丁醇钾和叔丁基二甲基氯硅烷的加入量分别为3.77g和4.56g。
具体地,在上述技术方案中,步骤S01中,对应于80ml的反应液,加入45-60ml的水稀释后,再加入乙醚萃取2-5次,每次加入45-60ml乙醚,并将有机相合并后用饱和NaCl水溶液洗涤。
具体地,在上述技术方案中,步骤S02中,对应于1.80g的2-(叔丁基二甲基甲硅烷基氧基)乙醇-d4,所述N,N-二异丙基乙基胺和三氟磺酸酐的加入量分别为1.42g和2.82g。
又进一步地,在上述技术方案中,步骤S1中,所述后处理的步骤,具体包括:
滴加10wt%的柠檬酸水溶液稀释后,再用乙酸乙酯萃取2-4次,将和并得到的有机相依次用饱和碳酸氢钠水溶液和饱和NaCl水溶液各洗涤一次,减压去除有机溶剂后,再用硅胶柱层析分离并用体积比为1:1的乙酸乙酯-正己烷洗脱。
又进一步地,在上述技术方案中,步骤S2中,所述再处理的步骤,具体包括:
加水稀释后,再用乙酸乙酯萃取2-4次,将和并得到的有机相依次用饱和碳酸氢钠水溶液和饱和NaCl水溶液各洗涤一次,减压去除有机溶剂后,将产物溶于乙酸乙酯后再逐滴加入正己烷直至有白色固体析出,随后将浑浊液置于0℃下静置12-20h,最后将析出物过滤后用正己烷洗涤2-5次。
本发明另一方面还提供了一种氘标记的依维莫司稳定同位素化合物,其是由上述制备方法制备得到,具有如下所示的分子结构:
本发明与现有技术相比,具有以下优点:
(1)本发明提供了稳定同位素标记的依维莫司及其合成方法,采用N,N-二异丙基乙基胺、三乙胺、吡啶和2,6-二甲基吡啶中的至少一种作为有机碱,与2-(叔丁基二甲基甲硅烷氧基)乙基三氟甲磺酸酯-d4同时逐滴加入到非同位素的雷帕霉素溶液中,在合适的反应温度和时间下,可确保制备得到的依维莫司-D4产物中不含其它难以分离的杂质及非同位素的依维莫司,从而避免了作为标准试剂在使用过程中像市场上含有杂质或非同位素的依维莫司的依维莫司标准试剂所产生的定量误差甚至是定性上的错误;
2)本发明所提供的氘标记的依维莫司稳定同位素化合物的制备方法反应过程简单,产品容易分离提纯,得到的产物的化学纯度和同位素丰度分别在99%和99%atom以上,满足作为定量检测依维莫司的标准试剂的要求,其使用价值高,具有良好的经济性。
附图说明
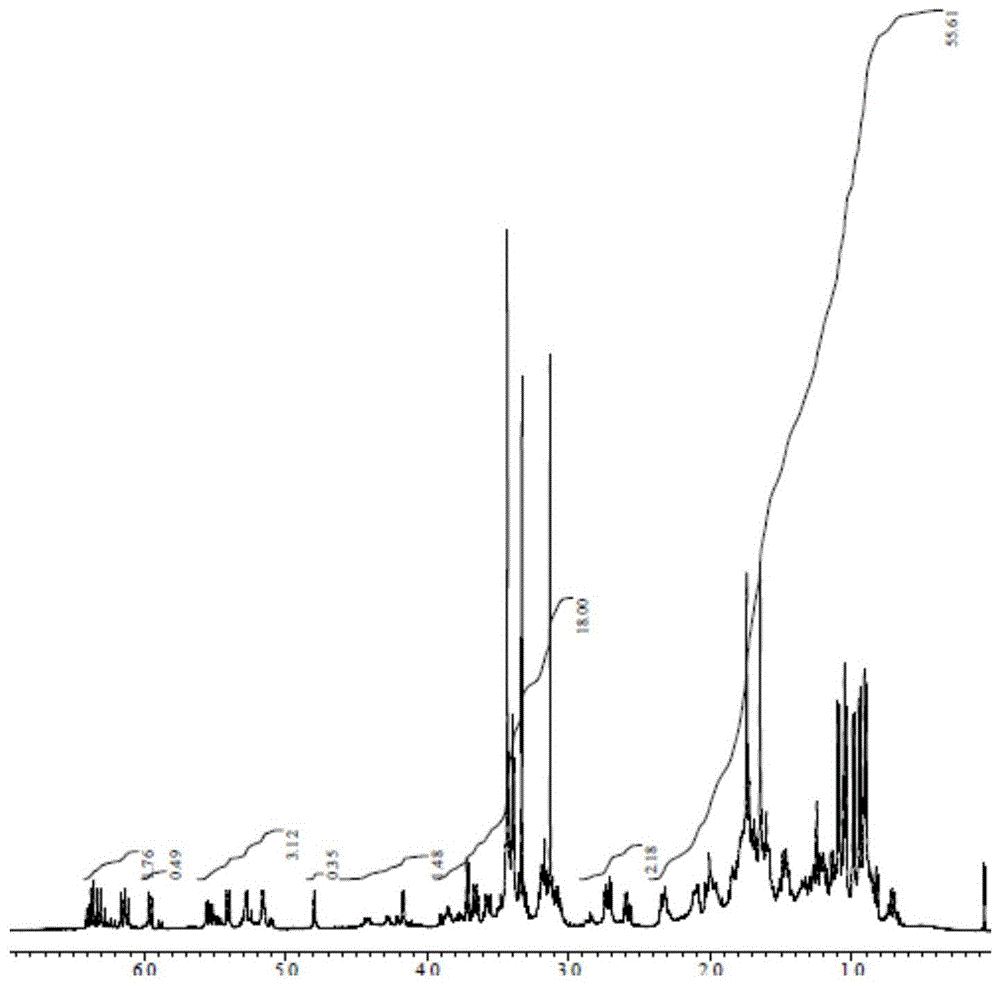
图1为本发明实施例1所制得的氘标记的依维莫司稳定同位素化合物(依维莫司-D4)的核磁共振氢谱图;
图2为本发明实施例1所制得的氘标记的依维莫司稳定同位素化合物(依维莫司-D4)的高效液相色谱图。
具体实施方式
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合实施例,对本发明进行进一步详细说明。
应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
实施例中,如无特别说明,所用手段均为本领域常规的手段。
本文中所用的术语“包含”、“包括”或其任何其它变形,意在覆盖非排它性的包括。例如,包含所列要素的组合物、步骤、方法、制品或装置不必仅限于那些要素,而是可以包括未明确列出的其它要素或此种组合物、步骤、方法、制品或装置所固有的要素。
实施例中未注明具体技术或条件者,按照本领域内的文献所描述的技术或条件,或者按照产品说明书进行。所用试剂或仪器未注明生产厂商者,均为可通过正规渠道商购买得到的常规产品。
在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视为在本文中具体公开。
此外,下面所描述的本发明各个实施方式中所涉及到的技术特征只要彼此之间未构成冲突就可以相互组合。
其合成路线如下:
实施例1
一种氘标记的依维莫司稳定同位素化合物(依维莫司-D4)的制备方法,其合成路线如下:
具体包括如下步骤:
S0、在0℃(冰水浴)和氮气保护下,依次向含有2.664g氘代乙二醇-D4的四氢呋喃溶液(60ml)中逐滴添加入含4.524g叔丁醇钾(tBuOK)的四氢呋喃溶液(18ml)和含5.472g叔丁基二甲基氯硅烷(TBDMS-Cl)的四氢呋喃溶液(18ml),然后移去冰水浴,搅拌3小时并自然升温至室温,加60ml水稀释后,用乙醚萃取三次(每次60mL),合并有机相并用饱和NaCl水溶液洗涤,减压去除有机溶剂后得到粗产物1(2.568g),将粗产物1经硅胶柱层析分离并用正己烷-乙醚(体积比2:1)进行洗脱,得到1.80g的2-(叔丁基二甲基甲硅烷基氧基)乙醇-d4(无色液体);在0℃(冰水浴)和氮气保护下,依次向含有1.80g的2-(叔丁基二甲基甲硅烷基氧基)乙醇-d4的正己烷溶液(40ml)中逐滴添加入1.42g N,N-二异丙基乙基胺和2.82g三氟磺酸酐(Trifluoromethanesulfonic anhydride),在0℃(冰水浴)下搅拌1.5h,减压去除有机溶剂后得到粗产物2(1.72g),再粗产物2经硅胶柱层析分离并用正己烷-乙醚(体积比4:1)进行洗脱,得到1.30g的2-(叔丁基二甲基甲硅烷氧基)乙基三氟甲磺酸酯-d4(无色液体);
S1、在氮气保护下,将含790mg雷帕霉素的甲苯溶液(10mL)搅拌升温至50℃,再将含300mg 2-(叔丁基二甲基甲硅烷氧基)乙基三氟甲磺酸酯-d4和0.45mL N,N-二异丙基乙基胺(DIPEA)的甲苯溶液(1mL)逐滴加入,在50℃下保温搅拌1h后,再次将含300mg 2-(叔丁基二甲基甲硅烷氧基)乙基三氟甲磺酸酯-d4和0.45mL N,N-二异丙基乙基胺(DIPEA)的甲苯溶液(1mL)逐滴加入,依此重复加入,总计加入1.2g 2-(叔丁基二甲基甲硅烷氧基)乙基三氟甲磺酸酯-d4和1.8mL N,N-二异丙基乙基胺(DIPEA),搅拌并加热,在氮气保护下50℃保温反应22h后自然冷却至室温(25℃),滴加25mL柠檬酸水溶液(10wt%)稀释后,再用乙酸乙酯萃取3次(每次25mL),将和并得到的有机相依次用饱和碳酸氢钠水溶液(25mL)和饱和NaCl水溶液(25mL)各洗涤一次,减压去除有机溶剂后得到粗产物3(842mg),再用硅胶柱层析分离并用体积比为1:1的乙酸乙酯-正己烷洗脱,得到600mg叔丁基二甲基甲硅烷氧基-依维莫司-d4(粘稠的灰白色油状物);
S2、在0℃冰水浴和氮气保护下,将1mL盐酸溶液(1mol/L)加入到含600mg叔丁基二甲基甲硅烷氧基-依维莫司-d4的甲醇溶液(5mL)中,搅拌反应1.5h(0℃冰水浴和氮气保护下),随后加入25mL水稀释后再用乙酸乙酯萃取3次(每次25mL),将和并得到的有机相依次用饱和碳酸氢钠水溶液(25mL)和饱和NaCl水溶液(25mL)各洗涤一次,减压去除有机溶剂后得到粗产物4(530mg),再将粗产物4(530mg)溶于乙酸乙酯(3mL)后再逐滴加入10mL正己烷直至有白色固体析出,随后将浑浊液置于0℃冰箱中静置16h,最后将析出物过滤后用正己烷洗涤3次(每次5mL),得到510mg氘标记的依维莫司稳定同位素化合物(依维莫司-D4,白色固体)。
实施例2
一种氘标记的依维莫司稳定同位素化合物(依维莫司-D4)的制备方法,其制备过程与实施例1类似,区别仅在于:
步骤S1中,往雷帕霉素的甲苯溶液中加入的是含300mg 2-(叔丁基二甲基甲硅烷氧基)乙基三氟甲磺酸酯-d4和0.30mL 2,6-二甲基吡啶的甲苯溶液(1mL),在50℃下保温搅拌1h后,重复加入,总计加入1.2g 2-(叔丁基二甲基甲硅烷氧基)乙基三氟甲磺酸酯-d4和1.2mL 2,6-二甲基吡啶,最后制得的粗产物3和叔丁基二甲基甲硅烷氧基-依维莫司-d4的质量分别为827mg和565mg;
步骤S2中,将1mL盐酸溶液(1mol/L)加入到含565mg叔丁基二甲基甲硅烷氧基-依维莫司-d4的甲醇溶液(5mL)中,最后制得的粗产物4和依维莫司稳定同位素化合物(依维莫司-D4)的质量分别为511mg和475mg。
实施例3
一种氘标记的依维莫司稳定同位素化合物(依维莫司-D4)的制备方法,其制备过程与实施例1类似,区别仅在于:
步骤S1中,往雷帕霉素的甲苯溶液中加入的是含300mg 2-(叔丁基二甲基甲硅烷氧基)乙基三氟甲磺酸酯-d4和0.21mL吡啶的甲苯溶液(1mL),在50℃下保温搅拌1h后,重复加入,总计加入1.2g2-(叔丁基二甲基甲硅烷氧基)乙基三氟甲磺酸酯-d4和0.84mL吡啶,最后制得的粗产物3和叔丁基二甲基甲硅烷氧基-依维莫司-d4的质量分别为815mg和481mg;
步骤S2中,将1mL盐酸溶液(1mol/L)加入到含481mg叔丁基二甲基甲硅烷氧基-依维莫司-d4的甲醇溶液(5mL)中,最后制得的粗产物4和依维莫司稳定同位素化合物(依维莫司-D4)的质量分别为433mg和411mg。
实施例4
一种氘标记的依维莫司稳定同位素化合物(依维莫司-D4)的制备方法,其制备过程与实施例1类似,区别仅在于:
步骤S1中,往雷帕霉素的甲苯溶液中加入的是含300mg 2-(叔丁基二甲基甲硅烷氧基)乙基三氟甲磺酸酯-d4和0.36mL三乙胺的甲苯溶液(1mL),在50℃下保温搅拌1h后,重复加入,总计加入1.2g 2-(叔丁基二甲基甲硅烷氧基)乙基三氟甲磺酸酯-d4和1.44mL三乙胺,最后制得的粗产物3和叔丁基二甲基甲硅烷氧基-依维莫司-d4的质量分别为796mg和343mg;
步骤S2中,将1mL盐酸溶液(1mol/L)加入到含343mg叔丁基二甲基甲硅烷氧基-依维莫司-d4的甲醇溶液(5mL)中,最后制得的粗产物4和依维莫司稳定同位素化合物(依维莫司-D4)的质量分别为309mg和292mg。
实施例5
一种氘标记的依维莫司稳定同位素化合物(依维莫司-D4)的制备方法,其制备过程与实施例1类似,区别仅在于:
步骤S1中,往雷帕霉素的甲苯溶液中加入的含300mg 2-(叔丁基二甲基甲硅烷氧基)乙基三氟甲磺酸酯-d4和0.45mL N,N-二异丙基乙基胺(DIPEA)的甲苯溶液(1mL)的次数不同,总计加入1.5g 2-(叔丁基二甲基甲硅烷氧基)乙基三氟甲磺酸酯-d4和2.25mL N,N-二异丙基乙基胺(DIPEA),最后制得的粗产物3和叔丁基二甲基甲硅烷氧基-依维莫司-d4的质量分别为824mg和589mg;
步骤S2中,将1mL盐酸溶液(1mol/L)加入到含589mg叔丁基二甲基甲硅烷氧基-依维莫司-d4的甲醇溶液(5mL)中,最后制得的粗产物4和依维莫司稳定同位素化合物(依维莫司-D4)的质量分别为530mg和503mg。
实施例6
一种氘标记的依维莫司稳定同位素化合物(依维莫司-D4)的制备方法,其制备过程与实施例1类似,区别仅在于:
步骤S1中,往雷帕霉素的甲苯溶液中加入的含300mg 2-(叔丁基二甲基甲硅烷氧基)乙基三氟甲磺酸酯-d4和0.45mL N,N-二异丙基乙基胺(DIPEA)的甲苯溶液(1mL)的次数不同,总计加入0.9g 2-(叔丁基二甲基甲硅烷氧基)乙基三氟甲磺酸酯-d4和1.35mL N,N-二异丙基乙基胺(DIPEA),最后制得的粗产物3和叔丁基二甲基甲硅烷氧基-依维莫司-d4的质量分别为817mg和515mg;
步骤S2中,将1mL盐酸溶液(1mol/L)加入到含515mg叔丁基二甲基甲硅烷氧基-依维莫司-d4的甲醇溶液(5mL)中,最后制得的粗产物4和依维莫司稳定同位素化合物(依维莫司-D4)的质量分别为463mg和439mg。
实施例7
一种氘标记的依维莫司稳定同位素化合物(依维莫司-D4)的制备方法,其制备过程与实施例1类似,区别仅在于:
步骤S1中,往雷帕霉素的甲苯溶液中加入的含300mg 2-(叔丁基二甲基甲硅烷氧基)乙基三氟甲磺酸酯-d4和0.45mL N,N-二异丙基乙基胺(DIPEA)的甲苯溶液(1mL)的次数不同,总计加入0.6g 2-(叔丁基二甲基甲硅烷氧基)乙基三氟甲磺酸酯-d4和0.9mL N,N-二异丙基乙基胺(DIPEA),最后制得的粗产物3和叔丁基二甲基甲硅烷氧基-依维莫司-d4的质量分别为809mg和433mg;
步骤S2中,将1mL盐酸溶液(1mol/L)加入到含433mg叔丁基二甲基甲硅烷氧基-依维莫司-d4的甲醇溶液(5mL)中,最后制得的粗产物4和依维莫司稳定同位素化合物(依维莫司-D4)的质量分别为390mg和369mg。
实施例8
一种氘标记的依维莫司稳定同位素化合物(依维莫司-D4)的制备方法,其制备过程与实施例1类似,区别仅在于:
步骤S1中,往雷帕霉素的甲苯溶液中加入的含300mg 2-(叔丁基二甲基甲硅烷氧基)乙基三氟甲磺酸酯-d4和0.45mL N,N-二异丙基乙基胺(DIPEA)的甲苯溶液(1mL)的次数为一次(没有重复加入),最后制得的粗产物3和叔丁基二甲基甲硅烷氧基-依维莫司-d4的质量分别为795mg和254mg;
步骤S2中,将1mL盐酸溶液(1mol/L)加入到含254mg叔丁基二甲基甲硅烷氧基-依维莫司-d4的甲醇溶液(5mL)中,最后制得的粗产物4和依维莫司稳定同位素化合物(依维莫司-D4)的质量分别为229mg和217mg。
图1和图2分别为本发明实施例1所制得的氘标记的依维莫司稳定同位素化合物(依维莫司-D4)的核磁共振氢谱图(以氘代氯仿CDCl
从图1中可以看出,化学位移3.82ppm到3.86ppm处未见乙二醇基的吸收峰,表明结构为依维莫司-D4(Everolimus-D4),不含有任何非同位素的依维莫司;从图2中可以看出,所制得的氘标记的依维莫司稳定同位素化合物(依维莫司-D4)样品的纯度达到99.38%。
分别计算实施例1-8的依维莫司-D4的产率,并分别通过高效液相色谱和质谱的方法检测所制备的氘标记的依维莫司稳定同位素化合物(依维莫司-D4)的化学纯度和同位素丰度,结果如下表1所示。
表1各实施例的产品的产率、化学纯度和同位素丰度的结果
从表1中可以看出,实施例2中的有机碱从N,N-二异丙基乙基胺改为2,6-二甲基吡啶,所有试剂比例不变,产品的产率降低至57%;实施例3中的有机碱从N,N-二异丙基乙基胺改为吡啶,所有试剂比例不变,产品的产率降低至50%;实施例4中的有机碱从N,N-二异丙基乙基胺改为三乙胺,所有试剂比例不变,产品的产率降低至35%;实施例5中的有机碱为N,N-二异丙基乙基胺,实施例1中四次投料改为五次,产率基本保持不变为61%;实施例6中的有机碱为N,N-二异丙基乙基胺,实施例1中四次投料改为三次,产品的产率降低至53%;实施例7中的有机碱为N,N-二异丙基乙基胺,实施例1中四次投料改为二次,产品的产率降低至45%;实施例8中的有机碱为N,N-二异丙基乙基胺,实施例1中四次投料改为一次,产品的产率降低至26%。所有实施例中最终产品的化学纯度和同位素丰度均达到合格要求。
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。
应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
- 一种氮-烷基(氘代烷基)芳杂环和烷基(氘代烷基)芳基醚类化合物的制备方法
- 一种氘代二苯氨基嘧啶类化合物的制备方法及其晶型
- 一种氘代二甲羟胺苯甲酸酯类化合物及其制备方法和应用
- 一种稳定同位素标记邻苯二甲酸二苯酯的合成方法
- 一种氘标记的雷帕霉素稳定同位素化合物及其制备方法
- 一种氘标记的去甲乌药碱稳定性同位素化合物的制备方法