一种高分子量聚酮合成的溶液聚合方法
文献发布时间:2024-04-18 19:58:30
技术领域
本发明属于高分子聚合物合成技术领域,具体涉及一种脂族聚酮合成的溶液聚合法工艺。
背景技术
高分子量聚酮(Polyketone,PK)是一种由烯烃类单体与一氧化碳交替共聚而成的聚合物。因其具有出色的冲击强度、耐化学性和气体阻隔性等特点,在薄膜、纤维、汽车零部件、电子和医疗器具等领域有广泛应用前景。
迄今,高分子量聚酮的工业合成均采用高活性的后过渡金属催化体系,一般由含双齿膦配体的钯系催化剂前体、一种强酸及一种氧化剂组成,并用甲醇作聚合反应的介质,与甲醇构成活性中心(KP 101675275;KP 20100019805;Chemical Review,1996,96:663-682),活性接近25kg PK·g
与沉淀聚合、淤浆聚合等非均相聚合方法不同,溶液聚合法将聚合反应与聚合物的析出分置于两个不同的过程。虽固态聚合物的获取及溶剂的回收能耗较大,但因聚合反应在均相溶液中进行,不仅反应器的传热能力强,而且不会产生聚合物的粘釜问题,并可较方便地从聚合产物中除去催化剂等杂质。因此,在一些聚合物的大规模生产中有较广的应用。
溶剂选择是实施溶液聚合的关键。除了聚合产物有良好溶解性外,溶剂对聚合反应的不良影响还应尽可能小,即尽可能保持高的聚合反应活性和聚合产物的分子量。这对聚酮的合成来说,是极具挑战性的。首先,聚酮的良溶剂极少,迄今仅发现部分氟醇、苯酚能溶解聚酮。其次,聚酮合成的具商业价值的高活性催化剂仅甲醇活化的钯系催化剂,而甲醇则是聚酮的沉淀剂。此外,商品化的聚酮分子量通常要求在100kDa以上,分子量越高,确保聚合体系为溶液态的聚合物临界浓度就越低。因此,迄今尚无高聚合活性下,合成高分子量聚酮的溶液聚合法工艺报道。
发明内容
有鉴于此,本发明的目的在于解决现有非均相聚合技术中聚合物粘釜及低堆密度等问题,并提供一种高聚合活性下合成高分子量聚酮的均相溶液聚合方法。
本发明具体采用的技术方案如下:
一种高分子量聚酮的溶液聚合制备方法,其具体做法为:在钯系催化体系的作用下,将烯烃类单体和一氧化碳通入有机混合溶剂中进行均相溶液聚合反应,从而制备出重均相对分子质量在50~600kDa之间的聚酮产物;
所述有机混合溶剂由第一有机溶剂和第二有机溶剂组成,第一有机溶剂在有机混合溶剂中的体积分数为65~95vol%,余量为第二有机溶剂;且第一有机溶剂为聚酮的良溶剂,第二有机溶剂为聚酮的不良溶剂;
所述钯系催化体系由钯系催化剂前体组成,或者由强酸和氧化剂中的至少一种和钯系催化剂前体组成。
基于上述技术方案,本发明还可以进一步提供以下优选方式。需说明的是,各优选方式在没有冲突的情况下,可以进行相互组合,不构成限制。
作为优选,所述的第一有机溶剂为含氟或苯酚类溶剂,第二有机溶剂为醇类溶剂。
进一步优选的,所述的第一有机溶剂为六氟异丙醇、间甲酚中的一种或它们的混合物。
进一步优选的,所述的第二有机溶剂为甲醇、乙醇中的一种或它们的混合物。
作为优选,所述的钯系催化剂前体为双齿膦配体和钯金属中心组成的有机金属化合物。
进一步优选的,所述的双齿膦配体选自1,3-双[二(2-甲氧基苯基)膦基]丙烷、1,3-双[二(2-乙氧基苯基)膦基]丙烷、2,2-二甲基-1,3-双(2-甲氧基苯基)膦基]丙烷、1,3-双[双(2-甲氧基-5-甲基苯基)膦基]丙烷、3,3-双[双-(2-甲氧基苯基)膦甲基]-1,5-二氧杂螺[5,5]十一烷中的一种。
进一步优选的,所述的钯金属中心选自三氟乙酸钯、乙酸钯、氯化钯中的一种或多种。
作为优选,所述的强酸为pK
进一步优选的,所述的强酸为对甲苯磺酸、三氟甲磺酸中的一种或它们的混合物。
作为优选,所述的氧化剂为对苯醌、2,3-二氯-5,6-二氰基苯醌中的一种或它们的混合物。
作为优选,所述的烯烃类单体为乙烯以及α-烯烃、苯乙烯和环状烯烃中的一种或多种。
进一步优选的,所述的α-烯烃包括丙烯、1-丁烯、1-己烯、1-辛烯中的一种或多种。
进一步优选的,所述的环状烯烃包括降冰片烯、环戊烯、环己烯中的一种或多种。
作为优选,所述的均相溶液聚合反应过程中,乙烯、其他烯烃类单体与一氧化碳的进料摩尔比为(0.25~4):(0~2):1。
作为优选,所述的钯系催化体系中,钯系催化剂前体、强酸和氧化剂的摩尔比值为1:(0~35):(0~80)。
作为优选,所述的均相溶液聚合反应温度为50~120℃,聚合压力为4~10Mpa,且整个聚合反应过程中聚合体系始终保持均相溶液状态。
作为优选,所述的均相溶液聚合反应过程中,催化剂前体的浓度为0.1~200μmol/L,强酸浓度为0~500μmol/L,氧化剂浓度为0~800μmol/L。
作为优选,均相溶液聚合反应的聚合活性可达35kgPK·g
作为优选,所述的聚酮产物在溶液聚合中所占的固含量为5~50wt%。
相对于现有技术而言,本发明的有益效果如下:
本发明采用对聚酮有良好溶解性的混合溶剂和钯系催化剂体系,能够进行聚酮的高聚合活性溶液聚合合成,所得聚合物具有高的分子量。与现有的以单一甲醇为介质的淤浆聚合法相比,本发明提供的溶液聚合方法避免了聚合物粘釜及低堆密度问题;同时可通过聚合结束后均相溶液中固态吸附剂的投入和过滤,实现催化剂体系中金属钯的回收和聚合物的除杂,进而进行聚合物溶液的脱挥、挤出造粒和溶剂回收,并根据需要在脱挥前的溶液或脱挥后的熔体中均匀地混入各种加工应用助剂,实现聚酮产品的全流程工业化。
附图说明
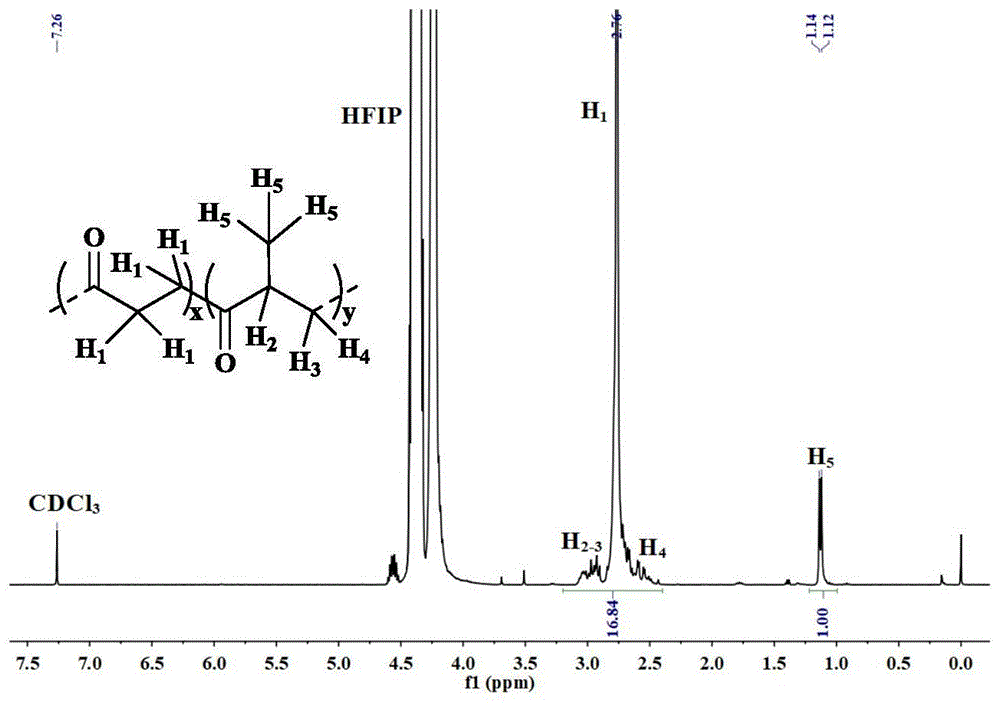
图1为实施例6中溶液聚合法制得的典型聚酮产物的常温核磁氢谱(
图2为实施例6中溶液聚合法制得的典型聚酮产物的常温核磁碳谱(
图3为实施例6中溶液聚合法制得的典型聚酮产物的GPC谱图。
具体实施方式
以下结合本发明的实施例,对本发明的技术方案进行描述。但所描述的实施例仅仅是本发明实施方法的一部分,实施范围不局限于这些实施例。
本发明提供了一种高分子量聚酮的溶液聚合制备方法,该方法是通过以下步骤实现的:
在搅拌釜式反应器中加入一定量的对聚酮有良好溶解性的混合有机溶剂,加入一定量的钯系催化剂前体、强酸和氧化剂,通入乙烯、其他烯烃类单体和一氧化碳,50~120℃、4~10MPa压力下进行高活性的均相溶液聚合反应。
上述制备方法中,整个反应体系中,催化剂前体的浓度(以反应釜内混合溶剂总体积计),为0.1~200μmol/L,强酸浓度为0~500μmol/L,氧化剂浓度为0~800μmol/L;所制得的聚酮的重均分子量在50~600kDa间、分子量分布指数在1.0~5.0间;整个聚合反应过程中,聚合体系始终保持均相溶液状态。均相溶液聚合反应的聚合活性可达35kgPK·g
上述制备方法中,乙烯、其他烯烃类单体与一氧化碳的进料摩尔比可以控制为(0.25~4):(0~2):1。
上述制备方法中,其他烯烃类单体可以为α-烯烃、苯乙烯和环状烯烃中的一种或多种。其中,α-烯烃可以是丙烯、1-丁烯、1-己烯、1-辛烯中的一种或多种。环状烯烃可以是降冰片烯、环戊烯、环己烯中的一种或多种。
上述制备方法中,聚合压力优选为4~6MPa,聚合温度优选为60~110℃。
上述制备方法中,钯系催化体系由钯系催化剂前体、强酸和氧化剂组成,但钯系催化剂前体是必要组分,强酸和氧化剂为可选组分,钯系催化剂前体、强酸和氧化剂的摩尔比值为1:(0~35):(0~80)。反应所用的钯系催化剂前体为双齿膦配体和钯金属中心组成的有机金属化合物,其中双齿膦配体选自1,3-双[二(2-甲氧基苯基)膦基]丙烷、1,3-双[二(2-乙氧基苯基)膦基]丙烷、2,2-二甲基-1,3-双(2-甲氧基苯基)膦基]丙烷、1,3-双[双(2-甲氧基-5-甲基苯基)膦基]丙烷、3,3-双[双-(2-甲氧基苯基)膦甲基]-1,5-二氧杂螺[5,5]十一烷;所述钯金属中心化合物选自三氟乙酸钯、乙酸钯、氯化钯中的一种或多种。另外,强酸(如有)为pK
上述制备方法中,聚合反应的有机混合溶剂由第一有机溶剂和第二有机溶剂构成,第一有机溶剂为含氟或苯酚类的聚酮良溶剂,第二有机溶剂为醇类的聚酮不良溶剂。其中第一有机溶剂可以选自六氟异丙醇(HFIP)、间甲酚中的一种或它们的混合物,第二有机溶剂可以选自甲醇、乙醇中的一种或它们的混合物。另外,该混合溶剂中,第一有机溶剂在混合溶剂中的体积分数为65~95vol%。
本发明中使用到的物料摩尔浓度均是以有机溶剂的总体积计,涉及到的当量比均相对于金属钯的摩尔量,烯烃类单体和一氧化碳的进料摩尔比是指单体进入反应釜时的摩尔浓度比。
实施例1
在单体混合储罐中按1:1的mol比进行乙烯/一氧化碳混合气的配置,并与250mL含有减压阀和内冷管的不锈钢高压釜相连接。加热高压釜被至120℃,交替进行三次抽真空和高纯氮置换,除去水和氧。将50mL六氟异丙醇(HFIP)、30mL甲醇和1mL浓度为30mmol/L的对甲苯磺酸HFIP溶液加入高压釜中;另将5μmol的钯系催化剂前体溶解于15mL HFIP中,并注射进高压釜中;剩余的HFIP也一并加入,使反应釜中HFIP的总加入量为100mL。搅拌(N=400rpm)并将高压釜内温度调至90℃。再将8g丙烯输送进高压釜中,并开启单体混合储罐与高压釜的连接阀输入单体混合气使釜内压力至5MPa;开始计时,并保持通气维持5MPa的恒定反应压力,在钯系催化体系的作用下,高压釜内的乙烯单体、少量丙烯和一氧化碳在有机混合溶剂中进行均相溶液聚合反应。聚合反应30分钟后结束,高压釜被冷却到25℃后,缓慢释放压力,开釜后看到生成的聚合物被完全溶解,表明为均相溶液聚合,不存在因聚酮产物析出而产生的粘釜、聚合物堆密度低等问题。将聚合后的反应溶液慢慢滴入进500mL的甲醇中,析出聚酮产物,然后用甲醇对产物洗涤三次,过滤后在80℃、真空下干燥6小时至恒重,称重得到产物9.1g,催化活性为34.2kgPK·g
本实施例中,上述实验所用的钯系催化体系由钯系催化剂前体和强酸组成,其中钯系催化剂前体为双齿膦配体和钯金属中心组成的有机金属化合物,催化剂前体的配体部分为3,3-双[双-(2-甲氧基苯基)膦甲基]-1,5-二氧杂螺[5,5]十一烷、钯金属中心为乙酸钯,强酸为对甲苯磺酸,无氧化剂;通入有机混合溶剂中的单体为乙烯、一氧化碳和少量丙烯;聚合溶剂(即混合有机溶剂)为HFIP和甲醇混合溶液。
实施例2
本实施例中,实验所用的钯系催化体系由钯系催化剂前体和氧化剂组成,其中钯系催化剂前体为双齿膦配体和钯金属中心组成的有机金属化合物,催化剂前体的配体部分为3,3-双[双-(2-甲氧基苯基)膦甲基]-1,5-二氧杂螺[5,5]十一烷、钯金属中心为乙酸钯,无强酸,加入氧化剂对苯醌,通入有机混合溶剂中的单体为乙烯、一氧化碳和少量丙烯;设备情况同实施例1。
具体操作为:反应开始前,催化剂加入阶段同时加入16.3g对苯醌,在进单体混合气阶段,将12g丙烯输送进高压釜中。聚合压力为5.5MPa,聚合温度为90℃,其它工艺参数、实验条件和操作步骤同实施例1,得到产物6.7g,催化活性为25.2kgPK·g
实施例3
本实施例中,实验所用的钯系催化体系由钯系催化剂前体和氧化剂组成,其中钯系催化剂前体为双齿膦配体和钯金属中心组成的有机金属化合物,催化剂前体的配体部分为3,3-双[双-(2-甲氧基苯基)膦甲基]-1,5-二氧杂螺[5,5]十一烷、钯金属中心为乙酸钯,无强酸,加入氧化剂对苯醌;通入有机混合溶剂中的单体为乙烯、一氧化碳和少量丙烯;设备情况同实施例1。
具体操作为:反应开始前,催化剂加入阶段同时加入对苯醌8.2g;在进单体混合气阶段,将8g丙烯输送进高压釜中;聚合压力为5MPa,聚合温度为80℃,其它工艺参数、实验条件和操作步骤同实施例1。得到产物5.9g,催化活性为22.2kgPK·g
实施例4
本实施例中,实验所用的钯系催化体系由钯系催化剂前体和强酸组成,其中钯系催化剂前体为双齿膦配体和钯金属中心组成的有机金属化合物,催化剂前体的配体部分为3,3-双[双-(2-甲氧基苯基)膦甲基]-1,5-二氧杂螺[5,5]十一烷、钯金属中心为乙酸钯,强酸为对甲苯磺酸,无氧化剂,通入有机混合溶剂中的单体为乙烯、一氧化碳和少量丙烯;设备情况同实施例1。
具体操作为:反应开始前,催化剂加入阶段,加入3mL浓度为30mmol/L的对甲苯磺酸HFIP溶液;在进单体混合气阶段,将4g丙烯输送进高压釜中,聚合压力为5.5MPa,聚合温度为90℃。其它工艺参数、实验条件和操作步骤同实施例1,开釜后看到生成的聚合物被完全溶解,表明为均相溶液聚合,不存在因聚酮产物析出而产生的粘釜、聚合物堆密度低等问题,最终得到产物4.1g,催化活性为15.4kgPK·g
实施例5
本实施例中,实验所用的钯系催化体系由钯系催化剂前体和强酸组成,其中钯系催化剂前体为双齿膦配体和钯金属中心组成的有机金属化合物,催化剂前体的配体部分为3,3-双[双-(2-甲氧基苯基)膦甲基]-1,5-二氧杂螺[5,5]十一烷、钯金属中心为乙酸钯,强酸为对甲苯磺酸,无氧化剂,通入有机混合溶剂中的单体为乙烯、一氧化碳和少量丙烯;设备情况同实施例1。
具体操作为:反应开始前,催化剂加入阶段,加入3mL浓度为30mmol/L的对甲苯磺酸HFIP溶液;釜内的聚合介质控制为HFIP 80mL、甲醇20mL,在进单体混合气阶段,将8g丙烯输送进高压釜中。聚合压力为5MPa,聚合温度为90℃,其它工艺参数、实验条件和操作步骤同实施例1,开釜后看到生成的聚合物被完全溶解,表明为均相溶液聚合,不存在因聚酮产物析出而产生的粘釜、聚合物堆密度低等问题,最终得到产物3.2g,催化活性为12.0kgPK·g
实施例6
本实施例中,实验所用的钯系催化体系单独由钯系催化剂前体组成,其中钯系催化剂前体为双齿膦配体和钯金属中心组成的有机金属化合物,催化剂前体的配体部分为3,3-双[双-(2-甲氧基苯基)膦甲基]-1,5-二氧杂螺[5,5]十一烷、钯金属中心为乙酸钯,无强酸,无氧化剂,通入有机混合溶剂中的单体为乙烯、一氧化碳和少量丙烯;设备情况同实施例1。
具体操作为:反应开始前,在进单体混合气阶段,将8g丙烯输送进高压釜中,聚合压力为5.5MPa,聚合温度为90℃,其它工艺参数、实验条件和操作步骤同实施例1,开釜后看到生成的聚合物被完全溶解,表明为均相溶液聚合,不存在因聚酮产物析出而产生的粘釜、聚合物堆密度低等问题,最终得到产物9.6g,催化活性为36.1kgPK·g
以本实施例为例,制得的聚酮产物的常温核磁氢谱(
实施例7
本实施例中,实验所用的钯系催化体系由钯系催化剂前体和氧化剂组成,其中钯系催化剂前体为双齿膦配体和钯金属中心组成的有机金属化合物,催化剂前体的配体部分为3,3-双[双-(2-甲氧基苯基)膦甲基]-1,5-二氧杂螺[5,5]十一烷、钯金属中心为乙酸钯,无强酸,加入氧化剂对苯醌;通入有机混合溶剂中的单体为乙烯、一氧化碳和少量丙烯;设备情况同实施例1。
具体操作为:反应开始前,催化剂加入阶段,加入16.3g对苯醌,在进单体混合气阶段,将8g丙烯输送进高压釜中,单体的配气比设为4/1(乙烯/CO=4/1,mol/mol)。聚合压力为5.5MPa,聚合温度为90℃,其它工艺参数、实验条件和操作步骤同实施例1,开釜后看到生成的聚合物被完全溶解,表明为均相溶液聚合,不存在因聚酮产物析出而产生的粘釜、聚合物堆密度低等问题,最终得到产物8.2g,催化活性为30.8kgPK·g
实施例8
本实施例中,实验所用的钯系催化体系由钯系催化剂前体和氧化剂组成,其中钯系催化剂前体为双齿膦配体和钯金属中心组成的有机金属化合物,催化剂前体的配体部分为3,3-双[双-(2-甲氧基苯基)膦甲基]-1,5-二氧杂螺[5,5]十一烷、钯金属中心为乙酸钯,无强酸,加入氧化剂对苯醌;通入有机混合溶剂中的单体为乙烯、一氧化碳和少量丙烯;设备情况同实施例1。
具体操作为:反应开始前,催化剂加入阶段,加入8.2g对苯醌,在进单体混合气阶段,将4g丙烯输送进高压釜中。聚合压力为5.5MPa,聚合温度为90℃,聚合时间为4小时,其它工艺参数、实验条件和操作步骤同实施例1,开釜后看到生成的聚合物被完全溶解,表明为均相溶液聚合,不存在因聚酮产物析出而产生的粘釜、聚合物堆密度低等问题,最终得到产物48.6g,催化活性为22.8kgPK·g
实施例9
本实施例中,实验所用的钯系催化体系由钯系催化剂前体和氧化剂组成,其中钯系催化剂前体为双齿膦配体和钯金属中心组成的有机金属化合物,催化剂前体的配体部分为3,3-双[双-(2-甲氧基苯基)膦甲基]-1,5-二氧杂螺[5,5]十一烷、钯金属中心为mol比1:1的三氟乙酸钯和乙酸钯的混合物,强酸为mol比1:1的对甲苯磺酸和三氟甲磺酸的混合物,无氧化剂;混合溶剂中的良溶剂为体积比1:2的六氟异丙醇和间甲酚混合物,不良溶剂为体积比2:1的甲醇和乙醇的混合物。通入混合溶剂中的单体为乙烯、一氧化碳和少量丙烯;设备情况同实施例1。
具体操作为:反应开始前,在进单体混合气阶段,将8g丙烯输送进高压釜中,聚合压力为5.5MPa,聚合温度为90℃,其它工艺参数、实验条件和操作步骤同实施例1,开釜后看到生成的聚合物被完全溶解,表明为均相溶液聚合,不存在因聚酮产物析出而产生的粘釜、聚合物堆密度低等问题,最终得到产物7.4g,催化活性为27.8kgPK·g
比较例
本实施例中,实验所用的聚合介质为100mL纯甲醇,不加入HFIP;钯系催化体系由钯系催化剂前体、强酸和氧化剂组成,其中钯系催化剂前体为双齿膦配体和钯金属中心组成的有机金属化合物,催化剂前体的配体部分为3,3-双[双-(2-甲氧基苯基)膦甲基]-1,5-二氧杂螺[5,5]十一烷,钯金属中心为乙酸钯,强酸为对甲苯磺酸,加入氧化剂对苯醌;通入有机混合溶剂中的单体为乙烯、一氧化碳和少量丙烯;设备情况同实施例1。
具体操作为:反应开始前,催化剂加入阶段,将80mL甲醇和5mL浓度为30mmol/L的对甲苯磺酸甲醇溶液及8.2g对苯醌加入高压釜中;另将5μmol的催化剂前体溶于甲醇中,并注射进高压釜中,控制釜内甲醇总加入量为100mL;在进单体混合气阶段,将8g丙烯输送进高压釜中。聚合压力为5.5MPa,聚合温度为90℃,聚合时间为0.5小时,其它实验条件和操作步骤同实施例1。开釜后发现产物明显结块,且附着在釜内搅拌桨及内壁上,较难从反应釜中排出,粘釜现象严重,得到产物5.1g,催化活性为19.2kgPK·g
以上所述的实施例只是本发明的一种较佳的方案,然其并非用以限制本发明。有关技术领域的普通技术人员,在不脱离本发明的精神和范围的情况下,还可以做出各种变化和变型。因此凡采取等同替换或等效变换的方式所获得的技术方案,均落在本发明的保护范围内。
- 一种低不饱和度高分子量高活性丁醇聚醚的合成方法
- 一种低分子量尼龙9T粉合成高分子量尼龙9T树脂的生产方法
- 一种高分子量脂肪族聚酮及其合成方法
- 一种高断裂伸长率的聚酮及其合成方法