改性氧化铝及其制备方法和应用
文献发布时间:2024-04-18 20:00:50
技术领域
本发明涉及催化剂技术领域,具体涉及一种改性氧化铝及其制备方法和应用、一种含有该改性氧化铝的负载型催化剂及其应用。
背景技术
催化剂载体是合成负载型催化剂的重要基石,其结构和理化性质对负载型催化剂的性质和应用范围存在很大影响。氧化铝是一种重要且常见的工业用催化剂载体,其反应活性、水热稳定性、耐磨性等性质是评价催化剂载体的重要指标。对氧化铝表面做修饰改性可增强氧化铝的某些性能,这对氧化铝应用范围的延伸扩展具有重要意义。
CN101448565A公开了一种具有高水热稳定性的氧化铝,通过用可溶硅无机化合物处理用作吸附剂和催化剂载体的过渡型氧化铝,改进了所述过渡型氧化铝的水热稳定性。
CN107774248A公开了一种硅改性的费托合成催化剂及其应用,该硅改性法合成的氧化铝催化剂载体在费托反应中具有优异的稳定性和耐磨性。
CN106582597A公开了一种硅改性氧化铝及其制备方法和应用,通过调节铝溶胶与硅溶胶混合体系的pH值,可实现在侧重改善孔容和侧重改善B酸含量之间进行灵活转换,材料表面B/L为40-95%,孔容为0.8-1.2cm
CN113562751A公开了一种改性拟薄水铝石及其制备方法,在制备改性拟薄水铝石时通过添加含磷化合物、含非金属助剂化合物、晶粒生长调节剂以及制备过程pH的分段控制,使得改性拟薄水铝石在煅烧后有特定的表面羟基分布,进而使催化剂具有优异的重油加氢活性和高稳定性。
上述现有技术均未涉及如何对氧化铝表面的复杂酸性位点进行调;通常不同种类、不同强度的酸性位点使氧化铝在精细化工领域的应用受到较大限制。因此,亟需一种具有低酸性的改性氧化铝。
发明内容
本发明的目的是为了克服现有改性氧化铝存在复杂的酸性位点,无法兼具耐磨性、高稳定性和酸量可调控等,以及用于精细化工领域存在活性低、寿命短等问题,提供一种改性氧化铝及其制备方法和应用,一种含有该改性氧化铝的负载型催化剂及其应用,该改性氧化铝具有低B酸密度,还兼具低磨损指数、高压碎强度、高比表面积和低平均孔径;同时,将该改性氧化铝用于催化剂载体,具有较高的活性。
为了实现上述目的,本发明第一方面提供一种改性氧化铝,所述改性氧化铝包括硅和氧化铝,其中,所述硅通过Si-O-Al化学键连接在所述氧化铝的表面,在所述氧化铝的表面的相邻硅通过Si-O-Si化学键连接;所述改性氧化铝的硅铝比<1;
其中,所述改性氧化铝的B酸密度≤2μmol/g。
优选地,所述改性氧化铝的B酸密度为0-2μmol/g,优选为0-1.4μmol/g,更优选为0-0.5μmol/g。
优选地,基于所述改性氧化铝的总重量,所述氧化铝的含量为50-90wt%;以SiO
优选地,所述氧化铝为γ-氧化铝。
本发明第二方面提供一种改性氧化铝的制备方法,该方法包括以下步骤:
(1)将铝源、酸性化合物和水进行第一混合,得到第一混合物;
(2)将所述第一混合物依次进行成型、第一干燥和第一焙烧,得到成型氧化铝;
(3)将所述成型氧化铝溶于水中,先加入碱性化合物调节pH为7-12,再加入硅源进行第二混合,得到第二混合物;
(4)将所述第二混合物进行固液分离,得到的改性氧化铝前体依次进行第二干燥和第二焙烧,得到改性氧化铝。
本发明第三方面提供一种第一方面提供的改性氧化铝,或者,第二方面提供的方法制得的改性氧化铝在催化剂载体、气液吸附、固相填充剂中的应用。
本发明第四方面提供一种负载型催化剂,所述负载型催化剂包括载体和负载在所述载体上的活性组分;
其中,所述载体选自第一方面提供的改性氧化铝,或者,第二方面提供的方法制得的改性氧化铝。
本发明第五方面提供一种第四方面提供的负载型催化剂在催化合成赖氨酸型抗菌单体、芳香胺的氨基烷基化反应、制备酰胺类化合物的衍生物中的应用。
相比现有技术,本发明具有以下优势:
(1)本发明提供的改性氧化铝,通过限定改性氧化铝中硅通过Si-O-Al化学键连接在氧化铝的表面和在氧化铝的表面的相邻硅通过Si-O-Si化学键连接,并结合硅铝比<1,实现对氧化铝的表面复杂酸性位点进行调控,在保证改性氧化铝兼具低磨损指数、高压碎强度、高比表面积和低平均孔径的前提下,有效降低了改性氧化铝的B酸密度,尤其通过调控改性氧化铝中硅和氧化铝的含量,更有利于调控改性氧化铝的B酸密度;
(2)本发明提供的改性氧化铝的制备方法,简化工艺流程,便于工业化生产;尤其通过调控所述第二混合中硅源投入量和混合体系的pH范围,实现对改性氧化铝的B酸密度的调控;
(3)本发明提供的改性氧化铝具有低酸浓度,能广泛应用于精细化工,尤其是应用于催化剂载体、气液吸附或固相填充剂;
(4)将本发明提供的改性氧化铝用于催化剂载体,制得的负载型催化剂具有较高的催化活性,尤其是用于催化合成赖氨酸型抗菌单体、芳香胺的氨基烷基化反应、制备酰胺类化合物的衍生物中,具有较高的转化率和选择性。
附图说明
图1(a)是实施例1制得的改性氧化铝S1的透射红外谱图;图1(b)对比例1制得的改性氧化铝DS1的透射红外谱图,其中,波数为1066cm
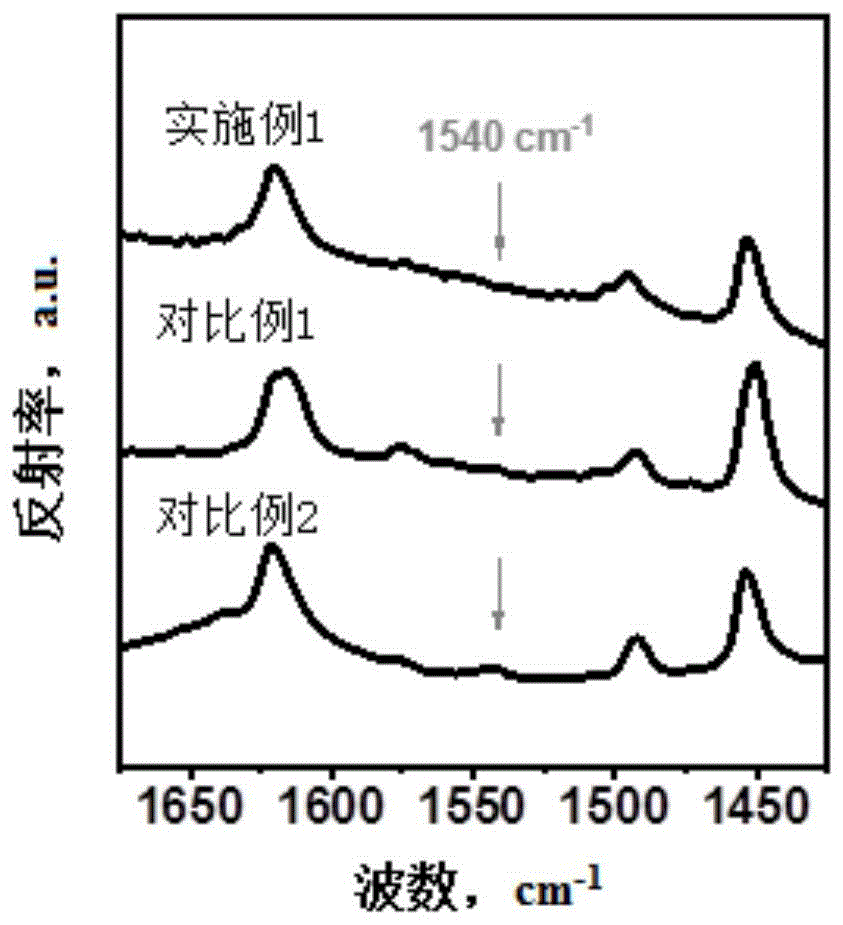
图2是实施例1和对比例1-2制得的改性氧化铝的吡啶红外表征谱图,其中,波数为1540cm
图3是实施例1制得的改性氧化铝的SEM图;
图4是实施例1和对比例2制得的改性氧化铝的XRD图。
具体实施方式
在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视为在本文中具体公开。
在本发明中,没有特殊情况说明下,“第一”和“第二”既不表示先后次序,也不表示对各个物料或步骤起限定作用,仅用于区分这不是同一物料或步骤。例如,“第一混合”和“第二混合”中“第一”和“第二”仅用于表示这不是同一混合。
本发明第一方面提供一种改性氧化铝,所述改性氧化铝包括硅和氧化铝,其中,所述硅通过Si-O-Al化学键连接在所述氧化铝的表面,在所述氧化铝的表面的相邻硅通过Si-O-Si化学键连接;所述改性氧化铝的硅铝比<1;其中,所述改性氧化铝的B酸密度≤2μmol/g。
本发明的发明人研究发现:氧化铝表面具有复杂的酸性位点,不同种类、强度的酸性位点使氧化铝在精细化工领域的应用受到较大限制。因此,在氧化铝的表面负载硅,并限定硅通过Si-O-Al化学键连接在氧化铝的表面,以及限定在所述氧化铝的表面的相邻硅通过Si-O-Si化学键连接,在保证改性氧化铝兼具低磨损指数、高压碎强度、高比表面积和低平均孔径的前提下,能够有效掩蔽氧化铝表面的酸性位点,进而调控改性氧化铝表面的B酸密度,使得改性氧化铝的B酸密度≤2μmol/g。
在本发明中,没有特殊情况说明下,所述硅通过Si-O-Al化学键连接在氧化铝的表面是指Si和氧化铝中Al共用部分O,从而使得以SiO
在本发明中,没有特殊情况说明下,B酸密度是指
在本发明中,B酸密度参数依据升温脱附的吡啶量来计算;B酸密度=测试吡啶红外光谱的改性氧化铝的酸量(单位为μmol)/改性氧化铝的质量(单位为g)。
在本发明的一些实施方式中,优选地,所述改性氧化铝的B酸密度为0-2μmol/g,例如,0μmol/g、0.1μmol/g、0.2μmol/g、0.3μmol/g、0.5μmol/g、0.8μmol/g、1μmol/g、1.4μmol/g、1.5μmol/g、2μmol/g,以及任意两个数值组成的范围中的任一值,优选为0-1.4μmol/g,更优选为0-0.5μmol/g。
在本发明的一些实施方式中,优选地,所述改性氧化铝的比表面积为100-220m
在本发明中,没有特殊情况说明下,比表面积参数采用全自动等温吸附仪测得;平均孔径参数采用全自动等温吸附仪,并配合BJH模型计算获得;磨损指数参数采用磨损指数分析仪测得;压碎强度参数采用颗粒强度测试仪测得。
在本发明中,没有特殊情况说明下,所述改性氧化铝除了硅和氧化铝外,不含有其他组分,即,所述硅和氧化铝的含量之和为100wt%。
在本发明的一些实施方式中,基于所述改性氧化铝的总重量,所述氧化铝的含量为50-90wt%,优选为70-80wt%;以SiO
在本发明的一些实施方式中,优选地,所述改性氧化铝的形状选自球状、条状,其中,所述球状包括并不局限于微球状、小球状。
在本发明的一些实施方式中,优选地,所述氧化铝选自γ-氧化铝。采用优选的条件,更有利于提高改性氧化铝的活性。
本发明第二方面提供一种改性氧化铝的制备方法,该方法包括以下步骤:
(1)将铝源、酸性化合物和水进行第一混合,得到第一混合物;
(2)将所述第一混合物依次进行成型、第一干燥和第一焙烧,得到成型氧化铝;
(3)将所述成型氧化铝溶于水中,先加入碱性化合物调节pH为7-12,再加入硅源进行第二混合,得到第二混合物;
(4)将所述第二混合物进行固液分离,得到的改性氧化铝前体依次进行第二干燥和第二焙烧,得到改性氧化铝。
在本发明的一些实施方式中,优选地,步骤(1),所述第一混合物中铝源的含量为0.01-10wt%,优选为0.05-5wt%;酸性化合物的含量为0.01-3wt%,优选为0.05-1wt%。在本发明中,所述铝源、酸性化合物和水的投料量/用量比满足上述限定即可。
在本发明中,对所述铝源的种类具有较宽的选择范围。优选地,所述铝源为可溶性铝盐,包括并不局限于γ-氧化铝、羟基氧化铝、拟薄水铝石、氯化铝、硝酸铝等。当所述铝源选自γ-氧化铝,步骤(1)-(2)是使得粉末态氧化铝表面酸化,形成水合羟基态,便于后续加入碱性化合物和硅源进行硅改性。
在本发明中,对所述酸性化合物的种类具有较宽的选择范围。优选地,所述酸性化合物选自盐酸、硝酸、硫酸和磷酸中的至少一种。在本发明中,所述酸性化合物以水溶液形式存在,优选酸性化合物溶液中酸性化合物的浓度为1-50wt%。
在本发明中,对所述第一混合的方式具有较宽的选择范围,只要将所述铝源、酸性化合物和水混合即可。优选地,所述第一混合的条件包括:温度为15-40℃,优选为20-30℃;转速为100-1000rpm,优选为300-1000rpm;时间为0.1-5h,优选为0.1-2h。
在本发明中,对所述成型的方式具有较宽的选择范围。优选地,步骤(2)中,所述成型的方式包括并不局限于油氨滴球成型、喷雾干燥成型、挤出成型。
在本发明中,所述第一干燥旨在除去所述第一混合物中水。优选地,步骤(2)中,所述第一干燥的条件包括:温度为80-120℃,时间为90-110℃;时间为1-20h,优选为1-12h。
在本发明中,对所述第一干燥的方式具有较宽的选择范围,只要所述第一干燥的条件满足上述限定即可。优选地,所述第一干燥的方式包括并不局限于喷雾干燥、鼓风干燥、真空干燥等。
在本发明的一些实施方式中,优选地,步骤(2)中,所述第一焙烧的条件包括:温度为700-1200℃,优选为800-1000℃;时间为1-10h,优选为1-5h。在本发明中,所述第一焙烧在马弗炉或管式炉中进行,焙烧气氛为非还原性气体,优选空气、氮气、氩气中的至少一种。
在本发明中,步骤(3)中,先将所述成型氧化铝溶于水中,加入碱性化合物调节pH,再加入硅源进行第二混合,旨在得到多羟基的硅酸和/或羟基水合硅。
在本发明的一些实施方式中,优选地,调节pH为8-12,例如,8、9、10、10.5、11.5、12,以及任意两个数值组成的范围中的任意值,优选为10.5-11.5。
在本发明的一些实施方式中,优选地,以Al
在本发明中,对所述硅源的种类具有较宽的选择范围。优选地,所述硅源为可溶性硅盐,优选选自有机硅盐和/或无机硅盐,包括并不局限于正硅酸乙酯、四甲基硅、氧化硅气凝胶和四氯化硅中的至少一种。
在本发明中,没有特殊情况说明下,可溶性是指易溶于水,或者,在助剂的作用下易溶于水。
在本发明的一些实施方式中,优选地,所述碱性化合物选自碳酸铵、碳酸氢铵和氨水中的至少一种。
在本发明的一些实施方式中,优选地,所述第二混合的条件包括:温度为20-70℃,优选为25-60℃;转速为100-1000rpm,优选为300-1000rpm;时间为1-20h,优选为6-12h。
在本发明中,所述固液分离的方式具有较宽的选择范围,只要将所述第二混合物进行固液分离,得到改性氧化铝前体即可;所述固液分离的方式包括并不局限于过滤、沉降等。
在本发明中,所述第二干燥旨在除去所述改性氧化铝前体中残留的水分。优选地,步骤(4)中,所述第二干燥的条件包括:温度为80-120℃,时间为90-110℃;时间为1-20h,优选为1-12h。
在本发明中,对所述第二干燥的方式具有较宽的选择范围,只要所述第二干燥的条件满足上述限定即可。优选地,所述第二干燥的方式包括并不局限于喷雾干燥、鼓风干燥、真空干燥等。
在本发明的一些实施方式中,优选地,步骤(4)中,所述第二焙烧的条件包括:温度为400-1000℃,优选为500-900℃;时间为1-10h,优选为1-5h。在本发明中,所述第二焙烧在马弗炉或管式炉中进行,焙烧气氛为非还原性气体,优选空气、氮气、氩气中的至少一种。
本发明第三方面提供一种第一方面提供的改性氧化铝,或者,第二方面提供的方法制得的改性氧化铝在催化剂载体、气液吸附、固相填充剂中的应用。
本发明第四方面提供一种负载型催化剂,所述负载型催化剂包括载体和负载在所述载体上的活性组分;
其中,所述载体选自第一方面提供的改性氧化铝,或者,第二方面提供的方法制得的改性氧化铝。
本发明提供的一些实施方式中,优选地,所述负载型催化剂包括改性氧化铝和活性组分,其中,所述活性组分选自Pd、Pt。
在本发明中,对所述负载型催化剂的制备方法具有较宽的选择范围,只要将所述活性组分负载在所述改性氧化铝的表面即可。
本发明第五方面提供一种第四方面提供的负载型催化剂在催化合成赖氨酸型抗菌单体、芳香胺的氨基烷基化反应、制备酰胺类化合物的衍生物中的应用。
根据本发明,将所述负载型催化剂用于上述应用时,能有效提高原料的转化率和产物的选择性,进而提高目标产物的收率。
根据本发明一种特别优选的实施方式,所述改性氧化铝包括硅和氧化铝;其中,所述硅通过Si-O-Al化学键连接在所述氧化铝的表面,在所述氧化铝的表面的相邻硅通过Si-O-Si化学键连接;所述改性氧化铝的硅铝比<1;其中,所述改性氧化铝的B酸密度为0-0.5μmol/g;
其中,所述改性氧化铝由以下方法制得:
(1)将铝源、酸性化合物和水进行第一混合,得到第一混合物;
(2)将所述第一混合物依次进行成型、第一干燥和第一焙烧,得到成型氧化铝;
(3)将所述成型氧化铝溶于水中,先加入碱性化合物调节pH为8-12,再加入硅源进行第二混合,得到第二混合物;
(4)将所述第二混合物进行固液分离,得到的改性氧化铝前体依次进行第二干燥和第二焙烧,得到所述改性氧化铝。
以下将通过实施例对本发明进行详细描述。
B酸密度参数依据升温脱附的吡啶量来计算;即,B酸密度=测试吡啶红外光谱的改性氧化铝的酸量(单位为μmol)/改性氧化铝的质量(单位为g)。
比表面积参数采用全自动等温吸附仪测得;
平均孔径参数采用全自动等温吸附仪,并配合BJH模型计算获得;
磨损指数参数采用磨损指数分析仪测得;
压碎强度参数采用颗粒强度测试仪测得。
实施例1-7和对比例1-6制得的改性氧化铝(S1-S7和DS1-DS6)的物性参数均列于表1。
实施例1
(1)将80g铝源(拟薄水铝石)、5g酸性化合物水溶液(30wt%的稀硝酸溶液)和800mL水在1500mL搅拌釜中进行第一混合(温度为25℃,转速为600rpm,时间为1h),得到第一混合物;
(2)将上述第一混合物进行喷雾干燥成型(温度为100℃,时间为5h)后,马弗炉静态空气800℃焙烧3h,得到微球状成型氧化铝;
(3)将上述成型氧化铝溶于800mL水中,先加入碱性化合物(氨水)调节pH至10,再加入60g硅源(正硅酸乙酯)进行第二混合(温度为25℃,转速为600rpm,时间为12h),得到第二混合物;
(4)将上述第二混合物进行固液分离,得到的改性氧化铝前体采用真空干燥箱在100℃干燥5h后,马弗炉静态空气600℃焙烧3h,得到改性氧化铝S1;
其中,改性氧化铝S1的透射红外谱图如图1(a)所示;由图1(a)可知,波数1066cm
其中,改性氧化铝S1的吡啶红外表征谱图如图2所示,由图2可知,改性氧化铝S1具有极低的B酸位点。
其中,改性氧化铝S1的SEM图如图3所示,由图3可知,改性氧化铝S1为微球状。
其中,改性氧化铝S1的XRD图如图4所示,由图4可知,改性氧化铝S1中氧化铝为γ-氧化铝。
实施例2
(1)将80g铝源(羟基氧化铝)、10g酸性化合物水溶液(10wt%的稀硝酸溶液)和800mL水在1500mL搅拌釜中进行第一混合(温度为20℃,转速为400rpm,时间为1h),得到第一混合物;
(2)将上述第一混合物进行喷雾干燥成型(温度为100℃,时间为5h)后,马弗炉静态空气800℃焙烧3h,得到微球状成型氧化铝;
(3)将上述成型氧化铝溶于800mL水中,先加入碱性化合物(氨水)调节pH至12,再加入60g硅源(正硅酸乙酯)进行第二混合(温度为25℃,转速为400rpm,时间为8h),得到第二混合物;
(4)将上述第二混合物进行固液分离,得到的改性氧化铝前体采用真空干燥箱在100℃干燥5h后,马弗炉静态空气600℃焙烧3h,得到改性氧化铝S2。
实施例3
(1)将40g铝源(拟薄水铝石)、4g酸性化合物水溶液(20wt%的稀硝酸溶液)和800mL水在1500mL搅拌釜中进行第一混合(温度为30℃,转速为400rpm,时间为1h),得到第一混合物;
(2)将上述第一混合物进行挤出成型和真空干燥(温度为100℃,时间为5h)后,马弗炉静态空气1000℃焙烧2h,得到条状成型氧化铝;
(3)将上述成型氧化铝溶于800mL水中,先加入碱性化合物(氨水)调节pH至12,再加入10g硅源(正硅酸乙酯)进行第二混合(温度为40℃,转速为400rpm,时间为6h),得到第二混合物;
(4)将上述第二混合物进行固液分离,得到的改性氧化铝前体采用真空干燥箱在100℃干燥5h后,马弗炉静态空气600℃焙烧3h,得到条状改性氧化铝S3。
实施例4
(1)将50g铝源(拟薄水铝石)、10g酸性化合物水溶液(20wt%的稀硝酸溶液)和800mL水在1500mL搅拌釜中进行第一混合(温度为30℃,转速为400rpm,时间为1h),得到第一混合物;
(2)将上述第一混合物进行油氨滴球成型和鼓风干燥(温度为100℃,时间为5h)后,马弗炉静态空气1000℃焙烧2h,得到小球状成型氧化铝;
(3)将上述成型氧化铝溶于800mL水中,先加入碱性化合物(氨水)调节pH至10,再加入10g硅源(四氯化硅)进行第二混合(温度为25℃,转速为400rpm,时间为6h),得到第二混合物;
(4)将上述第二混合物进行固液分离,得到的改性氧化铝前体采用鼓风烘干箱在100℃干燥5h后,马弗炉静态空气800℃焙烧3h,得到小球状改性氧化铝S4。
实施例5
(1)将80g铝源(γ-氧化铝)、5g酸性化合物水溶液(30wt%的稀盐酸溶液)和800mL水在1500mL搅拌釜中进行第一混合(温度为30℃,转速为800rpm,时间为1h),得到第一混合物;
(2)将上述第一混合物进行喷雾干燥成型(温度为100℃,时间为5h)后,马弗炉静态空气800℃焙烧3h,得到微球状成型氧化铝;
(3)将上述成型氧化铝溶于800mL水中,先加入碱性化合物(氨水)调节pH至12,再加入50g硅源(氧化硅气凝胶)进行第二混合(温度为25℃,转速为400rpm,时间为12h),得到第二混合物;
(4)将上述第二混合物进行固液分离,得到的改性氧化铝前体采用鼓风烘干箱在100℃干燥5h后,马弗炉静态空气800℃焙烧3h,得到微球状改性氧化铝S5。
实施例6
(1)将80g铝源(拟薄水铝石)、20g酸性化合物水溶液(10wt%的稀硫酸溶液)和800mL水在1500mL搅拌釜中进行第一混合(温度为30℃,转速为800rpm,时间为1h),得到第一混合物;
(2)将上述第一混合物进行挤出成型和鼓风干燥(温度为100℃,时间为5h)后,马弗炉静态空气800℃焙烧3h,得到条状成型氧化铝;
(3)将上述成型氧化铝溶于800mL水中,先加入碱性化合物(氨水)调节pH至12,再加入30g硅源(正硅酸乙酯)进行第二混合(温度为60℃,转速为800rpm,时间为6h),得到第二混合物;
(4)将上述第二混合物进行固液分离,得到的改性氧化铝前体采用真空干燥箱在100℃干燥5h后,马弗炉静态空气800℃焙烧3h,得到条状改性氧化铝S6。
实施例7
按照实施例1的方法,不同的是,步骤(3)中,调节pH为7,其余条件相同,得到微球状改性氧化铝S7。
对比例1
将800mL去离子水、80g拟薄水铝石和10g 10wt%稀硝酸水溶液在1500mL搅拌釜中进行混合(温度为25℃,转速为800rpm,时间为5h),得到的混合物进行固液分离,得到的固体进行喷雾干燥后,马弗炉静态空气800℃焙烧3h,得到微球状改性氧化铝DS1;
其中,改性氧化铝DS1的透射红外谱图如1(b)所示;由图1(b)可知,波数1066cm
其中,改性氧化铝DS1的吡啶红外表征谱图如图2所示,由2可知,改性氧化铝DS1具有较高的B酸位点。
对比例2
将800mL去离子水、80g拟薄水铝石和20g 5wt%稀硝酸水溶液在1500mL搅拌釜中进行混合(温度为25℃,转速为800rpm,时间为5h)后,加入60g正硅酸乙酯,加入氨水调节pH至12,并在25℃搅拌12h,得到的混合物进行喷雾干燥,马弗炉静态空气800℃焙烧3h,得到微球状改性氧化铝DS2。
其中,改性氧化铝DS2的吡啶红外表征谱图如图2所示,由图2可知,改性氧化铝DS2具有较高的B酸位点。
其中,改性氧化铝DS2的XRD图如图4所示,由图4可知,改性氧化铝DS2中氧化铝为γ-氧化铝。
对比例3
按照实施例1的方法,不同的是,没有步骤(1)-(2),即,直接将80g铝源(拟薄水铝石)溶于800mL水中,其余条件相同,得到改性氧化铝DS3。
对比例4
按照实施例1的方法,不同的是,没有步骤(2),即,直接将步骤(1)得到的第一混合物进行步骤(3),其余条件相同,得到改性氧化铝DS4。
对比例5
按照实施例1的方法,不同的是,步骤(3)中,直接将上述成型氧化铝、800mL水、碱性化合物(氨水)和60g硅源(正硅酸乙酯)进行第二混合(温度为25℃,转速为600rpm,时间为12h),并调节pH至10,其余条件相同,得到改性氧化铝DS5。
对比例6
按照实施例1的方法,不同的是,步骤(3)中,不加入氨水以调节pH为10,其余条件相同,得到改性氧化铝DS6。
表1
注:*-硅含量以SiO
续表1
通过表1的结果可以看出,相比对比例1-7,实施例1-6制得的改性氧化铝兼具低B酸密度、低磨损指数、高压碎强度、高比表面积和低平均孔径,即,本发明提供的改性氧化铝具有低B酸密度、耐磨性、高稳定性和优异孔结构。
测试例
将实施例1-7和对比例1-6制得的改性氧化铝(S1-S7和DS1-DS6)分别作为催化剂载体,浸渍于30wt%氨水配制的Pd(NH
其中,分别基于负载型催化剂(P1-P7和DP1-DP6)的总重量,活性组分Pd的含量为10wt%,改性氧化铝的含量为90wt%。
采用间歇式高压反应釜评价上述负载型催化剂的催化效果;具体操作包括:在50mL甲醇和0.1g负载型催化剂(P1-P7和DP1-DP6)存在下,将0.2g氨基己内酰胺和0.1g多聚甲醛进行临氢反应,其中,临氢反应的条件包括:氢压为0.3MPa,温度为80℃,时间为3h,得到反应产物,其中,反应产物包括二甲基氨基己内酰胺、甲基氨基己内酰胺和其它副产物。
其中,采用气相色谱分析催化反应结果,测试结果列于表2。
表2
通过表2的结果可以看出,将本发明提供的改性氧化铝作为载体,负载活性组分Pd,得到的催化剂用于氨基己内酰胺和多聚甲醛的临氢反应,具有较高的原料转化率和产物选择性,即,得到的二甲基氨基己内酰胺具有较高的收率。
以上详细描述了本发明的优选实施方式,但是,本发明并不限于此。在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,包括各个技术特征以任何其它的合适方式进行组合,这些简单变型和组合同样应当视为本发明所公开的内容,均属于本发明的保护范围。
- 一种缓凝型聚羧酸减水剂的制备方法
- 一种端胺基聚醚大单体及其制备抗泥型聚膦酸减水剂的方法
- 一种早强保坍型聚羧酸型减水剂及其制备方法
- 一种高强混凝土降粘型聚羧酸减水剂及其制备方法
- 一种两性抗泥型减水剂及其制备方法
- 一种缓凝型单体及其制备方法、缓凝型减水剂及其制备方法
- 一种缓凝型单体及其制备方法、缓凝型减水剂及其制备方法