5'-O-DMT-2'-O-丙炔基-尿苷的合成方法
文献发布时间:2024-04-18 20:01:23
技术领域
本发明属于核苷酸合成技术领域,尤其涉及一种5'-O-DMT-2'-O-丙炔基-尿苷的合成方法。
背景技术
寡核苷酸在癌症治疗和遗传学中具有广泛的应用前景,如:寡核苷酸是一种短的单链DNA或RNA,通常由几个到几十个核苷酸组成。它们可以与互补的DNA或RNA序列结合,形成稳定的双链结构,具体的,寡核苷酸可以与细胞内的DNA或RNA的内部或末端位置进行交联,这种交联可以阻止细胞的复制。因此,寡核苷酸可以被用作一种特殊的药物,与癌细胞的DNA或RNA结合,阻止癌细胞的复制,从而杀死癌细胞。这种治疗方法被称为基因靶向治疗,因为它针对的是特定的基因突变,而不是所有的健康或患病的细胞。
在遗传学中,寡核苷酸被用作探针,用于研究DNA和RNA的结构和功能。它们也可以被用于基因治疗,即通过修改DNA或RNA序列来治疗遗传性疾病。在这个过程中,寡核苷酸可以被设计成与异常基因序列结合,从而阻止有害基因的表达。
RNAi是一种生物体内常见的基因表达调控机制,它可以通过降解目标RNA来抑制特定基因的表达。在RNAi过程中,双链RNA(dsRNA)是关键的效应分子,它可以被细胞内的RNaseIII酶切割成多个小片段,这些小片段被称为siRNA(小干扰RNA)。siRNA可以与目标RNA结合,导致目标RNA被降解,从而抑制特定基因的表达。
在siRNA的合成过程中,2'的位置是一个很重要的修饰位点。在这个位置上,可以通过添加化学基团(如甲基、乙基等)来改变siRNA的理化性质和功能。这些修饰可以增加siRNA的稳定性和特异性,使其更加有效地结合目标RNA,从而更有效地抑制特定基因的表达。其中:糖修饰的寡核苷酸就是改变siRNA的理化性质和功能的非常重要的物质,如:糖修饰的寡核苷酸可以激活RNaseH的2'-脱氧-赤型-呋喃戊糖基核苷的亚序列,从而促进对目标核酸的降解。
因此,合成糖修饰的寡核苷酸是非常重要的,2'-O-丙炔基是一种有机化学基团,通常与核苷酸中的糖部分结合,这种基团的存在可以改变核苷酸的化学性质和生物学活性。5'-O-DMT-2'-O-丙炔基-尿苷的合成方法作为含有2'-O-丙炔基的医药中间体,也是合成糖修饰的寡核苷酸的中间体,其合成受到广泛关注。
发明内容
为解决上述问题,本申请提供了一种新的思路合成医药中间体5'-O-DMT-2'-O-丙炔基-尿苷。
为实现上述目的,本申请是通过如下方案来实现的:
本申请提供了一种5'-O-DMT-2'-O-丙炔基-尿苷的合成方法,包括如下步骤:
S1、在-5℃~5℃温度条件下,将吡啶加入第一反应容器内,向第一反应容器内持续通入惰性气体,在惰性气体气氛下,向第一反应容器内依次加入2'-O-丙炔基-尿苷、4,4'-双甲氧基三苯甲基氯,再在-5℃~5℃温度条件下充分搅拌反应完全,水洗有机相,减压浓缩有机相,柱层析纯化,得到目标产物5'-O-DMT-2'-O-丙炔基-尿苷。
作为本申请进一步的改进,步骤S1中,所述充分搅拌反应完全的时间为第一预设时间段,第一预设时间段可根据2'-O-丙炔基-尿苷、4,4'-双甲氧基三苯甲基氯以及吡啶的加入量而设定,其可以为但不仅仅限于1h~10h,具体的可以为但不仅仅限于1h、2h、3h、4h、5h、6h、7h等等。
作为本申请进一步的改进,步骤S1中,所述2'-O-丙炔基-尿苷和所述吡啶的体积摩尔比为0.2mol/L~0.6mol/L。
作为本申请进一步的改进,步骤S1中,所述4,4'-双甲氧基三苯甲基氯和所述2'-O-丙炔基-尿苷的体积摩尔比为0.8~1.5。
作为本申请进一步的改进,步骤S1中,通过HPLC控制搅拌反应的进程。
作为本申请进一步的改进,步骤S1中,应用于所述柱层析纯化的洗脱剂为二氯甲烷和甲醇的混合溶液,混合溶液中还含有体积百分含量为5%的三乙胺。
作为本申请进一步的改进,在步骤S1之前还包括步骤S2和步骤S3:
S2、将尿苷和N,N-二甲基甲酰胺加入第二反应容器中,充分溶解后,加入碳酸二苯酯和碳酸氢钠,加热到115℃~125℃,充分反应完全后,将温度降至20℃~30℃,固体析出,过滤,得到固体物质滤饼,采用第一有机溶剂对固体物质滤饼进行洗涤,得到2,2'-脱水尿苷;
S3、于-5℃~5℃温度下,将步骤S2中得到的2,2'-脱水尿苷加入第三反应容器中向第三反应容器内持续通入惰性气体,在惰性气体气氛下,向第三反应容器内依次加入第二有机溶剂、丙炔氧基硅烷,充分搅拌反应后,再在-5℃~5℃温度下,向第三反应容器内缓慢滴加三氟化硼乙醚,滴加完成后,于-5℃~5℃温度充分搅拌,使第三反应容器升温至115℃~125℃,且在115℃~125℃温度条件下,充分搅拌反应,反应完全后,依次进行减压浓缩、柱层析纯化、减压浓缩,得到2'-O-丙炔基-尿苷。
作为本申请进一步的改进,步骤S2中,所述碳酸氢钠和所述尿苷的摩尔比为1.5:1~3:1。
作为本申请进一步的改进,步骤S2中,所述碳酸二苯酯和所述尿苷的摩尔比为1.1:1~2:1。
作为本申请进一步的改进,步骤S2中,所述尿苷和所述N,N-二甲基甲酰胺的体积摩尔比为0.5mol/L~1.5mol/L。
作为本申请进一步的改进,步骤S2中,所述第一有机溶剂为乙醇、甲醇、异丙醇、丙酮、乙酸乙酯中的至少一种。优选的,当所述第一有机溶剂为甲醇时,所述洗涤的步骤为:于20℃~30℃条件下,将甲醇和步骤S2中的滤饼混合在一起,充分搅拌,再次过滤,以去除滤饼中的杂质和离子,得到纯净的2,2'-脱水尿苷。优选的,步骤S2中,通过HPLC控制所述反应的进程。
作为本申请进一步的改进,步骤S2中,所述充分反应完全的时间为第二预设时间段,第二预设时间段可根据尿苷、碳酸二苯酯以及碳酸氢钠的加入量而设定,其可以为但不仅仅限于5h~15h,具体的可以为但不仅仅限于5h、6h、7h、8h、9h、10h、11h、12h、13h、14h、15h等等。
作为本申请进一步的改进,步骤S3中,所述第二有机溶剂为N,N-二甲基乙酰胺。
作为本申请进一步的改进,步骤S3中,所述丙炔氧基硅烷为叔丁基二甲基(2-丙炔氧基)硅烷或丙炔氧基三甲基硅烷。优选的,所述叔丁基二甲基(2-丙炔氧基)硅烷和所述2,2'-脱水尿苷的摩尔比为1:4~1:6,或,所述丙炔氧基三甲基硅烷与所述2,2'-脱水尿苷的摩尔比为1:4~1:6。
作为本申请进一步的改进,步骤S3中,所述三氟化硼乙醚与2,2'-脱水尿苷的体积摩尔比为1:2~1:3。
作为本申请进一步的改进,步骤S3中,通过HPLC控制115℃~125℃温度条件下的反应进程。
作为本申请进一步的改进,步骤S3中,应用于所述柱层析纯化的洗脱剂为二氯甲烷和甲醇的混合溶液,混合溶液中还含有体积百分含量为5%的三乙胺。
作为本申请进一步的改进,步骤S3中:-5℃~5℃温度充分搅拌反应的时间为第三预设时间段,第三预设时间段可根据2,2'-脱水尿苷、丙炔氧基硅烷以及三氟化硼乙醚的加入量而设定,其可以为但不仅仅限于5min~1h,具体的可以为但不仅仅限于10min、20min、30min、50min、1h等等;115℃~125℃温度充分搅拌反应完全的时间为第四预设时间段,第四预设时间段可根据2,2'-脱水尿苷、丙炔氧基硅烷以及三氟化硼乙醚的加入量而设定,其可以为但不仅仅限于1h~10h,具体的可以为但不仅仅限于1h、2h、3h、4h、5h、6h、7h、8h、9h、10h等等。
作为本申请进一步的改进,步骤S1中,所述惰性气体可以为但不仅仅限于氮气、氩气、氦气、氖气等等;步骤S3中,所述惰性气体可以为但不仅仅限于氮气、氩气、氦气、氖气等等。
本申请的有益效果在于,本申请的合成5'-O-DMT-2'-O-丙炔基-尿苷的技术路线具有以下优势:
1)避免了异构体的产生,提高了产物的纯度和质量。
2)缩减了纯化的步骤,降低了生产成本,提高了生产效率。
3)整体流程设计合理,适合大规模生产,具有较高的实用性和经济性。
4)降低了能源消耗和环境污染,符合可持续发展的要求。
综上所述,本申请的合成技术路线具有优良的经济效益和工业化应用前景,对于相关领域的发展具有积极的推动作用。
附图说明
图1为实施例2制备出的2'-O-丙炔基-尿苷的核磁共振谱图;
图2为实施例2制备出的2'-O-丙炔基-尿苷进行的HPLC谱图;
图3为实施例2制备出的5'-O-DMT-2'-O-丙炔基-尿苷的核磁共振谱图;
图4为实施例2制备出的5'-O-DMT-2'-O-丙炔基-尿苷进行的HPLC谱图。
具体实施方式
为使本申请的目的、技术方案和优点更加清楚,下面将结合本申请具体实施例及附图对本申请技术方案进行清楚、完整地描述。显然,所描述的实施例仅是本申请一部分实施例,而不是全部的实施例,不用来限制本发明的范围。基于本申请中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本申请保护的范围。
为解决上述技术问题,本申请提供了一种新的思路合成医药中间体5'-O-DMT-2'-O-丙炔基-尿苷,技术方案如下:
5'-O-DMT-2'-O-丙炔基-尿苷的具体的合成步骤如下:
步骤一:在-5℃~5℃温度条件下,将1~1.1L吡啶加入第一反应容器内,氮气保护,且持续在氮气保护的条件下,向第一反应容器内依次加入354.29~357.83mmol(100~101g)2'-O-丙炔基-尿苷(化合物10)、425.15~428.1mmol(144~145g)4,4'-双甲氧基三苯甲基氯(化合物2),再在-5℃~5℃温度条件下搅拌反应3~5小时。通过HPLC控制上述实验的反应进程,反应完全后,用1L水洗有机相三遍,减压浓缩有机相,柱层析纯化,得到347.23mmol(203g)目标产物5'-O-DMT-2'-O-丙炔基-尿苷,收率98%。其中:柱层析纯化的过程中,所用的洗脱剂为二氯甲烷和甲醇的混合溶液,混合溶液中还含有5%的三乙胺,洗脱的过程中可以采用一次性添加洗脱剂,也可以采用多次添加洗脱剂的方式进行洗涤,按添加顺序,所述二氯甲烷和甲醇的体积比梯度降低,如:二氯甲烷和甲醇的体积比可以从20:1~10:1范围内梯度降低,具体的:如采用三次洗脱剂的方式,第一次添加二氯甲烷和甲醇的体积比为20:1,第二次添加二氯甲烷和甲醇的体积比为15:1,第三次添加二氯甲烷和甲醇的体积比为10:1。
步骤一中化合物10到目标产物的主要反应机理如下反应机理式Ⅰ:
式Ⅰ,
在吡啶作用下,4,4'-双甲氧基三苯甲基氯(化合物2)发生分解,生成4,4'-双甲氧基三苯甲基碳正离子,由于空间位阻效应,2'-O-丙炔基-尿苷(化合物10)的5'位置的羟基氧进攻碳正离子,得到目标产物。步骤三的反应中:吡啶作为溶剂,提供碱性环境,促进4,4'-双甲氧基三苯甲基氯的分解;4,4'-双甲氧基三苯甲基氯是一种重要的有机化合物,它主要被用作合成核苷的羟基保护基和去保护基,4,4'-双甲氧基三苯甲基氯作为一种羟基保护基,能够选择性保护核苷中的羟基,使其在后续的反应中不被反应试剂破坏,同时,在需要将保护基去除时,4,4'-双甲氧基三苯甲基氯也可以作为一种去保护基,通过特定的反应条件将其去除,从而得到目标产物;2'-O-丙炔基-尿苷作为底物,提供了尿苷部分的结构。投料的过程容易瞬间放热,导致反应体系变杂,-5℃~5℃的温度设定是为了确保反应体系的稳定性以及控制副反应的发生。
化合物10到目标产物的技术路线如式Ⅱ:
式Ⅱ,
在可实施的技术方案中,在步骤一之前还可包括如下步骤:
步骤二:将410~414.1mmol(100~101g)尿苷(化合物1)和400~450mL N,N-二甲基甲酰胺加入第二反应容器中,充分溶解后,加入447.2~451.9mmol(96~97g)碳酸二苯酯(化合物6)和7.82~8.42mmol(0.65~0.7g)碳酸氢钠,加热到115℃~125℃,反应8~10小时,通过HPLC控制上述实验的反应进程,反应完全后,将上述反应的温度降至20℃~30℃,固体析出,过滤,得到固体滤饼。于20℃~30℃条件下,将400~500mL甲醇和上述固体滤饼混合在一起,搅拌2~3小时,再次过滤,以便充分去除滤饼中的杂质和离子,得到88.2g白色粉末状产品(化合物7),收率95.2%。
步骤二中化合物1到化合物7的主要反应机理如下反应机理式Ⅲ:
式Ⅲ,
碱性碳酸氢钠中和化合物1中氮上的氢离子后,化合物1的电子迁移到羰基氧上,随后带负电核的氧进攻2'位的碳,随后电子迁移到2'位的羟基氧上,脱去羟基成为2,2'-脱水尿苷(化合物7),而脱去的羟基进攻碳酸二苯酯(化合物6)的羰基,碳酸二苯酯分解成为苯酚和碳酸一苯酯负离子。步骤一的反应中:尿苷作为反应的底物,提供尿苷部分的结构;碳酸氢钠作为缚酸剂,帮助维持合适的pH值,以促进反应的进行;碳酸二苯酯作为脱水剂,能与脱去的羟基反应,进而促进反应的进行;N,N-二甲基甲酰胺作为溶剂,帮助维持底物和催化剂的溶解状态,以促进反应的进行。115℃~125℃温度设定是为了促进反应的进行,打破尿苷、碳酸二苯酯等的初始结合,需要一定的能量,而高温可以提供这种能量。另外,高温还可能有助于增加体系的流动性,使得底物和试剂能够更好地混合和接触,从而促进反应的进行。
步骤三:于-5℃~5℃温度下,将0.442~0.446mol(100~101g)2,2'-脱水尿苷(化合物7)加入第三反应容器中,氮气保护,且持续在氮气保护的条件下,向第三反应容器内依次加入400~450ml N,N-二甲基乙酰胺、2.20~2.23mol(375~380g)叔丁基二甲基(2-丙炔氧基)硅烷或者2.20~2.23mol(382~387g)丙炔氧基三甲基硅烷,充分搅拌反应后,再在-5~5℃温度下,用50ml注射器吸取1.102~1.11mol(156.5~157.5g)三氟化硼乙醚(化合物9),并向第三反应容器内滴加,此处的三氟化硼乙醚指的是三氟化硼在乙醚中的溶解液,且三氟化硼在乙醚中的质量百分含量为47%,滴加时间为2h~4h,滴加完成后,于-5~5℃温度下搅拌15~30分钟,后使第三反应容器升温至115℃~125℃,且在115℃~125℃温度条件下,搅拌反应14h~16h。通过HPLC控制上述实验的反应进程,反应完全后,依次进行减压浓缩、柱层析纯化、减压浓缩,得到0.36mol(101.7g)白色固体产物2'-O-丙炔基-尿苷(化合物10),收率81.5%(以叔丁基二甲基(2-丙炔氧基)硅烷为原料),或,0.33mol(93.8g)白色固体产物2'-O-丙炔基-尿苷(化合物10),收率75.2%(以丙炔氧基三甲基硅烷为原料)。其中:柱层析纯化的过程中,所用的洗脱剂为二氯甲烷和甲醇的混合溶液,混合溶液中还含有5%的三乙胺,洗脱的过程中可以采用一次性添加洗脱剂,也可以采用多次添加洗脱剂的方式进行洗涤,按添加顺序,所述二氯甲烷和甲醇的体积比梯度降低,如:二氯甲烷和甲醇的体积比可以从20:1~10:1范围内梯度降低,具体的:如采用三次洗脱剂的方式,第一次添加二氯甲烷和甲醇的体积比为20:1,第二次添加二氯甲烷和甲醇的体积比为15:1,第三次添加二氯甲烷和甲醇的体积比为10:1。
步骤三中化合物7到化合物10的主要反应机理如下反应机理式Ⅵ:
式Ⅵ,
化合物7在路易斯酸BF3的作用下,导致2'的位置碳处于缺电子状态,从而使得与烷基(2-丙炔氧基)硅烷的氧原子发生S
由上述技术方案合成5'-O-DMT-2'-O-丙炔基-尿苷的技术路线如式Ⅴ:
式Ⅴ,
实施例1
5'-O-DMT-2'-O-丙炔基-尿苷的具体的合成步骤如下:
步骤一:在0℃温度条件下,将1L吡啶加入第一反应容器内,氮气保护,且持续在氮气保护的条件下,向第一反应容器内依次加入354.29mmol(100g)2'-O-丙炔基-尿苷(化合物10)、425.15mmol(144.05g)4,4'-双甲氧基三苯甲基氯(化合物2),再在0℃温度条件下搅拌反应4小时。通过HPLC控制上述实验的反应进程,反应完全后,用1L水洗有机相三遍,减压浓缩有机相,柱层析纯化,得到347.23mmol(203g)目标产物5'-O-DMT-2'-O-丙炔基-尿苷,收率98%。其中:柱层析纯化的过程中,所用的洗脱剂为二氯甲烷和甲醇的混合溶液,混合溶液中还含有5%的三乙胺,洗脱的过程中采用三次洗脱剂的方式,第一次添加二氯甲烷和甲醇的体积比为20:1,第二次添加二氯甲烷和甲醇的体积比为15:1,第三次添加二氯甲烷和甲醇的体积比为10:1。
实施例2
本实施例与实施例1的区别在于,在步骤一之前还包括如下步骤:
步骤二:将410mmol(100g)尿苷(化合物1)和400mL N,N-二甲基甲酰胺加入第二反应容器中,充分溶解后,加入450mmol (96.6g)碳酸二苯酯(化合物6)和8.18mmol(0.68g)碳酸氢钠,加热到120℃,反应9小时,通过HPLC控制上述实验的反应进程,反应完全后,将上述反应的温度降至25℃,固体析出,过滤,得到固体滤饼。于25℃条件下,将450mL甲醇和上述固体滤饼混合在一起,搅拌2.5小时,再次过滤,以便充分去除滤饼中的杂质和离子,得到88.2g白色粉末状产品(化合物7),收率95.2%。
步骤三:于0℃温度下,将0.442mol(100g)2,2'-脱水尿苷(化合物7)加入第三反应容器中,氮气保护,且持续在氮气保护的条件下,向第三反应容器内依次加入400ml N,N-二甲基乙酰胺、2.21mol(377g)叔丁基二甲基(2-丙炔氧基)硅烷或者2.21mol(384g)丙炔氧基三甲基硅烷,充分搅拌反应后,再在0℃温度下,用50ml注射器吸取1.105mol(156.9g)三氟化硼乙醚(化合物9),并向第三反应容器内滴加,此处的三氟化硼乙醚指的是三氟化硼在乙醚中的溶解液,且三氟化硼在乙醚中的质量百分含量为47%,滴加时间为3h,滴加完成后,于0℃温度下搅拌25分钟,后使第三反应容器升温至120℃,且在120℃温度条件下,搅拌反应15h。通过HPLC控制上述实验的反应进程,反应完全后,依次进行减压浓缩、柱层析纯化、减压浓缩,得到0.36mol(101.7g)白色固体产物2'-O-丙炔基-尿苷(化合物10),收率81.5%(以叔丁基二甲基(2-丙炔氧基)硅烷为原料),或,0.33mol(93.8g)白色固体产物2'-O-丙炔基-尿苷(化合物10),收率75.2%(以丙炔氧基三甲基硅烷为原料)。其中:柱层析纯化的过程中,所用的洗脱剂为二氯甲烷和甲醇的混合溶液,混合溶液中还含有5%的三乙胺,洗脱的过程中采用三次洗脱剂的方式,第一次添加二氯甲烷和甲醇的体积比为20:1,第二次添加二氯甲烷和甲醇的体积比为15:1,第三次添加二氯甲烷和甲醇的体积比为10:1。
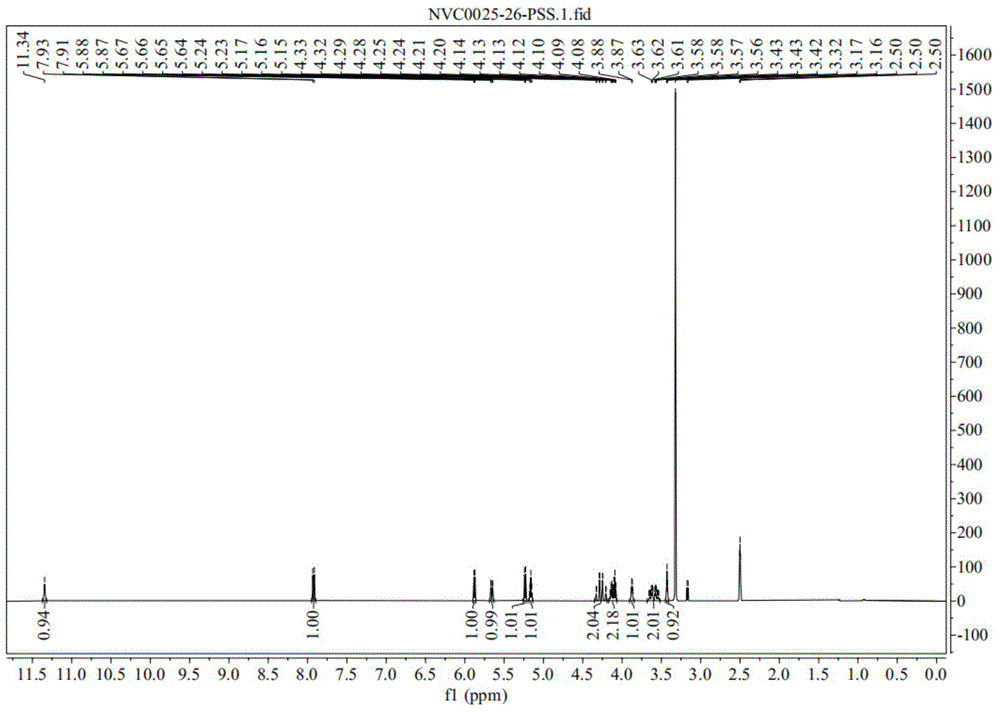
针对本实施例2制备出的2'-O-丙炔基-尿苷(化合物10)进行的1H NMR分析谱图如图1所示,具体如下:
1
针对本实施例2制备出的2'-O-丙炔基-尿苷进行的HPLC纯度分析如图2所示,HPLC纯度大于95%。
由本实施例2制备的5'-O-DMT-2'-O-丙炔基-尿苷进行的1H NMR分析谱图如图3所示,具体如下:
1
针对本实施例2制备出的5'-O-DMT-2'-O-丙炔基-尿苷进行的HPLC纯度分析如图4所示,HPLC纯度大于95%。
上述实施例1和实施例2中,使用的药品的来源如下:吡啶是国药集团化学试剂有限公司生产的批号为20230209的产品,2'-O-丙炔基-尿苷是苏州诺维康生物科技有限公司生产的批号为NVC0033-27的产品,4,4'-双甲氧基三苯甲基氯是芜湖华仁科技有限公司生产的批号为22042805的产品,尿苷是芜湖华仁科技有限公司生产的批号为20112006的产品,碳酸二苯酯是安徽泽升科技有限公司生产的批号为90WQRM4U的产品,碳酸氢钠是江苏强盛功能化学股份有限公司生产的批号为20230316的产品,甲醇是无锡晶科化工有限公司生产的批号为20230318的产品,2,2'-脱水尿苷是江苏艾康生物医药研发有限公司生产的批号为MPC42-1569533-2的产品,N,N-二甲基乙酰胺是上海泰坦科技股份有限公司生产的批号为P2637035的产品,叔丁基二甲基(2-丙炔氧基)硅烷是江苏艾康生物医药研发有限公司生产的批号为AZF23-1159477-1的产品,丙炔氧基三甲基硅烷是安徽泽升科技有限公司生产的批号为OWEE4RDR的产品,三氟化硼乙醚是安徽泽升科技有限公司生产的批号为OFPQR4XE的产品,二氯甲烷是无锡晶科化工有限公司生产的批号为20230308的产品,三乙胺是江苏强盛功能化学股份有限公司白茆分公司生产的批号为20230218的产品。
对比例1
为了佐证本申请的技术方案具有优异的技术效果,提供了本对比例,现有技术中采用的制备5'-O-DMT-2'-O-丙炔基-尿苷的技术方案如下步骤所述:
步骤一:在20℃~30℃下,向反应容器中加入410 mmol(100 g)尿苷(化合物1)和1L吡啶,分四次加入512 mmol(173.4g) 4,4'-双甲氧基三苯甲基氯(化合物2),在20~30℃下搅拌反应1~2小时。通过HPLC控制上述实验的反应进程,反应完全后,在40℃~50℃下,减压浓缩,用2L二氯甲烷溶清后,用2L水洗有机相三次,用100g~200g无水硫酸钠干燥1小时,在40℃~50℃下,减压浓缩,粗品通过柱层析纯化,洗脱剂为甲醇和二氯甲烷的混合溶液,混合溶液中甲醇的体积百分含量为0-10%,加压浓缩得到白色固体化合物3 (220g,402.5mmol,收率98%)。
步骤二:将4.03mmol(2.2g)化合物3和溶剂置于反应容器中,充分混合,溶剂为20ml苯和乙腈的混合溶剂,苯和乙腈的体积比为1:1,再向反应容器内依次加入4.43mmol的二丁基氧化锡(化合物4)、8.06mmol的氯丙炔(化合物5)、2.01mmol的四丁基碘化铵(TBAI),充分搅拌后,在95℃~105℃下微波反应4~6小时,通过HPLC控制上述实验的反应进程,反应完全后,减压浓缩,柱层析纯化,洗脱剂为甲醇和二氯甲烷的混合溶液,混合溶液中甲醇的体积百分含量为0-5%,加压浓缩得到白色固体目标产物(0.94g,1.612mmol,收率40%)。
上述两个步骤合在一起,也就是,对比例1的5'-O-DMT-2'-O-丙炔基-尿苷的合成的技术路线如式Ⅵ:
式Ⅵ。
综上,现有合成5'-O-DMT-2'-O-丙炔基-尿苷的技术路线中含有二丁基氧化锡(化合物4),二丁基氧化锡为剧毒药品,属于管制试剂,不利于产业化,且由于合成的过程中会产生5'-O-DMT-3'-O-丙炔基-尿苷异构体,合成收率较低。因此,本申请提供了一种新的思路合成5'-O-DMT-2'-O-丙炔基-尿苷,主要采用2'-O-丙炔基-尿苷为底物,通过加入4,4'-双甲氧基三苯甲基氯和吡啶,并通过设计的特定的工艺将2'-O-丙炔基-尿苷转化为5'-O-DMT-2'-O-丙炔基-尿苷。避免了异构体的生成,降低了纯化过程的难度,降低了成本。
虽然本说明书按照实施方式加以描述,但并非每个实施方式仅包含一个独立的技术方案,说明书的这种叙述方式仅仅是为清楚起见,本领域技术人员应当将说明书作为一个整体,各实施方式中的技术方案也可以经适当组合,形成本领域技术人员可以理解的其他实施方式。
上文所列出的一系列的详细说明仅仅是针对本发明的可行性实施方式的具体说明,它们并非用以限制本发明的保护范围,凡未脱离本发明技艺精神所作的等效实施方式或变更均应包含在本发明的保护范围之内。
- 电池或电池材料快速干燥方法、小容量热传导式真空干燥装置及其智能干燥系统
- 一种电池管理系统对电芯过放保护的SOP估算方法
- 一种基装材料订单的拆分方法、装置、设备和存储介质
- 干燥系统、干燥电芯的方法、电池的生产工艺和电池