一种评估花鲈生长和抗性性状综合育种值的方法及应用
文献发布时间:2023-06-19 19:33:46
技术领域
本发明涉及动物遗传育种领域,特别涉及一种评估花鲈生长和抗性性状综合育种值的方法及应用。
背景技术
花鲈(Lateolabrax maculatus),隶属鲈形目(Perciformes)、鮨科(Serranidae)、花鲈属(Lateolabrax),在黄渤海、东海、南海等区域均有分布。近年来,我国花鲈养殖年产量均超过15万吨,位列海水养殖鱼类产量前三位,已成为海水养殖支柱产业。然而,目前花鲈种业体系正面临着空前挑战:缺乏优良花鲈品种,尚未建立科学育种体系,长期近亲繁殖导致种质退化现象已经显现,良种化进程大大落后于其他主要海水养殖鱼类。因此急需针对花鲈养殖产业关注的经济性状,开展遗传选育工作。
鱼类育种的首要目标性状是生长性状,提高生长率,培育快速生长品种,可缩短养殖周期和成本,有效提高经济效益;此外,抗性性状也是育种工作关注的焦点,例如耐低氧、耐温、耐盐碱、抗病等都是显著影响养殖产业高质量发展的重要经济性状。目前,往往采用传统选育方式针对鱼类的单一性状进行遗传改良,难以满足花鲈遗传育种要求。
发明内容
鉴于此,本发明提出一种评估花鲈生长和抗性性状综合育种值的方法及应用,旨在解决背景技术中所提到的问题。
本发明将全基因组选择技术(GS)应用于花鲈的遗传育种中,首次建立了评估生长和抗性性状协同选育的综合育种值的方法。通过此方法对候选亲鱼的综合育种值进行评估可以挑选出兼具生长和抗性优势的核心育种群体,大大缩短育种年限,同时也为其他鱼类的多性状复合选育提供借鉴,具有广阔的应用前景。
本发明的技术方案是这样实现的:
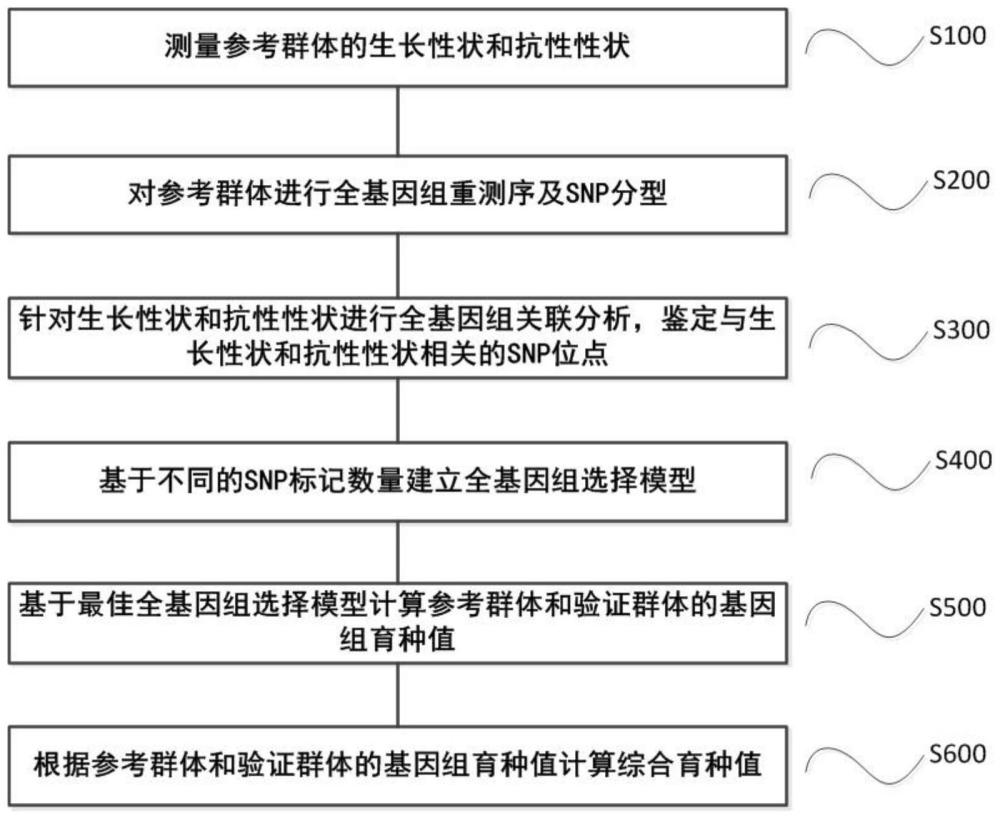
一种评估花鲈生长和抗性性状综合育种值的方法,包括以下步骤:
S1、测量参考群体的生长性状和抗性性状;
S2、对参考群体进行全基因组重测序及SNP分型;
S3、针对生长性状和抗性性状进行全基因组关联分析,鉴定与生长性状和抗性性状相关的SNP位点;
S4、基于不同的SNP标记数量建立全基因组选择模型;
S5、基于最佳全基因组选择模型计算参考群体和验证群体的基因组育种值;
S6、根据参考群体和验证群体的基因组育种值计算综合育种值。
进一步的,所述方法还包括:
根据基因组育种值与实际表型的相关性评估全基因组选择模型的预测准确性,选取最佳全基因组选择模型。
进一步的,S1步骤中,所述测量参考群体的生长性状和抗性性状的具体步骤包括:先对参考群体进行暂养驯化,暂养期间正常投喂;而后选取体质健壮、摄食状况良好的个体,给予胁迫因子处理,经环境胁迫死亡后记录其生长指标和死亡时间,分别作为生长和抗性性状的表型。更进一步的,所述暂养驯化时间为2周以上,所述胁迫因子为使用碳酸盐水体(26.16±0.5mmol/L)进行碱度胁迫。
更进一步的,S5步骤中,计算参考群体和验证群体的基因组育种值时基于所述选取最佳全基因组选择模型。
进一步的,S4、S5步骤中,采用五折交叉验证评估全基因组选择模型的预测准确性。
进一步的,S2步骤中,所述对参考群体进行全基因组重测序的具体步骤包括:
提取参考群体中每个个体的总基因组DNA;
对所述总基因组DNA进行检测;
对检测合格的总基因组DNA进行随机打断,纯化筛选符合要求的DNA片段,并将DNA片段连接测序接头,再通过滚环扩增制备DNB(DNA纳米球),对制备的DNB进行测序。
进一步的,S2步骤中,所述SNP分型的具体步骤包括:
对全基因组重测序数据进行质控和过滤;
将质控和过滤后的全基因组重测序数据比对至参考基因组数据,并消除全基因组重测序数据中PCR偏好性的影响;
对消除PCR偏好性后的全基因组重测序数据进行单核苷酸多态性检测,实现基因分型。
进一步的,S3步骤中,所述针对生长性状和抗性性状进行全基因组关联分析的具体步骤包括:
对全基因组重测序数据进行过滤;
填充测序缺失的SNP;
分别对生长性状和抗性性状进行全基因组关联分析。
进一步的,S6步骤中,所述根据参考群体和验证群体的基因组育种值计算综合育种值的步骤包括:
对参考群体和验证群体的基因组育种值进行归一化处理;
在归一化处理结果中分别赋予生长性状和抗性性状相应的权重,计算综合育种值。
进一步的,所述评估花鲈生长和抗性性状综合育种值的方法应用于花鲈育种。
与现有技术相比,本发明的有益效果是:
本发明提供的一种评估花鲈生长和抗性性状综合育种值的方法,建立了评估生长性状和抗性性状协同选育的综合育种值的方法,通过此方法对候选亲鱼的综合育种值进行评估可以挑选出兼具生长和抗性优势的核心育种群体,大大缩短育种年限,具有广阔的应用前景。
附图说明
图1为本发明实施例一种评估花鲈生长和抗性性状综合育种值的方法的步骤流程图;
图2为本发明实施例一种应用于花鲈体长和耐碱性状综合育种值评估的方法的步骤流程图;
图3为本发明实施例参考群体体长性状表型分布图;
图4为本发明实施例参考群体耐碱性状表型分布图;
图5为本发明实施例判断体长性状全基因组选择模型最佳标记的结果图;
图6为本发明实施例判断耐碱性状全基因组选择模型最佳标记的结果图;
图7为本发明实施例对参考群体体长性状和耐碱性状进行评估的结果图;
图8为本发明实施例对验证群体体长性状和耐碱性状进行评估的结果图。
具体实施方式
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合附图及实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
本发明实施例所用的实验方法如无特殊说明,均为常规方法。
本发明实施例所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
本发明实施例使用碳酸盐水体(26.16±0.5mmol/L),为碳酸氢钠和碳酸钠按照摩尔比16:5配制而成,试剂均为分析纯,厂家为国药集团化学试剂有限公司。
以下结合具体实施例对本发明的具体实现进行详细描述。
如图1所示,为本发明一个实施例提供的一种评估花鲈生长和抗性性状综合育种值的方法,包括以下步骤:
S100,测量参考群体的生长性状和抗性性状。
在本实施例中,首先建立参考群体,然后对建立的参考群体进行生长性状和抗性性状的测量。具体测量流程如下:
先对参考群体进行暂养驯化,暂养期间正常投喂;而后选取体质健壮、摄食状况良好的个体,给予胁迫因子处理,以个体失去身体平衡,且1分钟无法恢复到正常游泳状态这一特征作为环境胁迫的死亡标准。记录其生长指标(体长、体重等)和死亡时间,分别作为生长和抗性性状的表型;之后取每尾鱼的胸鳍置于无水乙醇中,-20℃条件下保存,用于基因组DNA提取。
S200,对参考群体进行全基因组重测序及SNP分型。
在本实施例中,从上述保存的胸鳍中提取参考群体的DNA,并进行全基因组重测序,然后对全基因组重测序数据进行SNP(single nucleotide polymorphism,单核苷酸多态性)分型。SNP分型即将每个鱼类个体的SNP鉴定出来。
S300,针对生长性状和抗性性状进行全基因组关联分析,鉴定与生长性状和抗性性状相关的SNP位点。
在本实施例中,通过对生长性状和抗性性状进行全基因组关联分析,鉴定出与生长性状和抗性性状相关的SNP位点。
S400,基于不同的SNP标记数量建立全基因组选择模型。
在本实施例中,选择不同的标记密度,并在linux环境下生成SNP标记的基因型数据,在R语言环境下使用rrBLUP包中的rrBLUP分析方法建立全基因组选择模型。具体的,是通过使用R包rrBLUP的mixed.solve函数通过岭回归最佳线性无偏预测构建模型。
S500,基于全基因组选择模型计算参考群体和验证群体的基因组育种值。
在本实施例中,使用全基因组选择模型计算参考群体的基因组育种值(GEBV),以及使用全基因组选择模型计算验证群体的基因组育种值,可以多次重复计算验证群体的基因组育种值,在分别计算验证群体生长性状和抗性性状的基因组育种值(GEBV)时,验证群体的个体要保持一致。
S600,根据参考群体和验证群体的基因组育种值计算综合育种值。
在本实施例中,建立了评估生长性状和抗性性状协同选育的综合育种值的方法,通过此方法对候选亲鱼的综合育种值进行评估可以挑选出兼具生长和抗性优势的核心育种群体,大大缩短育种年限,具有广阔的应用前景。
在本实施例的一种情况中,所述方法还包括:
根据基因组育种值与实际表型的相关性评估全基因组选择模型的预测准确性,选取最佳全基因组选择模型;
计算参考群体和验证群体的基因组育种值时基于所述选取最佳全基因组选择模型。
在本实施例中,通过选取最佳全基因组选择模型来提高育种效果。
在本实施例的一种情况中,采用五折交叉验证评估全基因组选择模型的预测准确性。
五折交叉验证即将群体随机分为五份,以其中四份作为训练集,用于模型构建,剩余一份作为验证集,用于检验,验证集即当作验证群体。
在本实施例的一种情况中,所述对参考群体进行全基因组重测序的步骤包括:
提取参考群体中每个个体的总基因组DNA;
对所述总基因组DNA进行检测;
对检测合格的总基因组DNA进行随机打断,纯化筛选符合要求的DNA片段,并将DNA片段连接测序接头,再通过滚环扩增制备DNB,对制备的DNB进行测序。
在本实施例中,使用TIANamp Marine Animals DNA Kit试剂盒参照说明书提取参考群体DNA,获得每个个体的总基因组DNA;通过Biodropsis BD-1000核酸分析仪检测样本DNA的质量和浓度,1%琼脂糖凝胶电泳检测其完整性,置于-20℃保存备用;对检测合格的DNA样品随机打断,纯化筛选符合要求的DNA片段,并将DNA片段连接测序接头,再通过滚环扩增制备DNB(DNA纳米球),然后使用BGISEQ-500测序仪进行测序。
在本实施例的一种情况中,所述SNP分型的步骤包括:
对全基因组重测序数据进行质控和过滤;
将质控和过滤后的全基因组重测序数据比对至参考基因组数据,并消除全基因组重测序数据中PCR偏好性的影响;
对消除PCR偏好性后的全基因组重测序数据进行单核苷酸多态性检测,实现基因分型。
在本实施例中,使用FastQC v0.11.9对原始数据(Raw data)质控,经SOAPnuke软件过滤后获得序列(clean data);使用BWA v0.7.17将clean data比对至花鲈参考基因组,经Samtools v1.14软件生成排序的bam文件,然后利用Picard v2.27.1软件消除测序中PCR偏好性的影响;使用GATK4 v4.2.6.1软件获得每个样本的g.vcf文件,再合并全部样本的g.vcf,获得全部测序个体的总vcf文件。经GATK基础过滤后,得到符合基本条件的SNP。
在本实施例的一种情况中,所述针对生长性状和抗性性状进行全基因组关联分析的步骤包括:
对全基因组重测序数据进行过滤;
填充测序缺失的SNP;
分别对生长性状和抗性性状进行全基因组关联分析。
在本实施例中,使用Plink v1.90软件对基因型数据进行了进一步的过滤;利用Begale v4.12软件填充部分测序缺失的SNP,获得高质量的基因型数据;使用GEMMAv0.98.3软件分别对生长性状和耐碱性状进行全基因组关联分析。
在本实施例的一种情况中,所述根据参考群体和验证群体的基因组育种值计算综合育种值的步骤包括:
对参考群体和验证群体的基因组育种值进行归一化处理;
在归一化处理结果中分别赋予生长性状和抗性性状相应的权重,计算综合育种值。
在本实施例中,采用离差标准化对生长性状和抗性性状的基因组育种值(GEBV)进行归一化;赋予生长性状和抗性性状不同的权重,计算综合育种值;计算参考群体的综合育种值与生长性状和抗性性状表型的相关性,以此评估本发明对鱼类生长性状和抗性性状预测的准确性;计算在10次随机抽样下,验证群体的综合育种值与生长性状和抗性性状表型的相关性,以此评估本发明对鱼类生长性状和抗性性状预测的准确性。
如图2所示,在一个实施例中,将所述评估花鲈生长和抗性性状综合育种值的方法应用于花鲈体长和耐碱性状育种。
目前我国花鲈种业体系正面临着空前挑战:缺乏优良花鲈品种,尚未建立科学育种体系,长期近亲繁殖导致种质退化现象已经显现,良种化进程大大落后于其他主要海水养殖鱼类。因此急需针对花鲈养殖产业关注的经济性状,开展遗传选育工作。育种的首要任务是提高花鲈生长率,培育快速生长品种,可缩短养殖周期和成本,有效提高经济效益;此外,花鲈属于广盐性鱼类,还能在一定范围内的盐碱水域中正常生长,可以针对不同地区的水质条件进行耐盐碱品种的选育,选育后可适应中等盐碱水域养殖。因此,通过遗传选育提高花鲈的生长速度以及耐盐碱能力对于花鲈养殖业的可持续发展和国内盐碱水域的高效利用均具有重要意义。
所述评估花鲈体长和耐碱性状综合育种值的方法包括以下步骤:
S10,建立参考群体,测量参考群体的体长性状和耐碱性状。
在本实施例中,2021年11月共获取黄渤海野生一龄花鲈500余尾,保证遗传多样性,鱼的体重规格为73.62±37.99g,体长规格为18.50±2.69cm。在室内水泥池经淡水驯化暂养两周,以用于碱度耐受性能的评估。暂养期间正常投喂。
选取327尾体质健壮、摄食状况良好的花鲈,转移至装有碳酸盐水体(26.16±0.5mmol/L)的养殖池中,进行72h碱度胁迫实验,以评估每尾花鲈的碱度耐受能力。自碱度胁迫开始,开始不间断地跟踪每尾鱼的健康状况。以1分钟无法恢复到正常游泳状态这一特征作为花鲈碱度胁迫的死亡标准,记录死亡时间作为耐碱性状指标,并将其捞出实验桶外,同时记录其体长作为生长性状指标,取胸鳍保存于无水乙醇中。如图3和图4所示,是参考群体的体长性状和耐碱性状表型分布图。
S20,对参考群体进行全基因组重测序及SNP分型。
在本实施例中,为消除因操作和个体健康状况而导致的死亡,最终选择301个体用于建库测序,使用TIANamp Marine Animals DNA Kit试剂盒参照说明书提取花鲈鳍条DNA,获得每个个体的总基因组DNA;DNA样品经琼脂糖凝胶电泳和Qubit检验其完整性和质量后,用于建库测序;对检测合格的DNA样品随机打断,纯化筛选符合要求的DNA片段,并将DNA片段连接测序接头,再通过滚环扩增制备DNB,然后使用BGISEQ-500测序仪进行测序获得原始数据(Raw data)。
在本实施例中,使用FastQC v0.11.9软件对原始数据(Raw data)进行质控,之后经SOAPnuke软件过滤后获得序列(clean data);使用BWA v0.7.17软件,建立花鲈参考基因组索引(index),并将过滤后的clean data比对到花鲈参考基因组上;使用Samtools v1.14软件将生成的sam文件转换并排序成bam文件;然后利用Picard v2.27.1软件的MarDuplicate模块标记文库构建过程中因PCR反应出现扩增偏差而产生偏好性的重复reads,降低这些重复序列对基因分型准确性的影响;使用GATK4 v4.2.6.1软件进行单核苷酸多态性的检测,利用其中的HaplotypeCaller模块进行基因分型,首先生成各样本的gvcf文件,再利用CombineGVCFs将所需样本进行合并以生成全部样本vcf文件,用于后续分析。为了保证SNP准确性,再通过GATK4软件的VariantFiltration模块进行初步过滤。
在本实施例中,这部分工作主要在linux系统下完成,以下是这部分工作的具体命令参数:
##数据过滤
a)Fastqc-t 12$i.fq($i为每个样本名称)#该步骤是数据质控,检查各样本原始数据质量;
b)SOAPnuke-1$i_1.fq-2$i_1.fq-T 4-n 0.1-l 5-q 0.5-Q 2-G-5 1-o$i_c1.fq-D$i_c2.fq#该步骤是数据过滤,获得各样本clean data。
##数据比对与格式转换
a)bwa-t 8-M genome$i_c1.fq$i_c2.fq|samtools view-bS->$i.bam#该步骤是将clean data比对至参考基因组,并转化为二进制bam文件;
b)samtools sort-@12-m 1G$i.bam-o$i_sort.bam#该步骤是对bam文件进行排序;
c)picard MarkDuplicates I=$i_sort.bam O=$i_picard.bam M=rm.bamREMOVE_DUPLICATES=ture#该步骤是去除PCR反应的扩增偏差。
##鉴定与过滤SNP
a)picard AddOrReplaceReadGroups I=$i_picard.bam O=$i.bamSO=coordinate ID=$i LB=$i PL=Illumina PU=run SM=$i#该步骤是为各样本加入标签,用于样本索引建立;
b)samtools index$i.bam#该步骤是建立样本索引;
c)samtools faidx genome#该步骤是建立参考基因组索引;
d)picard CreateSequenceDictionary R=genome.fa O=genome.Dict;
e)gatk HaplotypeCaller-R genome.fa-I$i.bam-ERC GVCF-O$i.g.vcf--genotyping-mode DISCOVERY--pcr-indel-model CONSERVATIVE--sample-ploidy 2--min-base-quality-score 10--kmer-size 10--kmer-size 25#该步骤是获得每个样本的g.vcf,包含每个样本的snp;
f)gatk CombineGVCFs-R genome.fa-O all.raw.vcf-V$i.g.vcf#该步骤是合并每个样本的g.vcf文件,生成vcf文件,包含全部样本的snp;
g)gatk VariantFiltration-R genome.fa-O all.filter.vcf-V all.raw.vcf--filter-name FilterQual--filter-expression"QUAL<30.0"--filter-name FilterQD--filter-expression"QD<13.0"--filter-name FilterMQ--filter-expression"MQ<20.0"--filter-name FilterFS--filter-expression"FS>20.0"--filter-nameFilterMQRankSum--filter-expression"MQRankSum<-3.0"--filter-nameFilterReadPosRankSum--filter-expression"FilterReadPosRankSum<-3.0"#该步骤是对鉴定的SNP进行初步筛选。
S30,针对体长性状和耐碱性状进行全基因组关联分析,鉴定与体长性状和耐碱性状相关的SNP位点。
在本实施例中,使用Plink v1.90软件对基因分型数据进行了进一步的过滤。主要的过滤参数为:最小等位基因频率(minor allele frequencies,MAF)大于0.05;基因型缺失率(missing rate)小于0.05;个体缺失率(missing rate)小于0.02;和不符合哈代温伯格定律(Hardy-Weinberg Law)(p<0.05);使用Beagle v5.4软件对过滤后的基因型数据进行填充,以预测测序丢失的部分SNP基因型,最终得到301个个体以及4660345个SNP位点;基于参考群体的体长和存活时间以及筛选后的SNP分型数据分别进行体长性状和耐碱性状的全基因组关联分析,所使用软件为linux环境下的GEMMA v0.98.3软件,以群体结构(PCA)作为固定效应,亲缘关系作为随机效应加入到混合线性模型的分析中,得到所有SNP位点的显著性P值,并将其由小到大排序。
在本实施例中,这部分工作主要在linux系统下完成,以下是这部分工作的具体命令参数:
##使用Plink v1.90软件对基因分型数据进行过滤
plink--vcf all.filter.vcf--maf 0.05--geno 0.05--mind 0.02–hwe 0.05--recode vcf-iid--out gwas.filtered;
##使用Beagle v5.4软件对过滤后的基因型数据进行填充
java-Xmx300m-jar beagle.28Jun21.220.jar gt=gwas.filtered.vcf out=gwas.imputed.vcf ne=301;
##使用Plink v1.90将VCF文件转换为Plink格式文件(ped,map)
plink--vcf gwas.imputed.vcf--make-bed--out gwas.imputed;
##使用shell脚本将性状的表型值加入到ped文件中
sh phe.sh#该步骤是执行脚本;
phe.sh内容如下:
awk'{print$1,$2,$3,$4,$5}'gwas.imputed.ped>ID
awk'{$1=null;$2=null;$3=null;$4=null;$5=null;$6=null;print$0}'gwas.imputed.ped>new.ped
paste-d""ID tl>tl.phe
paste-d""ID time>time.phe
paste-d""tl.phe new.ped>gwas.tl.ped
paste-d""time.phe new.ped>gwas.time.ped
cp gwas.imputed.map>gwas.tl.map
cp gwas.imputed.map>gwas.time.map;
##使用Plink v1.90软件对基因型数据进行PCA分析
plink--file gwas.imputed--pca 10--out pca;
##使用PCA结果的前三个主成分作为协变量
awk'{print$1,$2,$3,$4}'pca.eigenvec>c.txt;
##使用GEMMA v0.98.3软件计算亲缘关系G矩阵
plink--file gwas.imputed--make-bed--out gwas.imputed#该步骤是生成二进制文件;
gemma-0.98.1-linux-static-bfile gwas-gk 2-o kinship#该步骤是计算亲缘关系;
##使用GEMMA v0.98.3软件进行GWAS关联分析
plink--file gwas.tl--make-bed--out gwas.tl
plink--file gwas.time--make-bed--out gwas.time#该步骤是将文件转换为二进制文件;
gemma-0.98.1-linux-static-bfile gwas.tl-lmm-k./output/kinship.sXX.txt
-c c.txt-o gwas.tl#该步骤是体长性状的GWAS分析;
gemma-0.98.1-linux-static-bfile gwas.time-lmm-k
./output/kinship.sXX.txt-c c.txt-o gwas.time#该步骤是耐碱性状的GWAS分析;
##根据SNP位点的P值将其由小到大排序
awk'{print$2,$12}'gwas.tl.gemma.assoc.txt>sed-i'1d'>sort-k2-g>awk'{print$1}'>tl.sorted#体长性状;
awk'{print$2,$12}'gwas.time.gemma.assoc.txt>sed-i'1d'>sort-k2-g>awk'{print$1}'>time.sorted#耐碱性状。
S40,基于不同的SNP标记数量建立全基因组选择模型,根据基因组育种值与实际表型的相关性评估全基因组选择模型的预测准确性,选取最佳全基因组选择模型。
在本实施例中,按照已经排序好的SNP标记,分别选择50、100、150、200、400、800、1000、1200、1400、1600、3200、6400和12800个SNP标记作为选择标记,并在linux环境下生成SNP标记的基因型数据;在R语言环境下使用rrBLUP包中的rrBLUP分析方法建立全基因组选择模型,并利用五折交叉验证计算不同标记量下模型的预测准确性,五折交叉验证即将参考群体随机分为5份,其中4份为训练集,1份为测试集,利用训练集建立的模型来预测测试集的GEBV,预测准确性即为测试集的GEBV与实际表型值的相关系数的平方。结果表明:体长性状的预测准确性在1000个标记下已基本达到最高。耐碱性状的预测准确性在1400个标记下已基本达到最高;当预测准确性达到最高时,此时对应标记密度下建立的模型即为最佳的全基因组选择模型。如图5所示,是判断体长性状全基因组选择模型最佳标记的结果图,横坐标表示标记数量,左边的图中纵坐标代表模型拟合度,右边的图中纵坐标表示基因组育种值(GEBV)预测准确性。如图6所示,是判断耐碱性状全基因组选择模型最佳标记的结果图,横坐标表示标记数量,左边的图中纵坐标代表模型拟合度,右边的图中纵坐标表示基因组育种值(GEBV)预测准确性。
这部分工作主要在linux系统和R语言环境下完成,以下是这部分工作的具体命令参数:
##根据选择的SNP位点制作基因型数据(以体长性状示范)
head-n N tl.sorted>N.ID#该步骤是选取N个标记数量;
plink--bfile gwas.tl--extract N.ID--make-bed–out N#该步骤是获得N个标记的基因型二进制数据;
plink--bfile N--recodeA--out test#该步骤是将二进制数据转换为012型基因型数据;
head test.raw|cut-d""-f 1-9
cat test.raw|cut-d""-f1,7-|sed's/_[A-Z]//g'>genotype.txt#该步骤是转换为rrBLUP包可用的基因型数据;
##利用rrBLUP包构建全基因组选择模型并计算预测准确度
myGD<-read.table("genotype.txt",sep="",head=T)#该步骤是读入基因型数据;
myY<-read.table("P.txt",head=T)#该步骤是读入表型数据;
traits=1#该步骤是设定性状数为1;
cycles=30#该步骤是循环30次;
GEBV.accuracy=matrix(nrow=cycles,ncol=traits)#该步骤是构建GEBV数据矩阵;
for(r in 1:cycles){
n=287
testing=sample(n,round(n/5),replace=F)
training=-testing#该步骤是训练集和验证集的划分;
myGD_mat<-myGD[,-1]#删除第一列;
myGD_train<-as.matrix(myGD_mat[training,])#该步骤是训练集基因型数据的矩阵转换;
myGD_test<-as.matrix(myGD_mat[testing,])#该步骤是验证集基因型数据的矩阵转换;
myY_train<-myY[training,2]#该步骤是读入训练集的表型数据;
myY_test<-myY[testing,2]#该步骤是读入验证集的表型数据;
rrBLUP_model<-mixed.solve(y=myY_train,Z=myGD_train)#该步骤是构建模型;
pred_effects_testing<-myGD_test%*%rrBLUP_model$u#该步骤是计算育种值;
GEBV<-c(pred_effects_testing)
GEBV<-as.data.frame(GEBV)#该步骤是将GEBV转换为数据框格式;
rownames(GEBV)<-myY[testing,1]#该步骤是赋予验证集GEBV行名;
GEBV.accuracy[r,1]<-cor(GEBV,myY_test)#该步骤是计算GEBV与表型数据的相关性;
}
mean(GEBV.accuracy)#该步骤是计算平均相关性;
std.error(GEBV.accuracy)#该步骤是计算相关性的标准差。
S50,基于最佳全基因组选择模型计算参考群体和验证鱼类群体的基因组育种值。
在本实施例中,分别使用体长性状和耐碱性状的最佳全基因组模型,计算参考群体体长性状和耐碱性状的GEBV;分别使用体长性状和耐碱性状的最佳全基因组模型,并利用五折交叉验证随机抽取验证群体,并计算其体长性状和耐碱性状的GEBV,重复10次;在分别计算验证群体体长性状和耐碱性状的GEBV时,验证群体的个体要保持一致。
这部分工作主要在R语言环境下完成,以下是这部分工作的具体命令参数:
##计算参考群体的基因组育种值(以体长性状示范)
myGM<-read.table("snp.txt",sep="",head=T)
myGD<-read.table("genotype.txt",sep="",head=T)
myY<-read.table("TL.txt",head=T)
myX<-read.table("cv",sep="\t",head=F)
myGD_mat<-myGD[,-1]
myGD_TOTAL<-as.matrix(myGD_mat[,])
myY_TOTAL<-myY[,2]
rrBLUP_model<-mixed.solve(y=myY_TOTAL,myGD_TOTAL)pred_effect<-myGD_TOTAL%*%rrBLUP_model$upred_effect<-as.data.frame(pred_effect)
rownames(pred_effect)<-myY[,1]
##计算验证群体的基因组育种值(以体长性状示范)
myGD<-read.table("genotype.txt",sep="",head=T)
myY<-read.table("P.txt",head=T)
traits=1
cycles=10
GEBV.accuracy=matrix(nrow=cycles,ncol=traits)
for(r in 1:cycles){
n=287
testing=sample(n,round(n/5),replace=F)
training=-testingmyGD_mat<-myGD[,-1]
myGD_train<-as.matrix(myGD_mat[training,])
myGD_test<-as.matrix(myGD_mat[testing,])
myY_train<-myY[training,2]
myY_test<-myY[testing,2]
rrBLUP_model<-mixed.solve(y=myY_train,Z=myGD_train)
pred_effects_testing<-myGD_test%*%rrBLUP_model$u
GEBV<-c(pred_effects_testing)
GEBV<-as.data.frame(GEBV)
rownames(GEBV)<-myY[testing,1]
}#具体参数与上述同理。
S60,根据参考群体和验证群体的基因组育种值计算综合育种值,并对其体长性状和耐碱性状进行评估。
在本实施例中,采用离差标准化对体长性状和耐碱性状的基因组育种值进行归一化。离差标准化公式:x=(x-min)/(max-min);赋予体长性状0.6的权重,耐碱性状0.4的权重,计算综合育种值;公式为:GEBV综合=0.6*GEBV体长+0.4*GEBV耐碱;计算参考群体的综合育种值与体长性状和耐碱性状表型的相关性,结果表明综合育种值与体长性状和耐碱性状表型的相关系数分别为0.70(P<0.05)、0.63(P<0.05),均显著相关;计算在10次随机抽样下,验证群体的综合育种值与体长性状和耐碱性状表型的相关性,结果表明综合育种值与体长性状和耐碱性状表型的相关系数在0.5-0.8之间(P<0.05),表明本发明的对于体长性状和耐碱性状表型预测的准确性。如图7所示,是对参考群体体长性状和耐碱性状进行评估的结果图,横坐标表示个体的综合育种值,纵坐标代表个体的表型值。如图8所示,对验证群体体长性状和耐碱性状进行评估的结果图,横坐标表示循环次数,纵坐标代表验证鱼类群体的综合育种值和表型值的相关系数。
本实施例使用全基因组选择育种技术(GS)评估了花鲈体长性状和耐碱性状的综合育种值,理论上证明了GS在花鲈多性状协同改良中的可行性。包括:建立参考群体,并对参考群体进行体长性状和耐碱性状的表型测量;构建DNA文库进行全基因组重测序,并进行基因分型;对参考群体的体长性状和耐碱性状进行全基因组关联分析,分别鉴定出与体长性状和耐碱性状相关的SNP位点;选择不同的标记密度并以rrBLUP分析方法建立全基因组选择模型,通过基因组育种值(GEBV)与实际表型的相关性评估模型的预测准确性,选择最佳的全基因组选择模型;在最佳模型下,计算参考群体和验证群体体长性状和耐碱性状的GEBV,并对GEBV进行归一化,同时赋予体长性状0.6的权重,耐碱性状0.4的权重,计算综合育种值。在参考群体中,综合育种值与体长性状和耐碱性状的表型相关性分别为0.70(P<0.05)、0.63(P<0.05);在验证群体中,综合育种值与体长性状和耐碱性状的表型相关性在0.5-0.8之间(P<0.05)。结果表明本发明对于花鲈体长性状和耐碱性状的协同改良是可行的。通过此方法对亲鱼的综合育种值进行评估可以挑选出兼具体长和耐盐碱潜力的选育群体,大大缩短育种年限,同时也为其他鱼类的多性状协同改良供借鉴和基础,具有广阔的应用前景。
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
- 一种评估香猪表型性状的个体基因组育种值方法
- 一种基于SNP芯片的阈性状基因组育种值估计方法及应用