一种用于DTA预测的多模态信息融合模型及方法
文献发布时间:2024-04-18 19:53:33
技术领域
本发明涉及药物靶标结合亲和力预测技术领域,尤其指一种用于DTA预测的多模态信息融合模型及方法。
背景技术
药物发现是发现潜在新型药物的过程,涉及了药理学、化学、生物学等多种领域,通常需要耗费巨大的经济成本与时间成本。据统计,开发一种新药需要花费约26亿美元,而得到FDA的批准需要17年时间。多年来,随着计算机技术的发展,计算机辅助药物发现已成为一种趋势,所以迫切地需要开发出一种计算模型推进药物发现的进程。其中,成功识别药物-靶标相互作用是药物发现的关键步骤,而能进一步准确识别药物-靶标相互作用关系的亲和力对药物研发则更为重要。DTA代表了药物分子与靶标结合的强弱关系,一般来说,化合物分子与靶标结合越强,该化合物就越有可能影响靶标的生物学功能,也更有可能是一种合适的候选药物。因此,建立计算模型准确预测DTA可以加速药物分子的筛选过程,最大限度地减少不必要的体外筛选实验,对药物研发具有重要的意义。
目前已经提出了许多用于DTA预测的计算方法和模型,例如:传统的分子对接技术,其基于目标和化合物分子的3D结构,通过计算机模拟预测药物和靶标的结合模式和结合亲和力。许多成熟的分子对接算法是作为软件开发的,例如Gold和Dock,这些分子对接技术非常耗时。随着计算机技术的发展,出现了分子动力学模拟技术,如Elanie等人将快速几何对接算法与分子力学相互作用能量评估相结合,计算每个配体原子的潜力进行评分,更加灵活,预测结果更加准确,但代价是昂贵的计算和时间成本。
大多数早期的机器学习方法是基于通过结构相似性计算进行预测的矩阵计算,这大大降低了成本。例如,He等人提出了一种称为SimBoost的方法,该方法预测化合物和蛋白质结合亲和力的连续值。Li等提出了一种基于随机森林的分子对接方法,该方法通过应用Kronecker相似矩阵乘积进行预测。然而,这些方法过分依赖于分子的结构数据特征,并且获取这些数据既困难又费时。随着深度学习和大数据时代的飞速发展,卷积神经网络(CNN)、图神经网络(GNNs)以及它们的变体被应用于药物发现领域。由于药物和靶标的结构信息在DTA预测中起着极为关键的作用,因此现有的DTA预测方法大多基于药物和靶标的结构信息,它们可以分为基于字符串模态和基于图模态的方法。
基于字符串模态的方法是从序列数据中学习特征。例如,DeepDTA使用CNN对靶标序列和药物SMILES的一维表示进行特征提取。WideDTA在此基础上计算补充了蛋白质结构域、基序和最大共同亚结构词信息,并引入了一种基于词的序列表示法来进行DTA预测。相比之下,AttentionDTA则更加关注药物和靶标序列中重要的关键子序列,并引入了了一种双侧多头注意机制,以预测DTA。这些方法都只关注了药物SMILES和靶标信息的字符串模态,并且这种模态的信息忽略了空间结构以及氢原子信息。此外,在嵌入过程中只考虑了字符串的固定长度,这将导致一些有用信息的丢失。为了解决这一弊端,基于图模态的方法应运而生。GraphDTA提出将药物分子结构信息表示为图,并使用GNNs对药物分子图进行特征提取,使用CNN对靶标序列进行特征提取。DGraphDTA利用药物分子图和靶结构图进行DTA预测,通过图形卷积神经网络模型(GCN)进行特征提取。然而,药物分子图又缺失了字符串的上下文语义信息和原子的位置排列。并且该方法中靶标结构图只考虑了靶标的空间结构,而没有考虑靶标残基的排列顺序,忽略了肽链残基的位置信息。因此,有必要系统地考虑药物和靶标结构的多模态信息,以获得更好地预测DTA的完整信息。
多模态技术可以系统地考虑来自多种不同模态的信息。在过去的十年中,信息融合技术已经成功地实现了多模态信息的融合,多模态信息的利用引起了研究者们的注意。例如:Tuan等人提出了一种通过融合来自文本和视觉数据的多模态特征来检测假新闻的新方法。Mou等人提出了一种深度学习模型来融合多种模态的数据,包括眼睛数据、车辆数据和环境数据。由此可见,多模态信息融合技术已广泛应用于各个领域。同样,多模态信息的融合利用也可以应用在药物发现领域。例如:Deng等人开发了一个基于多模态变分图嵌入的方法Graph2MDA,其融合了微生物、药物的多方面属性和特征,以预测微生物-药物关联。Lyu等人考虑了药物与靶标、酶等多模态数据之间的潜在相关性,并设计了一个MDNN双通道框架来获取药物的多模态表征。可以看出,这些药物发现方法使用了以不同模态表达的药物不同属性的嵌入式表示,没有同时关注某一属性的多种模态信息。而且现有的DTA方法只考虑了药物和靶标的单一结构性质,没有考虑它们不同模式的多重属性信息。
发明内容
本发明所要解决的技术问题是提供一种用于DTA预测的多模态信息融合模型及方法,该模型可以嵌入药物和靶标中的字符串和图模态信息,并通过对比学习方法平衡不同模态的特征表示,以输出更丰富的用于DTA预测的信息。
为了解决上述技术问题,本发明采用如下技术方法:一种用于DTA预测的多模态信息融合模型,包括:药物分子结构信息编码器、靶标结构信息编码器、多模态平衡模块和药物靶标融合模块;
所述药物分子结构信息编码器使用Transformer模型对药物字符串模态信息进行编码,并使用GIN模型提取药物图模态信息特征;
所述靶标结构信息编码器使用Transformer模型对靶标字符串模态信息进行编码,并使用GCN模型提取药物图模态信息特征;
所述多模态平衡模块使用对比学习的方法将药物字符串和图模态信息进行平衡与整合,以及将靶标字符串和图模态信息进行平衡与整合;
所述药物靶标融合模块将多模态平衡模块得到的药物和靶标的两种模态特征连接起来,用于DTA预测。
作为本发明的另一面,一种用于DTA预测的多模态信息融合方法,包括:
步骤S1,字符串模态的嵌入;
将药物SMILES代码视为字符串,对其进行整数编码,融入该编码的位置编码得到向量表示,通过Transformer模型对该向量进行特征提取得到SMILES字符串的最终向量表示;
将靶标序列视为字符串,对其进行整数编码,融入该编码的位置编码得到向量表示,通过Transformer模型对该向量进行特征提取得到靶标字符串的最终向量表示;
步骤S2,图模态的嵌入;
将每个原子作为药物分子图中的节点,原子间的联系作为药物分子图的邻接矩阵,并将原子的属性作为药物分子图节点的属性特征;将药物分子图和其节点的特征向量作为输入,通过GIN模型进行节点嵌入,得到药物分子图的表示向量;
将每个残基作为靶标结构图中的节点,残基对间是否接触的概率作为靶标结构图的邻接矩阵,并将每个残基位置通过序列比对结果进行评分,作为靶标结构图节点的属性特征;将靶标结构图和其节点的特征向量作为输入,通过GCN模型进行节点嵌入,得到靶标结构图的表示向量;
步骤S3,多模态表示的对比学习和表示的融合;
通过最大化字符串模态和图模态的一致性来学习特征表示,分别得到药物和靶标的两种模态最终的表示之后,将其进行拼接,得到用于DTA预测的药物和靶标模态信息。
进一步地,步骤S1中,在对药物和靶标字符串进行整数编码之后,利用药物原子和靶标残基的排列信息来捕获字符串模态的位置信息,并通过Transformer模型从输入中学习不同级别的抽象特征,再应用最大池化层来获得药物和靶标字符串的最终向量表示。
进一步地,步骤S1中,采用如下公式表示字符串模态的位置信息:
PE
PE
其中,pos代表字符串中的字符,i是字符编码的维度,dmodel是该字符的编码。
再进一步地,所述Transformer模型包括一个MSA层和一个MLP块,所述MSA层的函数表示为:
z
其中,z
所述MLP块包含两个CNN层和一个归一化层,其函数表示为:
z
其中,z
再进一步地,所述GIN模型包括五个GIN层,每个GIN层后面均有一个批次归一化层,最后一个批次归一化层后连接一个全局最大池层,每个GIN层均通过多层感知器模型将节点特征x
其中,k代表GIN层,∈是可学习参数或固定标量,N(i)是i节点的邻居集合。
更进一步地,所述GCN模型包括三个GCN层,每个GCN层均由RELU函数激活,最后一GCN层后连接一个全局最大池层,每个GCN层均进行一次卷积操作,如下式:
其中,
优选地,步骤S3中,通过最大化字符串模态和图模态的一致性来学习特征表示时,将任意一个药物或靶标自身固定为锚点A,得到一组由该药物或靶标自身的多种模态组成的正样本P,将其它药物或靶标的所有模态的表示均视为负样本N,生成每个正对(A,P)和负对(A,N),采用对比学习强制锚点A的所有模态表示一致,并且与负样本的所有模态表示区别开来,其中,Loss的计算如下所示:
其中,对于每个样本i,P(i)是其正样本集合,|P(i)|是正样本个数,p是其中一个正样本,N(i)是负样本集合,n是其中一个负样本,T是温度系数。
相较于传统模型,本发明提供的多模态信息融合模型及方法具有更优异的特征捕获能力,能提供更丰富、完整的模态信息,以用于DTA预测。具体而言,本发明充分考虑了药物和靶标中的字符串和图模态信息,其中,字符串模态包括药物SMILES和靶标序列,图模态包括药物分子图和靶标结构图信息,选择不同的深度学习模型来提取不同模态的数据特征,并通过对比学习方法平衡不同模态的特征表示,还利用药物原子和靶标残基的排列信息来捕获串模态的位置信息,这均可以在SMILES和靶标序列中提取更多有用的特征信息,以有效提高DTA预测效果。
附图说明
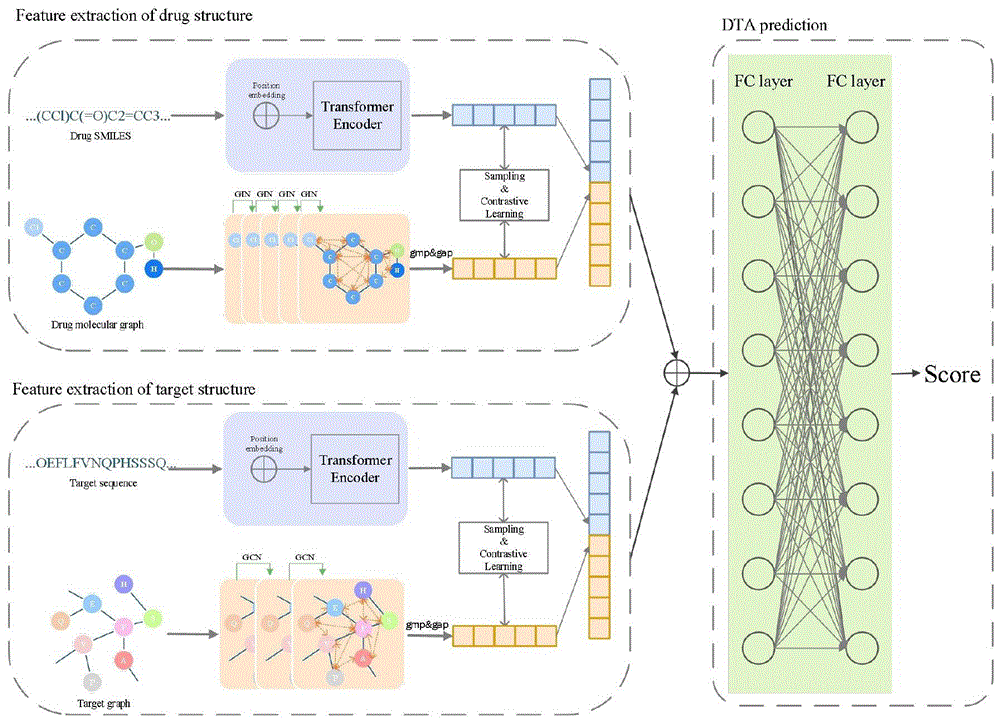
图1为本发明所涉多模态信息融合模型应用于DTA预测时的流程图;
图2为本发明所涉多模态信息融合方法中对比学习框架图;
图3为本发明评估实验中各模型在Davis数据集上的MSE和CI指标对比图;
图4为本发明评估实验中各模型在KIBA数据集上的MSE和CI指标对比图。
具体实施方式
为了便于本领域技术人员的理解,下面结合实施例与附图对本发明作进一步的说明,实施方式提及的内容并非对本发明的限定。
传统的基于生物实验的药物-靶标亲和力预测方法无法满足大数据时代药物R&D的需求,虽然基于深度学习的药物-靶标结合亲和力预测任务模型取得了重大成功,但是,这些模型仅仅考虑了药物和靶点信息的单一模态特征,信息不够完整和丰富。事实上,药物和靶标不同模态的信息可以互补,融合不同模态的信息可以获得更有价值的信息。基于此,本发明设计一种称为FMDTA的用于DTA预测的多模态信息融合模型,具体如下。
一、FMDTA
由于字符串模态和图模态的数据都有各自的优势和不足,所以本发明将两种模态的信息合并融合起来,以获取更完整的信息。FMDTA由四部分组成:药物分子结构信息编码器、靶标结构信息编码器、多模态平衡模块和药物靶标融合模块。其中:
药物分子结构信息编码器使用Transformer模型对药物字符串模态信息进行编码,并使用GIN模型提取药物图模态信息特征。
靶标结构信息编码器使用Transformer模型对靶标字符串模态信息进行编码,并使用GCN模型提取药物图模态信息特征。
多模态平衡模块使用对比学习的方法将药物字符串和图模态信息进行平衡与整合,以及将靶标字符串和图模态信息进行平衡与整合。
药物靶标融合模块将多模态平衡模块得到的药物和靶标的两种模态特征连接起来,用于DTA预测。
二、FMDTA的工作流程如图1所示,也即本发明提供的多模态信息融合方法,其主要包括三大步骤,分别为:
步骤S1,字符串模态的嵌入
S11,药物SMILES的嵌入
将每个药物SMILES代码视为字符串,通过整数编码,用整数作为字符来编码输入。此处,值得注意的是,模型在实际应用之前,需要进行训练,为了方便本发明提供的FMDAT进行训练,宜将每个SMILES字符串剪切或填充为100个字符的固定长度,这些整数序列用作返回128维向量表示的嵌入层的输入。使用Transformer模型从输入中学习到不同级别的抽象特征。
考虑到SMILES字符串的排列顺序对于药物分子的特征表达十分关键,因此本发明融入SMILES的位置信息,采用以下公式表示位置编码:
PE
PE
其中,pos代表SMILE字符串中的字符,i是字符编码的维度,dmodel是该字符的编码。
在得到位置编码之后,将药物SMLIES编码信息与位置编码相加得到完整的编码信息,再通过Transformer模型从输入中学习不同级别的抽象特征。
对于Transformer模型,本发明遵循原始的Transformer设计,它由一个多头注意力(MSA)层和一个MLP块组成。所述MSA层的函数表示为:
z
其中,z
在MSA层之后,MLP块包含两个CNN层和一个线性归一化层,其函数表示为:
z
其中,z
最后,应用最大池化层来获得药物字符串的最终向量表示。
S12,靶标序列的嵌入
将每个靶标序列视为字符串,将字符通过整数编码之后作为输入。同理,为了方便训练,将每个靶标序列剪切或填充为1000个残基的固定长度,这些整数序列用作返回128维向量表示的嵌入层的输入。同样的,考虑到靶标残基的排列顺序对于靶标的结构特征表达十分关键,通过位置信息编码后,再用Transformer模型从输入中学习到不同级别的抽象特征,再应用最大池化层来获得靶标字符串的最终向量表示。
步骤S2,图模态的嵌入
S21,药物分子图的嵌入
本发明中药物分子图数据来自GraphDTA[源于文献[15]Jiang,M.;Li,Z.;Zhang,S.;Wang,S.;Wang,X.;Yuan,Q.;Wei,Z.Drug–target affinity prediction using graphneural network and contact maps.RSC Advances 2020,10,20701–20712.]。本发明将每个原子作为药物分子图中的节点,将原子间的联系作为药物分子图的邻接矩阵。同时,将原子的相关属性作为药物分子图节点的属性特征。将药物分子图和其节点的特征向量作为输入,通过GIN(Graph Isomorphic Networks)模型进行节点嵌入。
具体来说,GIN模型包括五个GIN层,每个GIN层均通过多层感知器(MLP)模型将节点特征x
其中,k代表GIN层,∈是可学习参数或固定标量,N(i)是i节点的邻居集合。
该GIN模型中,每个GIN层后面均有一个批次归一化层,最后,加入全局最大池层,得到药物分子图的表示向量。
S22,靶标结构图的嵌入
本发明中靶标结构图数据来自DGraphDTA[源于文献Jiang,M.;Li,Z.;Zhang,S.;Wang,S.;Wang,X.;Yuan,Q.;Wei,Z.Drug–targetaffinity prediction using graphneural network and contact maps.RSCAdvances 2020,10,20701–20712.],本发明将每个残基作为靶标结构图中的节点,残基对间是否接触的概率作为靶标图的邻接矩阵,如此很好地保留了蛋白质的空间信息。同时将每个残基位置通过序列比对结果进行评分,并作为残基节点的特征向量。接着将靶标结构图和其节点的特征向量作为输入,通过GCN模型进行节点嵌入。
具体来说,GCN模型包括三个GCN层,每个GCN层都会通过进行一次卷积操作,如下式:
其中,
该GCN模型每个GCN层均由RELU函数激活,最后一个GCN层之后添加一个全局最大池层,得到靶标结构图的表示向量。
S3,多模态表示的对比学习和表示的融合
在从不同模态的数据中提取特征的过程中,一方面,由于不同药物或靶点的内部结构可能包含多个相似官能团或残基的信息,经过编码后,不同药物/靶点之间的特征变得模糊和相似。另一方面,由于不同模态的嵌入方法存在明显差异,导致同一药物/靶标的两种模态的特征表示存在显著差异,同一模态的不同药物/靶标的特征表示存在微小差异。因此,需要选择合适的融合方法来平衡不同模态之间的信息嵌入,使多模态的信息能够互补。
为了平衡不同模态间的信息嵌入,本发明采用对比学习的方法捕捉模态间信息的相互作用关系。如图2所示,在FMDTA中,我们通过最大化字符串模态和图模态的一致性来学习特征表示。具体而言,例如,对于药物i,将其自身固定为锚点A,得到一组由它自身的多种模态组成的正样本P,其它的药物的所有模态的表示均视为负样本N,生成每个正对(A,P)和负对(A,N)。然后,采用对比学习,强制锚点A的所有模态表示一致,并且与负样本的所有模态表示区别开来。在此对比学习过程中,通过计算损失迭代更新药物(靶标)的嵌入表示,使同一个药物(靶标)两种模态的表示更接近,同一模态的不同药物(靶标)和不同模态的不同药物(靶标)的表示差异性更大,以平衡药物(靶标)的嵌入表示,得到最终的嵌入表示,其中,Loss的计算如下所示:
其中,对于每个样本i,P(i)是其正样本集合,|P(i)|是正样本个数,p是其中一个正样本,N(i)是负样本集合,n是其中负样本,T是温度系数。
靶标不同模态嵌入信息的对比学习同理。
在得到药物和靶标的两种模态最终的表示之后,将其进行拼接,得到用于DTA预测的药物和靶标模态信息,随后通过两个全连接层即可进行预测DTA。
三、为了验证本发明所涉模型及方法的可行性、优异性,接下来,进行验证实验。
3.1实验数据
在本实验评估中,使用来自DeepDTA[源于文献
Davis数据集包含72种化合物和442种蛋白质,以及它们对应的亲和力值,亲和力值通过Kd值(激酶解离常数)来衡量,范围在5.0-10.8。化合物的SMILES字符串的平均长度为64,靶标序列的平均长度为788。
KIBA数据集包含来自不同来源(例如Ki、Kd和IC50)的组合激酶抑制剂生物活性,含有2,116种药物和229个靶点的结合亲和力,以KIBA分数衡量,范围从0.0到17.2。化合物的SMILES字符串平均长度为58,靶标序列的平均长度为728。数据信息汇总在表1中。
表1数据集数据统计
3.2评价指标
本实验评估使用mean squared Error(MSE)、the concordance index(CI)和
MSE是回归任务常用的评价指标,通过计算样本预测值和样本实际值之间差的平方和的平均值来衡量样本p
CI是评价预测值的排序和它们之间的真实值的排序一致性的指标,该值越高,则说明模型的预测效果越好。CI的计算公式如下:
其中,b
指数用于评估QSAR(定量结构活动关系)模型的外部预测性能。公式如下所示:
其中r
为了进行训练,本实验评估中使用一台带有2个Intel(R)Xeon(R)Gold 52152.5GHz CPU、256GB RAM和3个NVIDIA Corporation GV100GL GPU的服务器。用于实验的超参数如表2所示。
表2超参数
3.3评估策略
本实验采用5折交叉验证来验证模型的性能。在每折中以3:1:1的比例将数据集随机分为训练集、验证集和测试集。最后在测试集上评估通过验证选择的模型。
3.4模型对比
在本实验中,将经典的DTA预测模型分为了两类并与它们进行性能比较。其中只考虑了字符串模态的方法是DeepDTA、WideDTA和AttentionDTA。这些模型均采用CNN模型对字符串进行编码。
DeepDTA:由两个独立的CNN模块组成,分别用于学习药物的SMILES和靶标Sequence信息的特征表示,并将表示输入到三个全连接层中进行DTA预测。
WideDTA:在DeepDTA的基础上引入protein domains and motifs和maximumcommon substructure words信息,更好的进行DTA预测。
AttentionDTA:引入两侧多头注意力机制来关注在预测其亲和力时对药物和蛋白质序列很重要的关键子序列。
考虑到了图模态的方法是GraphDTA、DGraphDTA、DeepGLSTM和SAG-DTA,这些模型均采用GNNs对图进行编码。
GraphDTA:引入了药物分子图,利用GNNs学习药物的分子图特征,用CNN学习靶标Sequence的特征表示,并将这两部分拼接通过全连接层预测DTA。
DGraphDTA:在GraphDTA的基础上引入了靶标结构图,利用GNNs学习药物和靶标的特征,并将这两部分拼接通过全连接层预测DTA。
SAG-DTA:在GraphDTA的基础上通过自注意机制改进了药物分子图特征表示以进行DTA预测。
DeepGLSTM:一种基于图卷积网络和LSTM的方法,分别对药物和靶标进行编码从而预测DTA。
表3DAVIS数据集上DTA预测模型的结果
表3给出了FMDTA和基线方法在Davis数据集上的表现。为了保持对比的公平性,对于所有模型都使用相同的超参数和评价指标。可以看出,FMDTA在三个指标上都明显优于单纯依赖于字符串模态或图模态的方法,MSE为0.195,CI为0.909,R
为了进一步测试所提出方法的泛化性,本实验在KIBA数据集上使用与Davis数据集中相同的超参数来评估模型。实验结果如表4所示,可以看出FMDTA仍然表现优异,具体来说,FMDTA的MSE为0.133,CI为0.899,R
表4KIBA数据集上DTA预测模型的结果
3.5消融研究
3.5.1多模态信息融合的有效性分析
将本发明模型与DeepDTA、GraphDTA和DGraphDTA在Davis数据集上进行对比。为了保证对比的公平性,本发明模型与对比模型的相同模块采用相同的参数。其中,FMDTA(w/oPC)代表药物和靶标各自的两种模态简单拼接融合,不包含字符串位置信息编码和对比学习。
通过图3可以看出,融合两种模态的信息优于仅仅使用单模态信息。药物和靶标的结构图信息的结合可以获取到更完整的结构信息和空间结构信息,弥补了DeepDTA单纯利用字符串模态的局限。药物SMILES和靶标Sequence的字符串模态信息可以获取到字符串字符的排列信息以及上下文语义信息,弥补了DGraphDTA单纯利用结构图信息的局限。GraphDTA利用的是药物分子结构图和靶标Sequence信息,同样只利用了单一模态的信息,信息不够完整。这说明两种模态信息可以相互补充,融合两种模态的信息可以有效提高模型在DTA上的预测能力。同时,可以看出GraphDTA的效果优于DeepDTA和DGraphDTA,说明药和靶蛋白信息的模态不同,效果更好。
为了进一步验证多模态信息融合对DTA任务的有效性,将FMDTA(w/o PC)与DeepDTA、GraphDTA和DGraphDTA在KIBA数据集上进行了对比。图4显示了FMDTA(w/o PC)与单模态模型在KIBA数据集上的性能表现。可以看出,多模态信息的融合在KIBA数据集上同样表现优异,说明该方法是合理且有效的。
3.5.2字符串位置信息编码和多模态对比学习的有效性分析
本部分旨在证明字符串模态的位置信息编码的重要性和多模态对比学习的重要性。如前面所述,SMILES中原子的排列顺序和靶标sequence残基得到排列顺序对分子特征表示至关重要。同时,最大化字符串模态和图模态的一致性来学习特征表示也至关重要,此处部署了一项全面的消融研究来探索每个单独模块的必要性,并在Davis和KIBA数据集上进行了对比。
FMDTA的三个变体是:
(1)FMDTA(w/o PC):没有字符串模态编码组件和多模态对比学习组件的位置信息的FMDTA,其直接拼接药物和目标蛋白的两个单独模态。
(2)FMDTA(w/o C):无多模态对比学习组件的FMDTA提取字符串模态的位置信息但不考虑模态特征之间的交互作用。
(3)FMDTA(w/o P):无字符串模态位置信息编码的FMDTA,通过对比学习组件平衡融合字符串模态和图形模态特征。
表5关于位置信息和对比学习的消融研究结果
通过表5的结果可以看出,在Davis数据集上,加入字符串模态的位置编码模块,MSE指标提升了0.5%,CI提升了0.7%。在KIBA数据集上MSE指标改善不明显,而CI改善了0.3%。在Davis数据集上添加多模态对比表征学习模块后,MSE度量和CI分别显著提高了2%和1%。在KIBA数据集上,MSE和CI分别显著提高了0.4%和0.7%。位置编码模块和对比度学习模块的添加导致了更显著的改善,在Davis和KIBA数据集上,MSE分别改善了2.8%和1.5%,CI分别改善了0.7%和1.2%。由此,可以得出一些结论,如下:
(1)加入字符串编码的位置信息可以捕捉到更完整的信息,提高模型性能。
(2)对比学习允许来自不同模态的信息在编码期间相互作用,导致来自不同模态的特征的更平衡的表示和更灵活的模型。
上述实施例为本发明较佳的实现方案,除此之外,本发明还可以其它方式实现,在不脱离本技术方案构思的前提下任何显而易见的替换均在本发明的保护范围之内。
为了让本领域普通技术人员更方便地理解本发明相对于现有技术的改进之处,本发明的一些附图和描述已经被简化,并且为了清楚起见,本申请文件还省略了一些其他元素,本领域普通技术人员应该意识到这些省略的元素也可构成本发明的内容。