一种基于一代测序技术的数据质控方法和微生物鉴定方法
文献发布时间:2024-04-18 19:53:33
技术领域
本发明属于生物信息领域,特别是涉及一种基于一代测序技术的数据质控方法和微生物鉴定方法。
背景技术
第一代测序技术,其原理为双脱氧终止法,即利用特定标记的双脱氧三磷酸核苷酸(ddNTP)终止DNA链的复制,通过读取四种碱基的位置信息获得测序结果。一代测序由于精度高(99.999%),读长长(1000bp左右),被誉为测序检验的金标准。其原始输出结果为.ab1格式文件,里面包含了上机及测序的完整信息,通过一些图形界面软件可查看测序峰图、并导出序列结果(txt、fastq或fasta格式),进而开展下一步分析。
目前来讲,微生物一代测序的目的主要是通过对其16S区域测序来进行菌种鉴定,但由于PCR技术的限制,测序结果中首位两端的碱基质量通常较差;加之微生物送样中极易有其他杂菌干扰,需要通过测序来进行质控,因此,获得一条高质量测序序列对于后续分析鉴定极为重要。
现有通用的质控方法大都是通过第三方软件得到ab1文件的峰图,进而根据峰图去判断测序质量。但是该方法存在两大不足,一是该方法依赖于人工去识别峰图,具有一定的主观性,没有定量统一的标准,会导致结果的一致性较差。且人工方式通量较低,对于大批量的测序结果,需要花费大量时间才能完成判定;二是现有的第三方图形界面工具如FinchTV、SnapGene、DNAstar等只是解析展示峰图,无法判定结果质量(是否有杂菌污染,测序是否成功等)并截取合格序列片段至输出文件(只能导出全部测序结果)。由于原始下机序列首尾包含了大量的低质量序列,直接比对会导致结果的误差较大,影响判定。
同时,也存在一些程序语言专门处理一代测序数据,如python语言中的BioPython模块等。这些程序的优势在于可以通过设置循环或者并行来批量处理ab1文件。但其缺点在于只能实现对ab1文件的解析及格式转化,同样无法实现对测序质量的判定及合格序列的截取。
除此之外,也有一些方法,如专利CN113345522A中利用的sangeranalyseR和sangerseqR包实现了对一代测序数据的质量及杂峰控制。但该方法也存在一些不足:
1、质量控制模块中,只对两端的碱基进行剪切,但是一代测序结果中部甚至任一区域都可能会出现质量较差的碱基,对于这些碱基的质控也至关重要。
2、杂峰控制模块中,只是对双峰进行鉴定,但是一代测序结果中还有其他杂峰,这些杂峰同样也是判断菌株和测序质量的重要标准,不能忽视。
3、测序数据的质量会直接影响最终结果,例如当用低质量序列与已知物种比对时,会得出错误的判断结论。
发明内容
为了解决上述问题,本发明首先提出了一种基于一代测序技术的数据质控方法,通过处理一代测序结果,提高了处理的速度,并能获得到高质量的筛选序列,提高了比对结果的置信度。
为达到上述目的,本发明采用的技术方案是:一种基于一代测序技术的数据质控方法,包括步骤:
S1. 获取一代测序结果的指定文件夹下格式为ab1的全部文件路径,并设置循环逐一处理;
S2. 读取单个ab1文件,获得测序质量信息,并根据阈值滑窗截取合格片段;
S3. 循环处理S2中的每个合格片段,剔除具有混杂信号的片段,并在剩余片段中选取长度最大的片段;
S4. 判定S3结果中长度最大的片段的长度是否符合质控阈值;若是则质控成功;
其中,若S2、S3或S4任一步骤质控失败,则测序结果质量不合格,流程终止。
在本发明的一个具体方案中,所述步骤S1中,通过python中的os.walk()函数获取一代测序结果的指定文件夹下格式为ab1的全部文件路径。
在本发明的一个具体方案中,步骤S2中,使用python中的Biopython模块的SeqIO.read()函数解析ab1文件,并获取测序质量及四种碱基在每个位点的信号值。
在本发明的一个具体方案中,所述根据阈值滑窗截取合格片段,包括:从第一个碱基开始以一个碱基为单位向后延伸,若当前片段的测序质量符合阈值,则继续向后延长,直至片段质量不符合阈值或到序列末端;同时从当前片段的后一个碱基开始,继续进行滑窗判定,直至到达序列末端。
在本发明的一个具体方案中,所述根据阈值滑窗截取合格片段通过4个参数与设定阈值比较以判断片段是否合格:最低片段质量、最低碱基质量、离群碱基数量和连续离群碱基数量。
在本发明的一个具体方案中,可选的,对于最低片段质量的阈值设定包括两种选择模式,第一种为严格模式(strict mode),即该片段所有碱基的测序质量都满足阈值;第二种为宽松模式(loose mode),即该片段碱基的平均质量满足最低片段阈值。
特别地列举本发明的一些阈值,如,若片段末端位点在50bp前或800bp后,若质量不符合阈值将会被直接舍弃。
最低碱基质量:若片段中最低碱基质量低于设定阈值,则该片段质控失败。
离群碱基数量:离群碱基是指碱基质量低于片段最低质量阈值的碱基数量。
连续离群碱基数量:连续离群碱基数量是指在该片段中存在的连续碱基位点,其质量值都低于设定阈值。其数目若大于设定阈值,则该片段质控失败。
在本发明的一个具体方案中,在步骤S3中所述的混杂信号指同一个位点存在多个碱基信号或有多个碱基信号都很高,导致该位点存在噪音,无法分辨主信号类型。这种情况可能是由于样本制备中存在杂菌污染或PCR中的实验出错等因素导致。该特征可通过“最大碱基信号值/信号总值”反映。特别地,若一个位点只有两个碱基信号或两个碱基的信号都很高,则将该位点识别为双峰位点,该特征通过“第二大碱基信号值/最大信号值”反映。
在本发明的一个具体方案中,采用4个参数控制混杂信号剔除,包括主峰比例、次峰比例、混杂信号位点数量和连续混杂信号的最大片段长度。
在本发明的一个具体方案中,所述S2、S3或S4任一步骤质控失败包括以下任意一种情形:步骤S2中未截取到合格片段;步骤S3中剔除了所有片段;或步骤S4中长度最大的片段长度不符合质控阈值。
可以理解的是,本发明的质控方法可以用于任何与基因测序和基因比对的方法中。示例性的应用场景包括微生物鉴定。
因此,作为本发明的第二方面,本发明还提供了一种微生物物种鉴定方法,包括将质控成功的序列片段与指定参考物种的序列比对,获得一致性比较结果,所述质控成功的序列片段是通过前述任意一项所述的基于一代测序技术的数据质控方法进行质控得到的。
进一步的是,所述微生物为细菌,所述将质控成功的序列片段与指定参考物种的序列比对是将质控成功的序列片段与指定序列进行blast基因序列比对,获得一致性结果。
采用本技术方案的有益效果:
本发明相比于人工查看峰图,本发明的质控方法可直接使用程序化脚本批量处理大量测序数据,并获得高质量的测序结果,大大提高了工作效率和结果质量。
本发明相比于python中的一代测序数据处理函数,在其基础上增加了对一代测序数据质量和信号的判定及截取等功能,从而可以检测到因样本本身或测序导致的异常结果,并获得合格序列的高质量结果。
本发明将质控后高质量的序列与目标序列进行比对,提高了比对结果的置信度。
附图说明
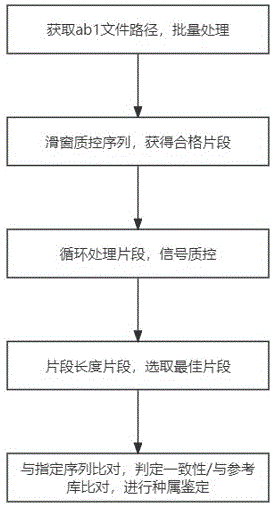
图1 为具体实例1采用本发明基于一代测序技术的数据质控方法进行微生物鉴定的流程示意图;
图2 为本发明具体实例1中合格序列的峰图文件示意图;
图3 为本发明具体实例1中不合格序列的峰图文件示意图;
图4 为本发明具体实例2对本发明方法测试结果与人工验证结果进行比较的测试结果(benchmark)示意图。
具体实施方式
为了使本发明的目的、技术方案和优点更加清楚,下面结合附图对本发明作进一步阐述。
一种基于一代测序技术的细菌批量质控方法,如图1所示,包括步骤:
S1. 获取一代测序结果的指定文件夹下格式为ab1的全部文件路径,并设置循环逐一处理;
S2. 读取单个ab1文件,获得测序质量信息,并根据阈值滑窗截取合格片段;
S3. 循环处理S2中的每个合格片段,剔除具有混杂信号的片段,并在剩余片段中选取长度最大的片段;
S4. 判定S3结果中长度最大的片段长度是否符合质控阈值;若是则质控成功;
若S2、S3或S4质控失败,则测序结果质量不合格,流程终止。
步骤S1获取一代测序结果的指定文件夹下格式为ab1的全部文件路径的方法不限。作为一种选择,通过python中的os.walk()函数获取指定路径下的全部ab1文件。
作为一种选择,在步骤S2中,使用python中的Biopython模块的SeqIO.read()函数解析ab1文件,并获取测序质量及四种碱基在每个位点的信号值。
采用滑窗法获取符合条件的序列片段,包括步骤:从第一个碱基开始以一个碱基为单位向后延伸,若当前片段的测序质量符合阈值,则继续向后延长,直至片段质量不符合阈值或到序列末端;同时从当前片段的后一个碱基开始,继续进行滑窗判定,直至到达序列末端。
作为一种选择,在所述步骤S2中,采用3个参数对测序质量进行控制,包括最低片段质量、最低碱基质量、离群碱基数量和连续离群碱基数量;
最低片段质量:对于片段测序质量的阈值设定可分两种模式,第一种为严格模式,必须片段所有碱基的测序质量都满足阈值;第二种为宽松模式,即片段碱基的平均质量满足最低片段阈值;
特别地列举本发明的一些阈值,如,若片段末端位点在50bp前或800bp后,若质量不符合阈值将会被直接舍弃。
最低碱基质量:若片段中最低碱基质量低于设定阈值,则该片段质控失败。
离群碱基数量:离群碱基是指碱基质量低于片段最低质量阈值的碱基数量。
连续离群碱基数量:连续离群碱基数量是指在该片段中存在的连续碱基位点,其质量值都低于设定阈值。其数目若大于设定阈值,则该片段质控失败。
在步骤S3中,混杂信号指同一个位点存在多个碱基信号或有多个碱基信号都很高,导致该位点存在噪音,无法分辨主信号类型,这种情况可能是由于样本制备中存在杂菌污染或PCR中的实验出错等因素导致。剔除具有混杂信号的片段时,混杂信号通过最大碱基信号值或信号总值反映;
若一个位点只有两个碱基信号或两个碱基的信号都很高,则将该位点识别为双峰位点,通过第二大碱基信号值或最大信号值反映。
作为一种选择,采用4个参数控制混杂信号剔除,包括主峰比例、次峰比例、混杂信号位点数量和连续混杂信号的最大片段长度;
主峰比例:为最大碱基信号值或信号总值,以此判定位点信号是否单一;
次峰比例:为第二大碱基信号值或最大信号值,判断是否有第二大信号干扰,进而进行杂菌污染鉴定;
混杂信号位点数量:若片段中杂峰位点数量或双峰位点数量超过指定阈值,则片段质控失败;
连续混杂信号的最大片段长度,连续混杂信号是指有连续的位点出现混杂信号,对连续位点的长度进行判定,若大于设定阈值,则片段质控失败。
在本发明的一个具体方案中,所述 S2、S3 或 S4 任一步骤质控失败包括以下任意一种情形:S2.未截取到合格片段;S3.剔除了所有片段;或 S4.长度最大的片段长度不符合质控阈值。
可以理解的是,本发明的质控方法可以用于任何与基因测序和基
因比对的方法中。示例性的应用场景包括微生物鉴定。 因此,作为本发明的第二方面,本发明还提供了一种微生物物种鉴定方法,包括将质控成功的序列片段与指定参考物种的序列比对,获得一致性比较结果,所述质控成功的序列片段是通过前述任意一项所述的基于一代测序技术的数据质控方法进行质控得到的。
在本发明的一个具体方案中,所述微生物选自细菌、古菌、真菌 和其他原核生物。在本发明的一个具体方案中,所述微生物为细菌。
所述将质控成功的片段与指定参考物种序列比对,是将质控成功的序列片段与指定序列(同种 16S 序列 或其他数据库)进行 blast 基因序列比对,获得一致性结果(coverage、 identity、gap、mismatch 等)。
实施例1
对某一细菌的测序结果进行质控分析和微生物鉴定。
质控方法
以擎科测序公司测序编号为:20220111-S-1-B12_16S-27F_TSS20220114-028-02268_E03的序列为输入文件,其峰图如图2所示。通过以下步骤完成:
通过使用python中的Biopython模块,通过python中的os.walk()函数获取一代测序结果的指定文件夹下格式为.ab1的全部文件路径,设置循环逐一处理。通过SeqIO.read()模块读取ab1测序文件路径并解析内容。获取每个位点中四种碱基的信号值及质量值,并以此计算出杂峰比例及双峰比例,并将结果输出至base_info.xlsx中,表格大致内容如下表1:
表1:输入序列基础信息汇总表
其中,Max_Peak_Ratio即为步骤S3中的主峰比例,而Double_Peak_Ratio为次峰比例。
以Site为1举例,Max_Peak_Ratio=singal_C/(signal_A + signal_T + signal_C+ signal_G);Double_Peak_Ratio=signal_A/singal_C。
这里需要注意的是,当输出碱基值不是最大信号值时,以输出碱基的信号值做为步骤S3中的“最大碱基信号值”。
将前述步骤的输出作为输入,进行滑窗判定,该功能由sanger_seq_quality_control.py中quality_control()函数的4个参数控制完成。结果输出至quality_control_out.xlsx中,如表2所示。
表2:质量控制结果汇总表
以序列的第一个位点作为表中所示start和end向后滑窗,若平均质量(loose模式)或每个碱基质量(strict模式)低于所设阈值,结束并从下个位点开始继续滑窗,对于单个碱基即停止滑窗的位点,其end=-1;同时,将会对该片段中最低碱基质量(min_quality)、离群碱基数量(outlier_num)、连续离群碱基数量(contiguous_outlier_num)进行控制,并截取满足条件的片段。
质量控制后的片段进行排序,并判断是否存在满足长度阈值的片段,该功能由参数最小片段长度(min_fragment_len)完成。
在本发明的一个优选方案中,参数设置为:min_window_quality=25,min_base_quality=10,mode='loose',quality_outlier_num=10,quality_contiguous_outlier_num=5,min_fragment_len=500;
即最低片段质量为25,最低碱基质量为10,宽松模式,离群碱基数量上限10,连续离群碱基数量上限5,最小片段长度为500。
综上,该实例中起始位点47,终止位点933的片段将进入下一轮分析。
对合格片段进行信号控制来判定杂峰。该功能同样由sanger_seq_quality_control.py中second_control()函数的4个参数完成,结果输出至signal_control_out.xlsx中,如表3所示。
表3:信号控制结果汇总表
本步骤将以主峰比例(Max_Peak_Ratio)、次峰比例(Double_Peak_Ratio)、混杂信号位点数量(single_peak_outlier_num)、连续混杂信号的最大片段长度(contiguous_peak_outlier_num)4个参数进一步滑窗处理序列片段,并截取得到合格片段,最终片段的长度同样需大于最小片段长度(min_fragment_len)。
在本发明的一个优选方案中,参数设置为:
Max_Peak_Ratio=0.8,Double_Peak_Ratio=0.33,single_peak_outlier_num=10,contiguous_peak_outlier_num=4;
即主峰比例为0.8,次峰比例为0.33,混杂信号位点数量上限10,连续混杂信号的最大片段长度上限4。
最终,截取得到起始位点为47,终止位点为920的序列作为最终合格序列,将其导出形成fasta格式文件。
微生物鉴定
利用blast.py将导出的fasta文件与指定序列文件或参考库进行blast比对,获得一致性或菌种鉴定结果。
可选的,本发明中的指定序列文件可为同一菌株的不同测序结果,若coverage>90%,identity>99%则认为比对结果一致;参考库为NCBI RefSeq数据库中的16S数据,取最高比对物种(species)作为菌种鉴定结果。
作为一个未通过质控的例子,将峰图如图3所示的序列信息为样本文件,采用类似如上的步骤和参数设置进行质控,最终该序列文件不符合质控要求而停止质控流程。
s实施例2
以经过多人交叉验证后的ab1文件作为测试数据,验证质控方法的精确度。
在该实例中,测试数据包括365条人工交叉验证为合格、307条人工验证为不合格的一代测序数据,按图1中的流程处理,并汇总实际分析标签与先验标签的比较结果。
如图4所示混淆矩阵,其中先验为合格,且分析为合格的结果称为TP(TruePositive);先验为合格,且分析为不合格的结果称为FP(False Positive);先验为不合格,且分析为合格的结果称为FN(Flase Negative);先验为不合格,且分析为不合格的结果称为TN(True Negative)。按照准确率(Accuracy)、精确率(Precision)、灵敏度(Sensitivity)、特异度(Specificity)的定义,分别计算得到:
TP = 340,FP = 25,FN = 17,TN = 290;
Accuracy = (TP+TN)/(TP+TN+FP+FN) = 0.9375;
Precision = TP/(TP+FP) = 0.9315;
Sensitivity = TP/(TP+FN) = 0.9524;
Specificity = TN/(TN+FP) = 0.9206;
进一步,F1 score = 2*Precision *Sensitivity/(Precision + Sensitivity)= 0.9418;
F1 score通常用于表示方法或模型的准确程度,越接近1越好,越接近0越差,本发明的F1 score为0.9418,表明其实际结果的准确度高,具有可行性。
以上显示和描述了本发明的基本原理、主要特征和本发明的优点。本行业的技术人员应该了解,本发明不受上述实施例的限制,上述实施例和说明书中描述的只是说明本发明的原理,在不脱离本发明精神和范围的前提下,本发明还会有各种变化和改进,这些变化和改进都落入要求保护的本发明范围内。