一类小分子化合物在制备新冠病毒与ACE2受体结合抑制剂药物中的应用
文献发布时间:2024-04-18 19:53:33
技术领域
本发明涉及生物医药技术领域,尤其涉及一类以人体血管紧张素转化酶2(ACE2)受体为作用靶点的小分子抑制剂。具体而言,该类抑制剂是公共数据库中记载的尚处实验阶段的化合物,通过计算机辅助药物设计中的虚拟筛选方法,这五种化合物被发现具有阻碍ACE2与新冠病毒刺突糖蛋白结合的新用途。分子对接测试与分子动力学模拟实验验证了其与上述作用靶点良好的结合亲和力与结合位点选择性,具有较大的成药潜力。
背景技术
新冠病毒的刺突糖蛋白通过与人体中的ACE2受体结合,从而入侵宿主细胞。不同于疫苗和大分子药物,小分子药物研发周期长、成本高、成功率低,如何加速先导药物的研发进程是目前亟待解决的问题。科学家们已经付出了很多努力来研发阻碍ACE2与刺突糖蛋白结合的小分子抑制剂,计算机辅助药物设计就是一种高效、快速、准确性高的方法,其中虚拟筛选是一种可以在疾病爆发初期快速发掘老药新用潜力的重要途径。在人体中,ACE2是心血管系统中一个重要的调节因子,在抗心室重塑、高血压、心力衰竭等方面发挥着积极作用,广泛分布于人体心脏和肾脏中。故由于ACE2兼具功能催化以及作为新冠病毒结合受体的特殊性,现有的靶向ACE2的药物发现过程中没有考虑药物对ACE2正常生理功能的影响,从而导致潜在的副作用。本发明旨在通过应用人工智能与传统计算机辅助药物设计方法,从已知数据库中筛选出一批成药性佳、副作用小的化合物来抑制ACE2与刺突糖蛋白的结合,进而用于后续实验测定或临床应用。
发明内容
本发明使用人工智能与传统计算机辅助药物设计方法,从公共数据库中筛选出了一类能够阻碍ACE2和新冠病毒结合的小分子抑制剂,赋予了它们新的用途。
本发明的技术方案:
一类小分子化合物在制备新冠病毒与ACE2受体结合抑制剂药物中的应用,这类小分子化合物的SMILES及数据库编号如下所示:
(1) [H][C@](CC(=O)NO)(CC1=CC(O)=CC=C1)C(=O)N[C@@]1([H])C2=C(C[C@@]1([H])O)C=CC=C2
DrugBank ID:DB06837
(2) OCCN(CC(=O)NO)S(=O)(=O)C1=CC=C(C=C1)C1=CC=CC=C1
DrugBank ID:DB08029
(3) CC1(OCC(CO1)OC(=O)NCCNC(=O)OC1COC(C)(OC1)C(O)=O)C(O)=O
DrugBank ID:DB07579
(4) [H][C@]12SCC(COC(C)=O)=C(N1C(=O)[C@H]2NC(=O)CSC1=CC=NC=C1)C(O)=O
DrugBank ID:DB01139
(5) OC(=O)C1=CC2=C(C=C1)C=C(C=C2)C(F)(F)P(O)(O)=O
DrugBank ID:DB08397
进一步地,最终得到的上述五种抑制剂通过如下技术方案筛选:
步骤1:对DrugBank数据库中的小分子化合物使用Lipinski五规则筛选出类药性化合物。
步骤2:构建深度学习模型,初步判断步骤1所得类药性化合物与ACE2和刺突糖蛋白结合界面的结合可能性。
步骤3:通过计算机辅助药物设计中的分子对接方法,计算深度学习模型预筛选后结合可能性较大的化合物与ACE2和刺突糖蛋白结合界面的结合亲和力大小。
步骤4:通过步骤2中的深度学习模型、结合位点打分模型以及步骤3中的两种分子对接结合亲和力打分,比较化合物对ACE2不同结合位点的选择性。
步骤5:通过分子动力学模拟方法,考察步骤4中结合位点选择性佳的化合物的动态结合稳定性,分析其结合机制,评估其结合强度,确定最终的候选抑制剂。
步骤6:评估步骤5中确定的候选抑制剂的药代动力学(ADME)和毒理学(T)性质(ADMET),以在药物发现早期了解其对人体的潜在影响及风险。
进一步地,步骤1具体包括:
步骤1.1:从DrugBank v5.1.8数据库中获取所有小分子化合物的3D结构文件。
步骤1.2:使用Lipinski五规则筛选出类药性化合物,即相对分子质量≤500,氢键供体数量≤5,氢键受体数量≤10,辛醇-水分配系数-2≤LogP≤5,结构中可旋转键数≤10。
进一步地,步骤2具体包括:
步骤2.1:使用人工智能方法构建卷积神经网络模型,构建小分子化合物中各原子的特征矩阵和原子间的邻接矩阵,蛋白质结合位点中各氨基酸的特征矩阵和氨基酸间的邻接矩阵,将各矩阵作为深度学习模型的输入,模型输出化合物与蛋白质某结合位点的可能性大小。通过Python代码对模型在DUD-E化合物结合亲和力诱饵数据库上的性能表现进行训练,经过多次迭代直到满足准确性要求。
步骤2.2:使用构建好的深度学习模型,快速对步骤1.2中获得的大量类药性化合物进行初筛,选出有结合潜力的化合物。
进一步地,步骤3具体包括:
步骤3.1:使用Autodock Vina分子对接软件,将步骤2.2中有结合潜力的化合物对接到ACE2和刺突糖蛋白的结合界面处。首先对配体化合物和蛋白质受体进行预处理,然后设置对接参数如下:对接区域为结合界面几何中心周围
步骤3.2:对接完成后,使用Vina和Vinardo两种结合亲和力打分来评估各化合物与ACE2和刺突糖蛋白的结合,确定二者在稳态下能否结合。
步骤3.3:选择步骤2.2和步骤3.2中结合潜力和结合稳定性(结合亲和力)排名均靠前的化合物,用于后续筛选步骤。
进一步地,步骤4具体包括:
步骤4.1:使用结合位点打分模型(文献Che X.H.,Chai S.Y.,Zhang Z.Z.,etal.Prediction of ligand binding sites using improved blind docking methodwith a machine learning-based scoring function[J].Chemical EngineeringScience,2022,261:10.中的),评价了步骤3.3中所得化合物与ACE2和刺突糖蛋白结合界面的结合倾向。
步骤4.2:为了对比化合物对ACE2不同结合位点的结合倾向,重新使用步骤2.1中构建的深度学习模型、步骤3.2中的两种对接结合亲和力打分以及步骤4.1中的结合位点打分模型,综合评价了化合物对ACE2催化位点的结合倾向。
步骤4.3:通过步骤4.2中四种不同打分模型的结果比对,从步骤3.3所选择的化合物中选择易于结合在ACE2和刺突糖蛋白结合界面而不易于结合在ACE2催化位点的化合物,用于后续筛选步骤,以尽可能降低化合物在结合过程中对ACE2正常催化功能的影响。
进一步地,步骤5具体包括:
步骤5.1:对步骤4.3中所选择的结合位点选择性佳的化合物进行了长时分子动力学模拟。首先根据步骤3.1中的对接结果将这些结合位点选择性佳的化合物分别与ACE2和刺突糖蛋白复合为新的配受体复合物,然后对其结构进行预处理,接着为复合物添加了立方体形的周期性边界条件,添加SPC显式水模型,添加适量的Na离子以平衡体系的负电荷,选择OPLS4力场。
步骤5.2:进行分子动力学模拟计算。首先对构建的模拟体系进行能量弛豫,然后进行了140ns的分子动力学模拟,前40ns采用NVT系综,后100ns采用NPT系综,时间步长为2fs,每100ps记录一帧体系结构,后1000帧的模拟数据被用于后续结果分析。模拟温度设定为300K,压力为标准大气压,恒温方法采用Nose-Hoover Chain方法,恒压方法采用Martyna-Tobias-Klein方法。
步骤5.3:对模拟结果质量进行分析,考察模拟是否符合预期设定。对模拟过程中配体和受体的动态变化情况进行分析,以考察化合物的结合稳定性和可能的抑制机制。
步骤5.4:使用MM-GBSA方法计算配受体在动态模拟过程中的结合自由能,以考察化合物的结合强度。
步骤5.5:最后重新将步骤4.3中选择的上述化合物与ACE2催化位点的对接构象和ACE2受体复合,使用步骤5.1-5.4中完全相同的参数和步骤,进一步对比这些化合物对ACE2不同结合位点的结合倾向,以此检验我们结合位点的选择性筛选结果。
步骤5.6:根据分子动力学模拟结果确定最终的候选小分子抑制剂。
进一步地,步骤6具体包括:
步骤6.1:通过admetSAR2.0在线网站评估了步骤5.6中确定的候选抑制剂在人体中吸收,分布,代谢,排泄,毒性五方面的性质。
步骤6.2:根据步骤6.1中的方法,选择五种性质中最为重要的数个评价指标,并对比各抑制剂的优缺点,给出未来应用于新冠病毒新药研发的建议。
本发明与现有技术相比,主要有以下创新性及有益效果:
(1)通过虚拟筛选方法从公共数据库中快速、准确筛选出一类能够阻碍ACE2与新冠病毒刺突糖蛋白结合的小分子抑制剂,并给出了它们的结合可能性、结合稳定性、抑制机制、结合强度以及药代动力学和毒理学性质,对新药研发具有指导意义。
(2)考察了靶向ACE2的新药发现过程中,小分子化合物对不同结合位点的选择性,在药物发现早期尽可能降低其副作用。
附图说明
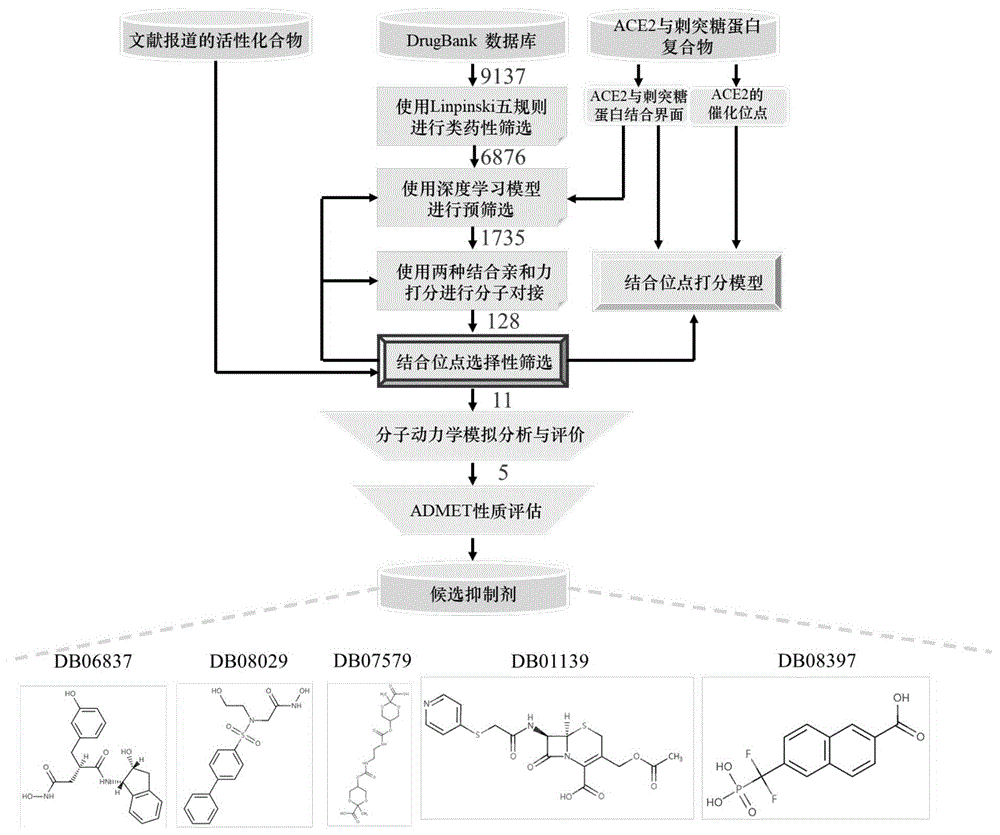
图1为本发明的虚拟筛选总流程及筛选出的小分子抑制剂结构图;
图2为用于预筛选的深度学习模型架构;
图3为该类化合物之一(DrugBank ID:DB06837)与ACE2和刺突糖蛋白结合界面的结合模式图。其中,A为通过分子动力学模拟得到的化合物与蛋白质受体的2D结合模式图;B和C分别为化合物与受体蛋白结合前后,ACE2与刺突糖蛋白间某关键氢键相互作用的改变。ACE2用红色卡通模型表示,刺突糖蛋白的受体结合域用橙色卡通模型表示,关键氨基酸残基用棒状表示。图中的键长单位为:
图4为该类化合物之一(DrugBank ID:DB06837)与ACE2催化位点的结合模式图。其中,A为通过分子动力学模拟得到的化合物与蛋白质受体的2D结合模式图;B为通过分子动力学模拟得到的化合物与蛋白质受体的3D结合模式图,ACE2用绿色卡通模型表示,ACE2催化中心距离Zn离子
具体实施方式
以下结合附图和实际应用方法对本发明的流程以及结果进行解释说明。本发明所得结果是在以本发明前述技术方案的前提下进行实施的,并给出了详细的实施方式和具体的操作过程,但本发明的保护范围不包含该技术方案在其他实例中的应用。但对本发明中筛选实例本身,虽然上述技术方案中的部分技术细节及特征可以被修改或等同替换,但均属于此技术方案应用于筛选结合位点选择性好的能阻碍ACE2与刺突糖蛋白结合的小分子抑制剂的精神和范畴,均应包含在本申请的保护范围之内。
基于附图1的虚拟筛选流程,包含9137个小分子化合物的DrugBank数据库被用于此次筛选,首先通过Lipinski五规则获得了满足要求的6876个类药小分子,然后使用构建的深度学习模型(附图2)预测了6876个类药小分子与ACE2和刺突糖蛋白结合界面的可能性大小,1735个化合物被模型认为有结合可能。将这些化合物通过分子对接方法对接到ACE2和刺突糖蛋白结合界面,Vina和Vinardo两种打分给出了它们的稳态时结合亲和力的大小。其中,1735个化合物中的前300个,其深度学习模型预测结果与对接打分呈现明显的正相关,因此选择了这300个化合物中Vina打分优于-6.4kcal/mol,Vinardo打分优于-4.8kcal/mol的共128个化合物进行结合位点选择性筛选。11种结合位点选择性好的化合物被挑选出来,如表1所示,同时为了便于与已知的活性化合物对比,来自文献(Razizadeh M.,NikfarM.,Liu Y.Small molecule therapeutics to destabilize the ace2-rbd complex:Amolecular dynamics study[J].Biophys J,2021,120(14):2793-2804.)的Nilotinib和SSAA09E2两种化合物的结合位点选择性也展示在表1。
表1不同化合物的结合位点选择性筛选结果
接着对这11种化合物分别与ACE2和刺突糖蛋白组成的新的配受体复合物的分子动力学模拟结果进行了分析。模拟质量分析显示,各体系在模拟过程中总体能量保持稳定,温度和压力在设定值上下波动,模拟过程符合预期设定。然后选取了分子动力学模拟后100ns的生产模拟过程对配体和受体的动态变化情况进行了分析。RMSD分析和可视化检查显示11种化合物中的7种能够在模拟过程中与受体蛋白稳定结合而不离开原本结合位点,故这7种化合物被用于后续分析。RMSF分析显示了各化合物的结合能够对ACE2和刺突糖蛋白结合界面处的关键氨基酸残基的构象灵活性产生特定影响,从而干扰ACE2与刺突糖蛋白的结合,但不会对蛋白质整体稳定性产生影响。附图3展示了配体化合物DB06837所在体系中配受体相互作用的详细分析结果。可以看出,该化合物的结合对ACE2与刺突糖蛋白间的相互作用产生了重要影响。例如,化合物茚满结构中的芳环与刺突糖蛋白的LYS417残基形成了长时(≥30ns)的π-阳离子相互作用,这种疏水相互作用阻碍了LYS417与ACE2的ASP30之间氢键的形成。当未结合任何配体时,LYS417侧链上的氨基氢与ASP30侧链上的羟基氧之间形成了一条氢键。而当DB06837稳定结合后,两残基间的距离被增大了,
除了上述定性分析,通过计算化合物与蛋白质受体间的结合自由能(△G)来定量表征了二者的动态结合强度。不同化合物在模拟过程中与受体的△G的平均值列在表2中。从表中可以看出,DB07579和DB08029的△G平均值与两活性化合物相当,这表明了我们的筛选方法在小分子抑制剂发现中的有效性,即筛选所得化合物不仅能够稳定结合在ACE2和刺突糖蛋白的结合界面处,且具有较高的结合强度。同时我们还观察到化合物DB08409、DB03313、DB01139和DB08397的△G虽然较活性化合物稍差,但在模拟过程中,它们的△G的波动较小,这意味着它们相较其他化合物的结合稳定性更佳,具有后续进行结构修饰及优化的潜力。
表2不同化合物与ACE2-刺突糖蛋白结合界面的结合自由能平均值
最后,对比了这些化合物对ACE2两不同结合位点的选择性,根据它们与ACE2催化位点的结合情况,将结果分为四类:
(1)化合物DB06837、DB08029和DB01139被发现结合在ACE2催化位点所在的狭长的“裂隙”内,但未见对以Zn离子为中心的催化位点存在显著影响,且它们与催化位点的结合自由能较它们与ACE2-RBD结合界面更差。
为了同附图3中的相互作用进行对比,化合物DB06837与ACE2催化位点的结合情况见附图4。
(2)化合物DB07579和DB08397被发现不能稳定结合在ACE2催化位点附近。
(3)化合物DB08409和DB03313被发现在模拟过程中与ACE2催化位点中的锌离子形成了稳定的配位作用,这势必会干扰底物与ACE2催化位点的结合。
(4)两活性化合物被发现结合在ACE2催化位点所在的狭长的“裂隙”内,虽未见对以Zn离子为中心的催化位点存在显著影响,但它们与催化位点的结合自由能较它们与ACE2-刺突糖蛋白结合界面更优,这可能导致其与ACE2-刺突糖蛋白结合界面的结合倾向变弱。
综上所述,前两类化合物是本文认为最有潜力的小分子抑制剂。化合物DB07579和DB08397不会结合在ACE2的催化位点附近,DB06837、DB08029和DB01139对ACE2催化功能的影响较小,同时它们又都能与ACE2-RBD结合界面形成稳定的相互作用,保持较高的结合强度,它们的结合位点选择性还优于两活性化合物。因此这五种化合物被最终确定并评估了它们的ADMET性质,结果如表3所示,每种性质中表现最好的抑制剂已加粗标注。表中有单位的性质是admetSAR2.0中回归模型的预测结果,没有单位的是分类模型的预测结果,此时数值表示预测可能性的大小,正负号表示是否具有该性质。
表3候选抑制剂及已知活性化合物的ADMET性质
总之,一类小分子化合物作为新冠病毒与ACE2受体结合抑制剂的新用途已被确定,同时他们还具有较好的ACE2结合位点选择性,其ADMET性质已被评价。本发明对于新冠病毒的新药研发具有较大参考价值。