一种复方厚朴提取物及其制备方法、质量控制方法
文献发布时间:2023-06-19 19:28:50
技术领域
本发明涉及饲料添加剂领域,具体涉及到一种复方厚朴提取物及其制备方法、质量控制方法。
背景技术
益生菌是指对机体健康有益的微生物,截至2019年,我国发布了食品领域可用的益生菌种类有35个种或亚种。现代中药发酵技术既继承了传统中药的炮制方法,又与现代生物工程技术相结合。益生菌发酵中药具有降低毒副作用,产生新的药效物质,扩大适应症,节约中药材资源、降低成本等优点。固体发酵不需要对单纯的营养基质进行灭菌,属于开放体系,温度、湿度、酸碱度、通气等发酵条件易于操作,但发酵后仍残留不易吸收消化的无效成分,不利于中药现代化。
半仿生提取法是山东中医药大学张兆旺、孙秀梅于1995年提出,目前应用于多种中药制剂,如芍甘止痛颗粒、寒痛定泡腾颗粒、黄连解毒颗粒等。本法是模仿口服药物在胃肠道的转运过程,采用选定pH的酸性水和碱性水依次连续提取。传统的半仿生提取法采用水煎煮或加热回流提取,提取温度高,与胃肠道环境差异大,且大多未考虑到幼龄动物消化系统的pH、酶种类及胃肠道蠕动能力等生理条件,且所提取的成分复杂。
《中国兽药典》是采用国家标准物质(金樱子对照药材)随行对照,薄层色谱法鉴别金樱子药材。所用浓硫酸属于强酸,三氯甲烷为易制毒,两者对操作人员、环境均不友好,并且薄层板喷完显色剂后需要在105℃加热,操作繁琐。
目前复方制剂中厚朴的质量控制主要是厚朴酚、和厚朴酚的薄层色谱鉴别及其含量测定。而厚朴中含多种化学成分,单一成分较难反映其质量。
母猪产程过长会造成很多不良影响,尤其易产生死胎和弱仔,仔猪出生后体质较差,生长缓慢,成活率较低。产程过长对母猪也有很大的伤害,如难产的母猪出现分娩综合征、产后高热不退、子宫及产道出血、精神状态较差、恢复较慢、感染子宫炎等疾病,甚至有可能导致母猪直接被淘汰。因此,亟待采取缩短母猪产程的可行措施,以减少母猪产程过长为生产者带来的损失,对于养猪业具有重大意义。
发明内容
本发明的目的是提供一种复方厚朴提取物及其制备方法、质量控制方法,用于解决现有技术中母猪产程过长的技术问题。
为达上述目的,本发明的一个实施例中提供了一种复方厚朴提取物,包括以下按重量份数配比的原料:
厚朴1份-60份,金樱子肉3份-50份,焦山楂1份-60份,
焦六神曲4份-63份,焦麦芽1份-60份,生姜82份-120份,
大枣肉90份-101份。
本发明优选的方案之一,复方厚朴提取物包括以下按重量份数配比的原料:厚朴52份-57份,金樱子肉29份-33份,焦山楂12份-18份,焦六神曲35份-36份,焦麦芽26份-28份,生姜102份-103份,大枣肉96份。
基于本发明公开的一种复方厚朴提取物,本发明还公开了一种复方厚朴提取物的制备方法,包括以下步骤:
步骤(1):将焦六神曲粉碎制得焦六神曲细粉,将厚朴、金樱子肉、焦山楂以及焦麦芽粉碎,制成原料微粉;
步骤(2):向生姜、大枣肉中加水进行榨汁处理,制得姜枣汁;
步骤(3):取步骤(1)制得的焦六神曲细粉和原料微粉以及步骤(2)制得的姜枣汁,混合均匀得到发酵底物;
步骤(4):加入复合益生菌粉,与发酵底物混合均匀,发酵,得到发酵产物;
步骤(5):向步骤(4)制得的发酵产物中多次加入添加剂进行搅拌提取,提取完成后滤过,滤液离心,浓缩,得到发酵原料浓缩液;
步骤(6):取步骤(5)制得的发酵原料浓缩液,调pH,经大孔吸附树脂柱,依次用水、乙醇洗脱,收集洗脱液,并将洗脱液浓缩,干燥,粉碎,总混,分装,得到复方厚朴提取物。
本发明优选的方案之一,步骤(4)中复合益生菌粉按质量计,包括安卡红曲霉1份,植物乳杆菌0.8份-3.6份,两歧双歧杆菌1份-1.7份,并且复合益生菌粉接种量与发酵底物的质量比为1:46-130。
本发明优选的方案之一,步骤(5)中发酵原料需经三次搅拌提取,其中第一次搅拌提取加入的添加剂为介质和酶的混合物,其中介质为乳酸、盐酸、柠檬酸和水,酶为胃蛋白酶、凝乳酶和脂肪酶,提取时间为0.5h;第二次搅拌提取加入的添加剂为介质和酶的混合物,其中介质为碳酸氢钠和水,酶为乳糖酶、淀粉酶、胰蛋白酶,提取时间为1h;第三次搅拌提取加入的添加剂为介质,其中介质为碳酸钠和水,提取时间为2h。
本发明还公开了一种复方厚朴提取物的质量控制方法,包括金樱子肉的检测方法以及厚朴特征图谱的检测方法。
本发明优选的方案之一,金樱子肉的检测方法包括以下步骤:
步骤a:制备供试品溶液:取本品1g研细,加溶剂进行提取,再经滤过、滤液蒸干,残渣加溶剂溶解并稀释,得到供试品溶液;
步骤b:制备对照品溶液:取金樱子对照药材1g,加溶剂进行提取,再经滤过、滤液蒸干,残渣加溶剂溶解并稀释,得到对照品溶液;
步骤c:测定:吸取供试品溶液和对照品溶液,分别点于同一硅胶G薄层板上,置于展开剂中展开,再取出,晾干,检视。
本发明优选的方案之一,步骤a中提取溶剂为乙酸乙酯,提取方式为超声提取;步骤b中提取溶剂为乙酸乙酯,提取方式为超声提取以及加热回流提取中的任意一种;步骤c中展开剂为石油醚、乙酸乙酯以及甲酸的混合物,其中石油醚、乙酸乙酯以及甲酸的体积比为100:20:1。
本发明优选的方案之一,厚朴特征图谱的检测方法包括以下步骤:
步骤a:制备参照物溶液:称取和厚朴酚对照品,加溶剂制得参照物溶液;
步骤b:制备供试品溶液:称取复方厚朴提取物,置于具塞锥形瓶中,加入丙酮,超声处理,滤过,滤液水浴蒸干,残渣加水溶解,经D101大孔吸附树脂柱,依次用水、乙醇洗脱,收集乙醇洗脱液,滤过,取滤液,得到供试品溶液;
步骤c:测定:分别吸取参照物溶液和对照品溶液,注入液相色谱仪中进行测定。
本发明优选的方案之一,步骤c中色谱条件为:色谱柱为C18,填充剂为十八烷基硅烷键合硅胶,流动相为甲醇和0.4%甲酸的混合溶液,其中甲醇为流动相A,0.4%甲酸溶液为流动相B,流速为1ml/min,柱温为30℃,检测波长为230nm。
综上所述,本发明的有益效果为:
1、本发明通过益生菌固体发酵使有效成分溶出更完全,条件更温和,从而避免了热敏性有效物质的破坏。
2、本发明采用半仿生提取法,模拟了断奶仔猪胃肠道的生理条件,保证了获取的有效成分对断奶仔猪更为有利,避免了断奶仔猪与断奶前、成年的胃肠道生理条件(pH、酶的种类)的显著差异造成不良影响。
3、本发明采用低速搅拌提取,模拟了断奶仔猪胃肠蠕动功能较弱的特点,防止了杂质的溶出。
4、本发明的提取液经大孔吸附树脂分离纯化,富集了有效成分(如总糖、总黄酮),保证了制剂的有效性。
5、本发明能缩短母猪的产程,对内江断奶仔猪的腹泻治疗有显著疗效,并具有增强免疫力、调整胃肠道运动的药理作用。
6、本发明采用薄层色谱法鉴别了复方厚朴提取物中的金樱子肉,该方法专属性强、操作简便、重现性好。
7、本发明采用高效液相色谱法建立了复方厚朴提取物中厚朴的特征图谱,可用于全面评价厚朴质量。
附图说明
图1为本发明一个实施例的复方厚朴提取物(共9批中试)中一部分的金樱子肉的薄层色谱鉴别结果示意图;
图2为本发明一个实施例中图1的另一部分的薄层色谱鉴别结果图;
图3为本发明一个实施例的复方厚朴提取物按《中国兽药典》2020年版二部中“金樱子”项下薄层色谱鉴别方法试验结果图;
图4为本发明一个实施例的复方厚朴提取物特征图谱检测方法的厚朴专属性考察结果图;
图5为本发明一个实施例的建立的复方厚朴提取物中厚朴HPLC对照特征图谱;
图6为本发明一个实施例的15批复方厚朴提取物中厚朴特征图谱叠加图。
具体实施方式
一种复方厚朴提取物,包括以下按重量份数配比的原料:厚朴1份-60份,金樱子肉3份-50份,焦山楂1份-60份,焦六神曲4份-63份,焦麦芽1份-60份,生姜82份-120份,大枣肉(新鲜)90份-101份。
优选的,一种复方厚朴提取物,包括以下按重量份数配比的原料:厚朴52份-57份,金樱子肉29份-33份,焦山楂12份-18份,焦六神曲35份-36份,焦麦芽26份-28份,生姜102份-103份,大枣肉(新鲜)96份;
一种复方厚朴提取物的制备方法,包括以下步骤:
步骤(1):将焦六神曲低温粉碎制成细粉,制得焦六神曲细粉,再将厚朴、金樱子肉、焦山楂以及焦麦芽超微粉碎,制成原料超微粉;
步骤(2):向生姜、大枣肉中加水进行榨汁处理,制得姜枣汁;
步骤(3):取步骤(1)制得的焦六神曲细粉和原料超微粉以及步骤(2)制得的姜枣汁,加入益生菌,混合均匀,得到发酵底物,其中加入的姜枣汁的量为46%,
步骤(4):加入复合益生菌粉适量,与发酵底物混合均匀,发酵,得到发酵产物,其中,复合益生菌粉按质量计,包括安卡红曲霉(Monacus anka AS 3.782,CCTCC编号为AF93208)1份,植物乳杆菌(Lactobacillus plantarum,CCTCC编号为AB 2010210)0.8份-3.6份,两歧双歧杆菌(Bifidobacterium bifidum ATCC29521,CCTCC编号为AB 94055)1份-1.7份,并且复合益生菌粉接种量与发酵底物的质量比为1:46~130,发酵的温度为21℃-37℃,优选为33℃-34℃,发酵的时间为72h-120h;
步骤(5):取步骤(4)发酵产物,加入乳酸、盐酸、柠檬酸和水(并调节pH至4.6),胃蛋白酶、凝乳酶、脂肪酶,在37℃下搅拌(转速为45rpm)提取0.5h,滤过,药渣加入碳酸氢钠和水(并调节pH至7.2),乳糖酶、淀粉酶、胰蛋白酶,在37℃下搅拌(转速为45rpm)提取1h,滤过,药渣再加入碳酸钠和水(并调节pH至6.1),在37℃下搅拌(转速为45rpm)提取2h,滤过;滤液离心,浓缩,得到发酵原料浓缩液;
步骤(6):取步骤(5)制得的发酵原料浓缩液,调节pH至中性,经AB-8型大孔吸附树脂柱,依次用水、80%乙醇洗脱,收集洗脱液,并将洗脱液浓缩,干燥,粉碎,总混,分装,得到复方厚朴提取物。
一种复方厚朴提取物的质量控制方法,包括金樱子肉的检测方法以及厚朴特征图谱的检测方法。
金樱子肉的检测方法包括以下步骤:
步骤a:制备供试品溶液:取本品1g研细,加50ml乙酸乙酯溶剂进行超声提取20min,再经滤过、滤液蒸干,残渣加乙酸乙酯溶解并稀释至0.5ml,得到供试品溶液;
步骤b:制备对照品溶液:取金樱子对照药材1g,加50ml乙酸乙酯溶剂进行超声提取或者加热回流,优选加热回流提取1h,再经滤过、滤液蒸干,残渣加乙酸乙酯溶解并稀释至0.5ml,得到对照品溶液;
步骤c:测定:吸取供试品溶液和对照品溶液各5ul,分别点于同一硅胶G薄层板上,置于展开剂中展开,展至8cm,再取出,晾干,置紫外光灯(365nm)下检视,供试品色谱中,在与对照药材色谱相应的位置上,显一个相同的黄色荧光斑点;其中展开剂为石油醚、乙酸乙酯以及甲酸的混合物,并且石油醚、乙酸乙酯以及甲酸的质量比为100:20:1。
厚朴特征图谱的检测方法包括以下步骤:
步骤a:制备参照物溶液:精密称取和厚朴酚对照品,加甲醇制得每1ml含和厚朴酚30.26ug的参照物溶液;
步骤b:制备供试品溶液:精密称取复方厚朴提取物0.8g,置于具塞锥形瓶中,加入25ml丙酮,超声处理20min,滤过,滤液水浴蒸干,残渣加50ml水溶解,经D101大孔吸附树脂柱(内径为1cm,柱高为15cm),依次用50ml水、50ml65%乙醇洗脱,收集65%乙醇洗脱液,微孔滤膜滤过,取滤液,得到供试品溶液;
步骤c:测定:分别吸取参照物溶液和对照品溶液,注入液相色谱仪中进行测定;
采用高效液相色谱进行分析,色谱条件如下:
色谱柱:以十八烷基硅烷键合硅胶为填充剂,特别地,色谱柱为WondasilC18superb(柱长250mm,内径为4.6mm,粒度为5um);
流动相:以甲醇为流动相A,以0.4%甲酸溶液为流动相B;
梯度洗脱:按下表中的规定进行梯度洗脱,其中下表中的%指的是体积百分比:
流速:1ml/min
柱温:30℃
检测波长:230nm
复方厚朴提取物的对照特征图谱包括16个特征峰,分别为峰1~16。其中,与和厚朴酚参照物相对应的峰9为S峰,计算各特征峰与S峰的相对保留时间,各峰相对保留时间为:
实施例1
一种复方厚朴提取物,包括以下按重量份数配比的原料:
厚朴52份,金樱子肉33份,焦山楂18份,焦六神曲35份,焦麦芽26份,生姜102份,大枣肉(新鲜)96份。
一种复方厚朴提取物的制备方法,包括以下步骤:
步骤(1):取焦六神曲35g,低温粉碎成细粉,再取厚朴52g、金樱子肉33g、焦山楂18g、焦麦芽26g等4味药,超微粉碎,制成原料超微粉,控制超微粉粒径D(v,0.9)≤5um;
步骤(2):另取生姜102g、大枣肉96g,加水适量趁鲜榨汁,制成姜枣汁。
步骤(3):取步骤(1)制得的焦六神曲细粉和原料微粉以及步骤(2)制得的姜枣汁,混合均匀,得到发酵底物;
步骤(4):加入复合益生菌粉适量,与发酵底物混合均匀,发酵,得到发酵产物;其中,复合益生菌粉按质量计,包括安卡红曲霉(Monacus anka AS 3.782,CCTCC编号为AF93208)1份,植物乳杆菌(Lactobacillus plantarum,CCTCC编号为AB 2010210)0.8份,两歧双歧杆菌(Bifidobacterium bifidum ATCC 29521,CCTCC编号为AB 94055)1份,并且复合益生菌粉的接种量与发酵底物的质量比为1:110,发酵的温度为33℃,发酵的时间为72h;
步骤(5):取步骤(4)发酵产物,加入乳酸、盐酸、柠檬酸和水(并调节pH至4.6),胃蛋白酶、凝乳酶、脂肪酶,在37℃下搅拌(转速为45rpm)提取0.5h,滤过,药渣加入碳酸氢钠和水(并调节pH至7.2),乳糖酶、淀粉酶、胰蛋白酶,在37℃下搅拌(转速为45rpm)提取1h,滤过,药渣再加入碳酸钠和水(并调节pH至6.1),在37℃下搅拌(转速为45rpm)提取2h,滤过。滤液离心,浓缩,即得发酵原料浓缩液;
步骤(6):取步骤(5)的发酵原料浓缩液,调节pH至中性,通过AB-8型大孔吸附树脂柱,依次用水、80%乙醇洗脱,收集洗脱液,浓缩,干燥,粉碎,总混,分装,即得。
实施例2
复方厚朴提取物中金樱子肉的薄层色谱鉴别实验研究:
1:标准物质与样品
/>
2:试验条件的筛选
2.1甲酸用量的考察
结果:表明甲酸仅加入0.1即可。
2.2展开剂种类的筛选
2.3供试品制备方法的选择
厚朴提取物供试品溶液(甲醇超声提取,乙酸乙酯萃取)的制备:称取厚朴提取物1g,加甲醇50ml,超声处理20min,滤过,滤液蒸干,残渣加水50ml使溶解,用乙酸乙酯萃取3次,每次30ml,合并乙酸乙酯液,水浴蒸干,残渣加乙醇使溶解并稀释至1ml,即得。
厚朴提取物供试品溶液(乙酸乙酯超声提取)的制备:称取厚朴提取物1g,研细,加乙酸乙酯50ml,超声处理20min,滤过,滤液蒸干,残渣加乙酸乙酯使溶解并稀释至0.5ml,作为供试品溶液。
金樱子对照药材溶液(甲醇超声提取,乙酸乙酯萃取)的制备:称取金樱子对照药材1g,加甲醇50ml,超声处理20min,滤过,滤液蒸干,残渣加水50ml使溶解,用乙酸乙酯萃取3次,每次30ml,合并乙酸乙酯液,水浴蒸干,残渣加乙醇使溶解并稀释至1ml,即得。
金樱子对照药材溶液(乙酸乙酯超声提取)的制备:称取金樱子对照药材1g,加乙酸乙酯50ml,超声处理20min,滤过,滤液蒸干,残渣加乙酸乙酯使溶解并稀释至0.5ml,作为供试品溶液。
金樱子对照药材溶液(乙酸乙酯加热回流提取)的制备:称取金樱子对照药材1g,加乙酸乙酯50ml,加热回流提取1h,滤过,滤液蒸干,残渣加乙酸乙酯使溶解并稀释至0.5ml,作为对照药材溶液。
结果如表1所示:
表1:供试品制备方法考察结果
由表1可知,金樱子对照药材采用乙酸乙酯加热回流提取,复方厚朴提取物采用乙酸乙酯超声提取所制备的供试品的薄层色谱行为良好。
3:试验方法与结果
供试品溶液的制备:取本品1g,研细,加乙酸乙酯50ml,超声处理20min,滤过,滤液蒸干,残渣加乙酸乙酯使溶解并稀释至0.5ml,作为供试品溶液。
对照药材溶液的制备:另取金樱子对照药材1g,加乙酸乙酯50ml,加热回流提取1h,滤过,滤液蒸干,残渣加乙酸乙酯使溶解并稀释至0.5ml,作为对照药材溶液。
缺金樱子肉的阴性溶液的制备:取缺金樱子肉的阴性1g,研细,加乙酸乙酯50ml,超声处理20min,滤过,滤液蒸干,残渣加乙酸乙酯使溶解并稀释至0.5ml,作为阴性溶液。
薄层色谱试验法:照薄层色谱法(《中国兽药典》附录0502)试验,吸取上述两种溶液各5ul,分别点于同一硅胶G薄层板上,以石油醚(60~90℃)-乙酸乙酯-甲酸(10:2:0.1)为展开剂,展至约8cm,取出,晾干,置紫外光灯(365nm)下检视。
结果:9个批次的复方厚朴提取物中试样品的供试品色谱中,在与金樱子对照药材色谱相应的位置上,均显一个相同黄色的荧光斑点,且阴性无干扰,表明该方法专属性强、重复性好,结果见图1和图2,其中1~3为复方厚朴提取物(中试批号依次为20220301、20220302、20220303);4和9为金樱子对照药材;5~6为复方厚朴提取物(中试批号依次为20220304、20220305);7~8为厚朴提取物(中试批号依次为20220401、20220402);10~11为复方厚朴提取物(中试批号依次为20220501、20220502);12为缺金樱子肉的阴性。
实施例3
复方厚朴提取物中厚朴特征图谱的方法建立,具体试验如下:
1:材料
厚朴(厚朴)对照药材(批号121285-201303,中国食品药品检定研究院);厚朴酚对照品(中国食品药品检定研究院,纯度99.0%,批号110729-202015);和厚朴酚对照品(中国食品药品检定研究院,纯度99.8%,批号110730-201915);
收集15批次的厚朴饮片,经鉴定为木兰科植物厚朴Magnolia officinalisRehd.et Wils.或凹叶厚朴Magnolia officinalis Rehd.et Wils.var.biloba Rehd.etWils.的干燥干皮、根皮及枝皮。按照本发明处方、工艺制成相对应的成品,详细信息见表2。
表2:厚朴饮片(15批)来源及其成品信息
注:其中7个批次(批号20220101、20220102、20220103、20210104、20220201、20220202、20220203)为本实验室小试制备样品,8个批次(批号220220301、20220302、20220303、20220304、20220401、20220402、20220501、20220502)为本公司中试生产样品。
2:色谱条件
LC-20A岛津高效液相色谱仪(SPD-20A型紫外可见光检测器,SIL-20AxR型自动进样器,柱温箱CTO-20AC,DGU-20A5R脱气机);Wondasil C18 superb(4.6x250mm,5um,S/N:9F3706-04);柱温:30℃;流速:1ml/min;进样量10ul;以甲醇为流动相A,以0.4%甲酸溶液为流动相B,按表3中的规定进行梯度洗脱;检测波长为230nm。
表3梯度洗脱程序
3:样品溶液的制备
厚朴饮片供试品溶液的制备:取厚朴饮片粉末约0.1g,精密称定,置具塞锥形瓶中,精密加入60%甲醇50ml,超声处理20min,放冷,再称定重量,用60%甲醇补足减失的重量,摇匀,微孔滤膜滤过,取续滤液,即得
厚朴对照药材参照物溶液的制备:取厚朴(厚朴)对照药材粉末约0.1g,精密称定,置具塞锥形瓶中,精密加入60%甲醇50ml,超声处理20分钟,放冷,再称定重量,用60%甲醇补足减失的重量,摇匀,微孔滤膜滤过,取续滤液,即得。
和厚朴酚对照品溶液的制备:精密称取和厚朴酚对照品15.16mg(中国食品药品检定研究院,纯度99.8%,批号110730-201915),置于100ml棕色量瓶中,加甲醇溶解并稀释至刻度,精密量取2ml,置于10ml棕色量瓶中,加甲醇溶解并稀释至刻度,即制成浓度为30.26ug/ml。
厚朴酚对照品溶液的制备:精密称取厚朴酚对照品20.75mg,置于50ml棕色量瓶中,加甲醇溶解并稀释至刻度,精密量取1ml,置于10ml棕色量瓶中,加甲醇溶解并稀释至刻度,即制成浓度为41.09ug/ml。
复方厚朴提取物供试品溶液的制备:取复方厚朴提取物约0.8g,精密称定,置具塞锥形瓶中,加丙酮25ml,超声处理20min,滤过,滤液水浴蒸干,残渣加水50ml超声使溶解,通过D101大孔吸附树脂柱(内径为1cm,柱高为15cm),依次用水50ml,65%乙醇50ml洗脱,收集65%乙醇洗脱液,微孔滤膜滤过,取续滤液,即得。
缺厚朴的阴性溶液的制备:按照本发明的处方(去掉厚朴)、工艺制成缺厚朴的阴性样品,称取约0.8g,制备方法同“复方厚朴提取物供试品溶液的制备”。
4:测定方法
分别精密吸取厚朴饮片溶液、对照药材参照物溶液、对照品参照物溶液、复方厚朴提取物供试品溶液、缺厚朴阴性溶液各10ul,注入高效液相色谱仪,按照规定色谱条件进行测定。
5:方法学考察
5.1:专属性考察
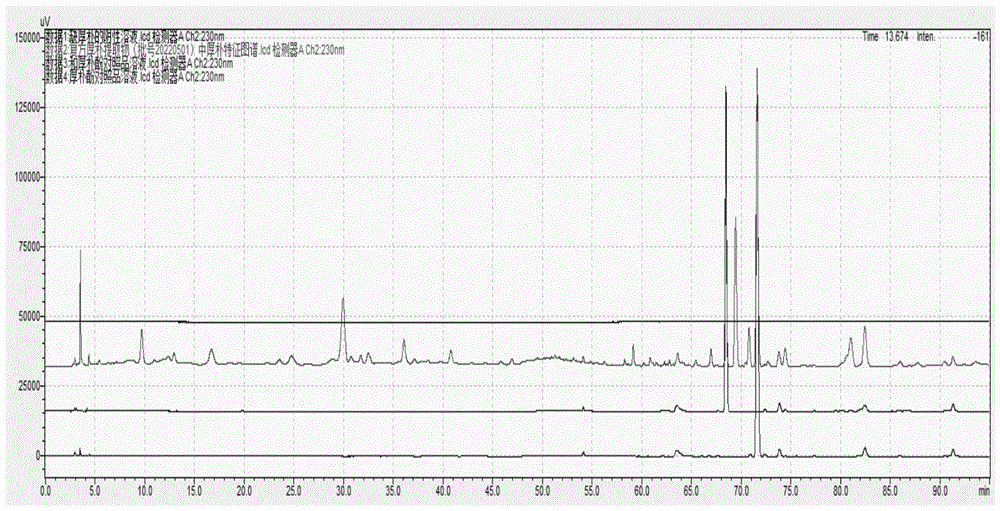
结果表明:阴性对厚朴的检测不存在干扰,专属性良好。结果见图4,其中从上至下分别为:数据1:缺厚朴的阴性溶液;数据2:复方厚朴提取物(批号20220501);数据3:和厚朴酚对照品溶液,数据4:厚朴酚对照品溶液。
5.2:精密度
取复方厚朴提取物(批号20220301)供试品溶液,在上述色谱条件下连续进样6针,记录色谱图。以峰9(和厚朴酚)为S峰,计算各特征峰与S峰的相对保留时间、相对峰面积的RSD,结果见表4。
表4:精密度考察结果
/>
结果表明:6针供试品溶液中各特征峰的相对保留时间RSD%、相对峰面积RSD%均小于2.0%,故该方法的仪器精密度良好。
5.3:重复性
取复方厚朴提取物(批号20220301)制备6份供试品溶液,在确定的色谱条件下进行分析,记录色谱图。
以峰9(和厚朴酚)为S峰,计算各特征峰与S峰的相对保留时间、相对峰面积的RSD,结果见表5。
表5:重复性考察结果
/>
/>
结果表明:6针平行样品中各特征峰的相对保留时间RSD%、相对峰面积RSD%均小于2.0%,说明本发明方法的重复性良好。
5.5:稳定性
取复方厚朴提取物(批号220220301)分别在室温条件下放置0h、2h、4h、6h、8h、10h、12h、18h、24h后,在确定的色谱条件下进行分析,记录色谱图。
以峰9(和厚朴酚)为S峰,计算各特征峰与S峰的相对保留时间、相对峰面积的RSD,结果见表6。
表6:稳定性考察结果
/>
结果表明:供试品溶液在24h内,16个特征峰的相对保留时间RSD%、相对峰面积RSD%均小于2.0%,稳定性良好。
6:特征图谱的建立
6.1:共有峰的选择
将15批复方厚朴提取物的HPLC图谱与厚朴对照药材、15批厚朴饮片进行比较,最终选取其中16个峰,面积较大,分离度较好,且理化性质稳定,三者所共有的色谱峰作为共有峰(即峰1~16),结果见图5,其中峰9(S峰)为和厚朴酚,峰12为厚朴酚。
6.2:共有峰的标定及参照峰的选择
将和厚朴酚、厚朴酚对照品色谱图与厚朴饮片、厚朴对照药材、复方厚朴提取物中相应位置的色谱峰比较,可知样品图谱中9号特征峰为和厚朴酚,12号特征峰为厚朴酚。由于各批次样品图谱中和厚朴酚的色谱峰分离良好,峰面积较大且为所有样品共有,故确定和厚朴酚为参照峰(S峰),结果见图4。
6.3:复方厚朴提取物相似度评价
将15批厚朴饮片HPLC色谱图导入国家药典委员会“中药色谱指纹图谱相似度评价系统》(2012.130723版本)”软件,设参照图谱,对色谱图进行多点校正、峰匹配,采用平均数法生成15批厚朴饮片的对照特征图谱,结果见图5。计算各特征峰与S峰的相对保留时间,相对保留时间为:0.14(峰1)、0.24(峰2)、0.36(峰3)、0.44(峰4)、0.52(峰5)、0.59(峰6)、0.86(峰7)、0.97(峰8)、1.00(峰9)、1.02(峰10)、1.03(峰11)、1.05(峰12)、1.07(峰13)、1.08(峰14)、1.21(峰15)、1.33(峰16)。
对复方厚朴提取物供试品S1~S15中的16个共有色谱峰进行多点校正,峰匹配,并计算15批复方厚朴提取物与厚朴饮片的相似度,结果见表7、图6。
表7:复方厚朴提取物(共15批)与厚朴饮片的相似度计算结果
由以上可知:15批复方厚朴提取物与厚朴饮片对照特征图谱相似度较高,均大于0.90。复方厚朴提取物供试品色谱中应呈现16个与对照特征图谱相对应的色谱,其中峰9、峰12保留时间应与和厚朴酚、厚朴酚对照品色谱峰的保留时间相对应。表明本发明的特征图谱检测方法稳定可靠,可用于复方厚朴提取物中厚朴的质量控制。
对比例1
称取厚朴52g、金樱子肉33g、焦山楂18g、焦麦芽26g、焦六神曲35g,生姜102g、大枣肉(新鲜)96g,以饮片直接投料,加水煎煮3次,滤过,合并滤液,浓缩,减压干燥,粉碎,总混,即得成品。
对比例2
参考《中国兽药典》2020年版二部中“金樱子”项下的薄层鉴别方法试验。
复方厚朴提取物供试品溶液的制备:取复方厚朴提取物2g,加乙醇50ml,超声处理30分钟,滤过,滤液蒸干,残渣加水20ml使溶解,用乙酸乙酯萃取3次,每次30ml,合并乙酸乙酯液,水浴蒸干,残渣加甲醇使溶解并稀释至2ml,即得。
对照药材溶液的制备:称取金樱子对照药材药材2g,制法同复方厚朴提取物供试品溶液。
吸取上述溶液5ul,分别点于同一硅胶G薄层板上,以三氯甲烷-乙酸乙酯-甲醇-甲酸(5:5:1:0.1)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰。
结论:供试品色谱中,在与金樱子对照药材色谱相对应的位置上,无相同颜色的斑点,结果见图3,其中1~2为金樱子对照药材,3为复方厚朴提取物(批号20220701),4为复方厚朴提取物(批号20220702)。
试验例1
分别测定实施例1、对比例1成品中的总糖与总黄酮含量,方法如下:
(1)总糖的测定方法
对照品溶液的制备:取经105℃干燥至恒重的无水葡萄糖70mg,精密称定,置100ml量瓶中,加水溶解并稀释至刻度,摇匀,即得(每1ml中含无水葡萄糖0.7mg)。
标准曲线的制备:精密量取对照品溶液0.5ml、1.0ml、1.5ml、2.0ml、2.5ml,分别置50ml量瓶中,各加水至刻度,摇匀。分别精密量取上述溶液2ml,置具塞试管中,各精密加4%苯酚溶液1ml,混匀,迅速精密加入硫酸7ml,摇匀,置40℃水浴中保温30min,取出,置冰水浴中放置5min,取出,以相应试剂为空白,照紫外-可见分光光度法(通则0401)在490nm的波长处测定吸光度,以吸光度为纵坐标,浓度为横坐标,绘制标准曲线。
供试品溶液的制备:取实施例1成品、对比例1成品各约0.2g,精密称定,置具塞锥形瓶中,精密加水50ml,称定重量,静置1小时,加热回流1小时,放冷,再称定重量,用水补足减失的重量,摇匀,滤过,精密量取续滤液1ml,置100ml量瓶中,加水至刻度,摇匀,精密量取25ml,置50ml量瓶中,加水至刻度,摇匀,密量取2ml,置具塞试管中,照标准曲线的制备项下的方法,自“各精密加4%苯酚溶液1ml”起,依法测定吸光度。
从标准曲线上读出供试品溶液中总糖的重量(ug),并计算样品中总糖含量。
(2)总黄酮的测定方法
对照品溶液的制备:取芦丁对照品适量,精密称定,加乙醇制成每1ml含芦丁0.15mg的溶液,即得。
标准曲线的制备:精密量取对照品溶液1ml、2ml、3ml、4ml、5ml、6ml,分别置25ml量瓶中,各加水至6ml,加5%亚硝酸钠溶液1ml,使混匀,放置6min,加10%硝酸铝溶液1ml,摇匀,放置6min,加氢氧化钠试液10ml,再加水至刻度,摇匀,放置15分钟,以相应的试剂为空白,照紫外-可见分光光度法(通则0401),以500nm的波长处测定吸光度,以吸光度为纵坐标,浓度为横坐标,绘制标准曲线。
供试品溶液的制备:取实施例样品、对比例1样品各0.10g,精密称定,置具塞锥形瓶中,精密加入稀乙醇25ml,密塞,摇匀,超声处理5min,放置3h以上,滤过,精密量取续滤液2ml,置25ml量瓶中,用水稀释至刻度,摇匀,作为供试品溶液。精密量取供试品溶液2ml,置25ml量瓶中,照标准曲线的制备项下的方法,自“加水至6ml”起,依法测定吸光度,同时精密量取供试品溶液2ml,置25ml量瓶中,加水至刻度,摇匀,作为空白溶液。
从标准曲线上读出供试品溶液中芦丁的量,计算含量。
成品的含量测定结果见表8。
表8:实施例1、对比例1成品中总糖与总黄酮含量的比较结果
根据表8可知,实施例1有效物质总糖、总黄酮分别为43.83%、41.26%,均显著高于常规制法(对比例1)。原因分析可能为固体发酵使有效成分更易于溶出,大孔吸附树脂能有效除去无效杂质。
试验例2
雄性昆明小鼠72只,体重18-22g,随机分为6组,每组各12只。分别为空白对照组、模型组、对比例1组、实施例1组(低、中、高剂量组)。灌胃给药,实施例1剂量分别为21.5mg、43mg、86mg/kg,对比例1剂量为100mg/kg,空白对照组、模型组给予等体积生理盐水,连续给药30天。模型组小鼠在给药第21腹腔注射环磷酰胺,隔天腹腔注射一次,共5次。
MTT法测定小鼠脾淋巴细胞增殖情况,足跖增厚法测定迟发型变态反应。采用统计分析软件SPSS22.0处理,单因素方差分析,组间进行多重比较,以P<0.05认为差异有统计学意义,结果见表9-11。
表9:实施例1、对比例1对小鼠体液免疫功能的影响
注:与空白对照组比较,*P<0.05
表10:实施例1、对比例1对小鼠细胞免疫功能的影响
注:与空白对照组比较,*P<0.05
表11:实施例1、对比例1对小鼠足趾肿胀程度的影响
注:与空白对照组比较,*P<0.05
由表9-11可知,与空白对照组比较,实施例1高剂量组(86mg/kg)能显著升高小鼠迟发型变态反应能力、淋巴细胞转化能力、抗体生产细胞数和血清半数溶血值,表明本发明的复方厚朴提取物能够增强小鼠免疫力的功能。现代研究认为胃肠道的淋巴组织是具有免疫功能的周围淋巴组织的重要组成部分,消化系统疾病多与免疫功能失调有关。
试验例3
昆明小鼠60只,体重18-22g,随机分为5组,每组各12只。分别为空白对照组、模型组、实施例1组(低、中、高剂量组)。灌胃给药,实施例1剂量分别为30mg、60mg、120mg/kg,空白对照组、模型组给予等体积生理盐水,连续给药30天。模型组用复方地芬诺酯造模。实验结束前禁食不禁水16h,于测定当天再给予一次受试样品,30min后各实验组和模型组给予复方地芬诺酯,空白对照组给予蒸馏水,30min后各组再给予指示剂,25min后断颈处死动物,计算墨汁推进率,测定结果见表12。
表12:实施例1对小鼠小肠推进度的影响
注:与模型组比较,#P<0.05
SD大鼠48只,按体重大小随机分为4组,每组各12只。分别为空白对照组、实施例1组(低、中、高剂量组)。以灌胃方式给样,空白对照组给蒸馏水,每天1次,连续30天。实施例1剂量分别为45mg、90mg、180mg/kg。实验期间每周测2次体重和食物摄入量,并根据体重变化调整给样量。实验结束时计算动物增重、摄食量和食物利用率,结果见表13。
表13:实施例1对大鼠体重增加量、摄食量、食物利用率的影响
注:与空白对照组比较,*P<0.05
由表13可知,与模型组比较,实施例1各剂量组均能调整小肠的推进作用;由表6可知,与空白对照组比较,实施例1中剂量组(90mg/kg)、高剂量组(180mg/kg)能增加大鼠体重、摄食量、食物利用率。
试验例4
将诊断为腹泻的内江断奶仔猪随机分成2组,每组20头(公母各半),实施例1组给药剂量为110mg/kg,每日一次,连续使用三天,空白对照组不作处理,测定仔猪腹泻头数、腹泻天数等指标,计算腹泻率和腹泻指数,结果见表14。
表14:实施例1对内江断奶仔猪腹泻的治疗结果
注:与空白对照组比较,*P<0.05
由表14可知,空白对照组、实施例1组的内江断奶仔猪腹泻率分别为79.2%、6.67%,腹泻指数分别为1.95、0.35。实施例1组的腹泻率、腹泻指数均显著低于空白对照组(P<0.05)。说明本发明的复方厚朴提取物治疗内江断奶仔猪腹泻有明显效果。
试验例5
选取胎次相近的健康怀孕95~96天的母猪60头,随机分为3组,即实施例1组、对比例1组、空白对照组,每组20头。按照0.1g/kg体重/天,饮水或拌料给药,连续饲喂5天。记录母猪产程时间,即从母猪启动分娩开始,至最后一头仔猪出生后胎衣排出时间,结果见表15。
表15:实施例1对母猪产程影响结果
注:与空白对照组、对比例1组比较,*P<0.05
由表15可知,与空白对照组、对比例1组比较,实施例1组的母猪产程显著降低(P<0.05)。说明怀孕母猪饲喂本发明的复方厚朴提取物能在产程中获益。
虽然结合附图对本发明的具体实施方式进行了详细地描述,但不应理解为对本专利的保护范围的限定。在权利要求书所描述的范围内,本领域技术人员不经创造性劳动即可做出的各种修改和变形仍属本专利的保护范围。
- 一种太子参提取物的制备方法及其制备的太子参提取物和检测方法
- 一种从厚朴中分离制备和厚朴酚的方法
- 一种用于空气消毒剂的中药提取物及其制备方法、环保复合型空气消毒剂及其制备方法
- 一种二冬汤对照提取物及其制备方法和质量控制方法
- 一种枇杷清肺饮对照提取物及制备方法和质量控制方法