一种基因检测技术及应用
文献发布时间:2024-04-18 19:58:26
技术领域
本发明属于生物医疗技术领域,具体涉及一种基因检测技术及其应用。
背景技术
作为解析基因突变的一种重要手段,核酸分子检测在疾病的诊断和治疗中发挥了越来越重要的作用。目前主流的核酸检测技术有实时荧光定量PCR、基因芯片和二代测序技术。
利用实时荧光定量PCR技术平台和经过特殊设计、靶向基因突变位点的PCR引物,可实现对单碱基变异的精准检测。根据探针的类型,实时荧光定量PCR大体可分为Taqman探针法和荧光染料法。在临床基因检测方面,实时荧光定量PCR技术的应用是一次由定性分析向定量检测的飞跃,被广泛用于肿瘤突变基因检测和靶向药物应用筛选等方面。但该技术的检测通量较低,制约了其在多位点、或多样本筛选中的应用,
为了提高检测通量,近年来发展了包括微阵列基因芯片、二代测序在内的多个高通量核酸检测技术平台,为发展临床核酸检测带来了新的希望。微阵列基因芯片技术的原理是利用寡核苷酸的原位合成、显微打印等技术,将大量探针有序地固定于尼龙膜、硅片、玻璃等固相支持物表面,使其与荧光标记的被检测核酸样本进行杂交,从而对样本中的目标核酸分子进行定性、定量检测。与实时荧光定量PCR相比,基因芯片技术最大优点在于其极高的检测通量,每一小块芯片就可容纳一万多个检测探针点。但该技术的检测灵敏度低、定量准确性不高,并且需较为昂贵的仪器和数据分析软件,因此主要应用于基础研究中。
二代测序又称为高通量测序技术(High-throughput sequencing),能一次精确测定几十万到几百万条核酸分子的序列,测序读长在40至100多个碱基之间。目前主流的二代测序技术包括Illumina sequencing、454pyrosequencing、ABI SOLiD sequencing、以及最近发展出来的单分子测序技术等。二代测序技术兼顾了高通量、高灵敏度、高准确性等多方面的技术优势,在临床核酸检测中具有较大的应用潜力。不足之处是这些技术需要复杂和昂贵的设备平台、以及受过特殊训练的操作人员,导致其检测成本过高、技术应用的不够灵活,不适合疾病筛查这类大规模的应用。另外,二代测序的操作流程复杂,检测周期长,不能满足许多对时效性要求较高的临床检测的需要。
综上所述,实时荧光定量PCR具有较高的检测灵敏度和准确性,缺点是检测通量低。基因芯片的检测通量高,但在灵敏度和准确性上很难满足临床应用的需要。以二代测序为代表的高通量方案兼顾了检测的灵敏度、准确性和通量的要求,但对实验设备和技术人员的要求较高、检测周期长,成本负担大。并且无论是基因芯片还是二代测序技术,在样本前处理过程中均涉及对核酸片段的PCR扩增。所以就技术特点而言,它们既继承了PCR技术的优势,能够实现对微量核酸样本检测;也继承了PCR技术不足的方面,使得它们无法从根本上突破PCR技术的检测瓶颈。例如,在高野生型同源基因的背景下,对低丰度变异序列的检测灵敏度较低的问题,这些技术限制极大地制约了包括实时荧光定量PCR、基因芯片、以及二代测序技术在某些临床检测中的应用。
发明内容
作为恶性肿瘤一种重要的致病机制,肿瘤细胞的基因组变异主要表现为基因融合、以及蛋白编码序列的碱基突变。这也表明在基因组序列水平,肿瘤细胞和正常细胞是可以被区分开来的。在肿瘤的发生和发展过程中,肿瘤细胞可能自发的、或因诊疗操作等因素从实体肿瘤部位脱落,游离到外周血液中,这部分以痕迹量存在于外周血液中的肿瘤细胞即被称为循环肿瘤细胞。通常情况下,在10毫升血液中只存在几个到几十个循环肿瘤细胞,而具备完整正常基因组的白细胞则有4000万-1亿个。从技术层面上来看,要检测到痕迹量的循环肿瘤细胞就需要在较高正常的同源基因的背景下,检测到极低丰度的突变肿瘤基因,这种检测能力取决于检测技术对差异性非常微小的基因序列的区分能力。因此,对肿瘤患者的早期筛查而言,对低丰度变异基因的精准检测技术具有关键性的作用。
对低丰度、微量、甚至痕迹量的变异基因的检测,需要克服海量的正常细胞基因组背景的干扰,这是检测难度所在。临床上,目前主要的检测手段有实时荧光定量PCR和高通量测序。荧光定量PCR对检测环境和条件的要求较高,仅适合在大型医疗机构中使用;高通量测序技术更加复杂,一般需要由第三方检测机构的专门技术人员来实施,检测周期长,检测成本高,不能很好地满足临床应用的需要。因此,该技术领域还存在较大的发展空间。
虽然在理论上,可以通过设计突变基因特异的PCR引物,在一定程度上实现差异基因的区分检测。但在实际操作中,这种技术方案的选择性尚不足以完成对循环肿瘤细胞检测、以及类似的对低丰度同源基因检测的应用,这主要是源于PCR技术对基因错配具有一定的容忍度、以及其强大的扩增效率。
为了解决这个问题,本发明提供了一种新颖的技术方案。利用序列特异性单链DNA作为引导分子,引导热稳定性RNase H2蛋白对同源基因序列进行差异性切割。进一步的,本发明将热稳定RNase H2蛋白的差异性切割能力与DNA聚合酶的模板扩增能力结合起来,实现对微量同源基因的检测。更进一步的,本发明将热稳定RNase H2蛋白的差异性切割能力与PCR聚合酶强度的模板扩增能力结合起来,实现对痕迹量同源基因的检测。
本发明的有益效果
利用热稳定性RNase H2核酸酶对序列差异性的识别能力,本发明将其用于微量核酸分子的检测,能够显著提高检测灵敏度,具有临床应用价值。
附图说明
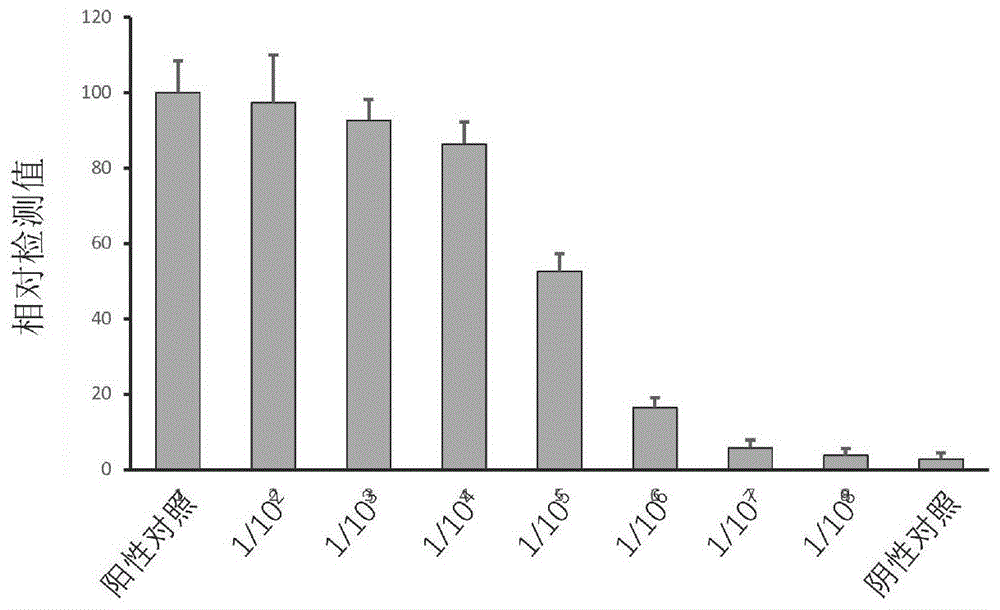
图1、对EML4-ALK融合基因1基因组DNA的检测。横轴为不同的被检测样本,其中“阳性对照”为表达EML4-ALK融合基因1的肿瘤细胞,“阴性对照”为表达野生型EML4、ALK基因的HEK293T细胞。按照一定的比例,将两种细胞进行混合,得到含有不同“肿瘤细胞/HEK293T细胞”比例的混合细胞样本,其中样本“1/10
图2、对
图3、对
图4、对
图5、对
图6、对
图7、对
图8、对
图9、对
图10、对
图11、对
图12、对
图13、对
图14、对
具体实施方式
下面结合具体实施例,进一步阐述本发明。应当理解,这些实施例仅用于说明本发明而不能用于限制本发明的范围。下列实施例中未注明具体条件的实验,通常按照常规的实验条件,如分子克隆:实验室手册(New York:Cold Spring Harbor Laboratory Press,1989,Sambrook等人著)中所描述的实验条件,或按照设备或试剂盒制造厂商所建议的实验条件进行。
实施例一:PaRNase H2蛋白的表达及纯化
Pyrococcus abyssi是一种嗜热菌,其编码的PaRNase H2(Gene ID:1495420;Locus tag:PAB_RS02765;蛋白编号:WP_010867642)蛋白能够耐受95℃的高温,适合于本发明目的的应用。本实施例以PaRNase H2蛋白为例,说明热稳定性RNase H2蛋白的表达和纯化方案。
通过检索基因数据库,获得PaRNase H2蛋白的cDNA序列。对原始序列进行密码子优化,合成全长的cDNA序列,在其氨基末端加上六个连续的组氨酸标记,用于辅助表达蛋白的纯化。按照novagen公司提供的《PET系统操作手册》描述的实验方案,将带有组氨酸标记的PaRNase H2蛋白序列插入到原核表达载体pet20b中,构建PaRNase H2表达载体。将该表达载体转入大肠杆菌,进行扩增,对获得的表达质粒进行基因测序验证。
获得测序正确的原核表达载体,再次转入大肠杆菌宿主细胞中,将其中的一个单克隆在37℃条件下培养过夜。取400uL过夜培养液,加入到100mL含有氨苄青霉素的LB培养基中,在37℃条件下震荡培养3小时左右,使菌液的OD600值达到0.3-0.6。加入0.5mM的IPTG溶液,在37℃条件下培养过夜,诱导蛋白表达。
按照常规的实验方案,利用镍柱纯化带His标签的PaRNase H2蛋白,将纯化的PaRNase H2蛋白进行定量,贮存于-20℃条件下备用,贮存液含有10mM Tris pH 8.0、1mMEDTA、100mMNaCl、0.1%Triton X-100、50%甘油。
本实施例研究结果表明,我们制备了纯化的PaRNase H2蛋白。
实施例二:PaRNase H2核酸酶的切割活性研究
为了研究PaRNase H2蛋白的活性,设计了一系列包含核糖核苷酸的单链引导核酸分子,分别命名为Seq01-Fr(SEQ ID NO:0101)、Seq02-Fr(SEQ ID NO:0102)、Seq03-Fr(SEQID NO:0103)、Seq04-Fr(SEQ ID NO:0104)、Seq05-Fr(SEQ ID NO:0105)、Seq06-Fr(SEQ IDNO:0106)、Seq07-Fr(SEQ ID NO:0107)、Seq08-Fr(SEQ ID NO:0108)、Seq09-Fr(SEQ IDNO:0109)、Seq10-Fr(SEQID NO:0110)。这些单链引导核酸分子的长度为26个核苷酸,其第21位为核糖核苷酸(用带下划线的核苷酸表示),3’末端为双脱氧核糖核苷酸(ddA,ddG,ddT,ddC),其他位置为脱氧核糖核苷酸。针对每条单链引导核酸分子序列,设计一条单链靶DNA分子,分别命名为Seq01-T(SEQ ID NO:0201)、Seq02-T(SEQ ID NO:0202)、Seq03-T(SEQID NO:0203)、Seq04-T(SEQ ID NO:0204)、Seq05-T(SEQ ID NO:0205)、Seq06-T(SEQ IDNO:0206)、Seq07-T(SEQ IDNO:0207)、Seq08-T(SEQ ID NO:0208)、Seq09-T(SEQ ID NO:0209)、Seq10-T(SEQ ID NO:0210)。单链靶DNA分子的长度为35个核苷酸,均由脱氧核糖核苷酸组成,能够与单链引导核酸分子形成碱基互补配对,具体如下。
Seq01-Fr(SEQ ID NO:0101)5’------GTTCGAGAAGCCCTTCGCCT
Seq01-T(SEQ ID NO:0201)3’-CGCTCCAAGCTCTTCGGGAAGCGGATGAGGCGGTA
Seq02-Fr(SEQ ID NO:0102)5’------TCAGGAGAACGTTAGTATAG
Seq02-T(SEQ ID NO:0202)3’-CTTTAAGTCCTCTTGCAATCATATCAATCGTTTAA
Seq03-Fr(SEQ ID NO:0103)5’------AGGATCAAAATTACTTAGTA
Seq03-T(SEQ ID NO:0203)3’-CGTTATCCTAGTTTTAATGAATCATTAAATATAAA
Seq04-Fr(SEQ ID NO:0104)5’------CATAAACTGGGTGGGCAGAG
Seq04-T(SEQ ID NO:0204)3’-ATCTAGTATTTGACCCACCCGTCTCGGTGGCGGGT
Seq05-Fr(SEQ ID NO:0105)5’------GGTGTTAACACGGGCTAAAG
Seq05-T(SEQ ID NO:0205)3’-GCCCGCCACAATTGTGCCCGATTTCGCCATCATCC
Seq06-Fr(SEQ ID NO:0106)5’------CATTTTTGAGTATGTCCCCG
Seq06-T(SEQ ID NO:0206)3’-AGCAAGTAAAAACTCATACAGGGGCCACGGGCAGA
Seq07-Fr(SEQ ID NO:0107)5’------TTCAGATCCTCTGGATTAAC
Seq07-T(SEQ ID NO:0207)3’-ACTGCAAGTCTAGGAGACCTAATTGCTCACTGCAG
Seq08-Fr(SEQ ID NO:0108)5’------CTGTCACCATGTAGGTACCG
Seq08-T(SEQ ID NO:0208)3’-GTCTTGACAGTGGTACATCCATGGCTGATCCCAGC
Seq09-Fr(SEQ ID NO:0109)5’------AGACAAATGTCCCAACGGGC
Seq09-T(SEQ ID NO:0209)3’-GACGTTCTGTTTACAGGGTTGCCCGTGGTCATCAC
Seq10-Fr(SEQ ID NO:0110)5’------AAGCGGATCTCACGCGTTGT
Seq10-T(SEQ ID NO:0210)3’-CTAAGTTCGCCTAGAGTGCGCAACATTTTACTCGG
注:带下划线的核苷酸为核糖核苷酸;ddA、ddG、ddT、ddC为具有链终止作用的双脱氧核糖核苷酸;G-C3spacer、T-C3spacer为带有C3 spacer结构的链终止核苷酸;其他的为脱氧核糖核苷酸。
RNase H2核酸酶是逆转录病毒整合酶超家族的一个成员,在基因组序列的切除修复中发挥了重要作用。该核酸酶保守存在于真核细胞以及原核细胞中,能够识别DNA双链中出现的单个核糖核苷酸掺入,并在该核苷酸的5’端进行切割。对于由单链引导核酸分子与单链靶DNA分子形成的双链结构,PaRNase H2蛋白能够识别、结合这种双链结构,并在单链引导核酸分子的核糖核苷酸位点的5’端进行切割。为了研究PaRNase H2蛋白的切割作用,我们开展了以下研究。
1、切割反应体系的优化
按照以下的组成,设置9个切割反应体系,每个反应中添加不同浓度的PaRNase H2核酸酶。不同反应体系中,PaRNase H2核酸酶添加的终浓度分别为0ng/uL、0.005ng/uL、0.01ng/uL、0.025ng/uL、0.05ng/uL、0.075ng/uL、0.1ng/uL、0.15ng/uL、0.2ng/uL。针对每个酶浓度,进行三次独立的重复实验。
其中,5×Reaction Buffer含有:50mM Tris-Cl(pH8.0),250mM NaCl,20mMMgCl2,50ug/mL BSA,0.05%Triton X-100。将反应体系至于70℃水浴中,反应20分钟;加入EDTA至终浓度20mM终止反应。将反应产物上样到20%的尿素变性PAGE凝胶中,进行电泳分离。用GelRed染色液对PAGE凝胶进行染色,用凝胶成像仪对染色结果成像,用ImageJ软件对分离的核酸条带进行定量分析。
以未添加PaRNase H2核酸酶的反应体系为参照,将其中单链引导核酸分子电泳条带的量定义为100%,计算不同反应体系中经PaRNase H2核酸酶切割后剩余的单链引导核酸分子的量。计算结果如下:
从这个结果可以看出,当PaRNase H2核酸酶的终浓度为0.05ng/uL时,即能够有效切割浓度为4uM的单链引导核酸分子。所产生的切割反应产物的浓度已高于PCR反应常用的引物浓度,使得该切割反应有可能与PCR反应偶联。为了保证切割反应的充分性,将PaRNaseH2核酸酶的添加量固定为0.1ng/uL。
2、对不同核酸链的切割作用的研究
按照以上描述的方案,以单链引导核酸分子Seq01-Fr、Seq02-Fr、Seq03-Fr、Seq04-Fr、Seq05-Fr、Seq06-Fr、Seq07-Fr、Seq08-Fr、Seq09-Fr、Seq10-Fr与其对应的单链靶DNA分子所形成的双链为底物,分析PaRNase H2核酸酶的切割活性。针对每个单链引导核酸分子,进行三次独立的重复实验。切割反应体系如下:
针对每条单链引导核酸分子与其对应的单链靶DNA分子形成的双链底物,设置一个阴性对照反应,阴性对照反应中不添加PaRNase H2核酸酶,反应总体积仍保持为10μL。将所有反应体系至于70℃水浴中,反应20分钟;加入EDTA至终浓度20mM终止反应。将反应产物上样到20%的尿素变性PAGE凝胶中,进行电泳分离。用GelRed染色液对PAGE凝胶进行染色,用凝胶成像仪对染色结果成像,用ImageJ软件对分离的核酸条带进行定量分析。
以阴性对照反应中单链引导核酸分子电泳条带的量定义为参照,计算PaRNase H2核酸酶对不同双链底物的切割效率。具体如下:
实验结果显示,对于由不同单链引导核酸分子形成的双链结构,PaRNase H2核酸酶均能进行有效的切割,表明该切割作用是具有普适性的。另外,PaRNase H2核酸酶对不同双链分子的切割效果存在差异,提示双链分子的核酸序列、或其二级结构特征影响了PaRNase H2核酸酶的切割作用。
实施例三:PaRNase H2核酸酶对融合基因片段的选择性切割
为了实现对肿瘤细胞来源的融合基因的检测,首先需要能将其与正常细胞来源的野生型基因片段区分开来,具有对融合基因片段的选择性区分能力。对基于PaRNase H2核酸酶的检测方案来说,就需要其能实现对融合基因、野生型基因片段的差异性切割。
1、对融合基因位点片段的切割作用研究
为了研究PaRNase H2核酸酶对融合基因片段的选择性切割能力,分别获得单链野生型上游基因位点片段Seq21-T(SEQ ID NO:0221)、单链野生型下游基因位点片段Seq22-T(SEQ IDNO:0222)、单链融合基因位点片段Seq23-T(SEQ ID NO:0223)。针对融合基因位点,设计单链引导核酸分子Seq23-Fr(SEQ ID NO:0123)。
Seq21-T(SEQ ID NO:0221)3'-GAGGACCACGAAGGCCGCCATGTGTTAGTACTACGGCCTCTTTCGGTC
Seq22-T(SEQ ID NO:0222)3'-AACAAACGACTAAACTATATGTGGAAATCCAGGAAAGGGTCCACACCC
Seq23-Fr(SEQ ID NO:0123)5'------GGTGCTTCCGGCGGTACAC
Seq23-T(SEQ ID NO:0223)3'-GAGGACCACGAAGGCCGCCATGTGAAATCCAGGAAAGGGTCCACACCC
注:带下划线的核苷酸为核糖核苷酸;G-C3spacer为带有C3 spacer结构的链终止核苷酸;其他为脱氧核糖核苷酸。
按照实施例二描述的方案,以Seq23-Fr分别与Seq21-T(SEQ ID NO:0221)、Seq22-T(SEQID NO:0222)、Seq23-T形成的双链为底物,分析PaRNase H2核酸酶对这些双链结构的切割活性。针对每个双链底物,进行三次独立的重复实验。
实验结果显示,对Seq23-Fr/Seq21-T中Seq23-Fr的切割效率为8.8±1.6%,对Seq23-Fr/Seq22-T中Seq23-Fr的切割效率为4.6±2.1%,对Seq23-Fr/Seq23-T中Seq23-Fr的切割效率为92.6±4.2%。这个结果表明PaRNase H2核酸酶能对与融合基因片段、野生型基因片段结合的单链引导核酸分子产生差异性切割作用,对与融合基因片段结合的单链引导核酸分子产生更强的切割作用。
2、对不同融合序列的切割作用研究
为了进一步分析PaRNase H2核酸酶对融合基因片段的选择性切割能力,我们设计了如下九组融合基因及野生型基因序列。以Seq31-T(SEQ ID NO:0231)、Seq31-Fr(SEQ IDNO:0131)、Seq41-T(SEQ ID NO:0241)、Seq51-T(SEQ ID NO:0251)这一组为例进行说明:Seq31-Fr为单链引导核酸分子,Seq31-Fr为融合基因位点片段,Seq41-T、Seq51-T分别为两个野生型基因位点片段。单链引导核酸分子分别与融合基因位点片段、以及两个野生型基因位点片段形成可能的双链结构,并引导PaRNase H2核酸酶作用于这些双链底物,在核糖核苷酸杂合位点的5’端进行切割。
Seq31-Fr(SEQ ID NO:0131)5’------GGTTCGAGAAGCCCTTCGGC
Seq31-T(SEQ ID NO:0231)3’-TCGCTCCAAGCTCTTCGGGAAGCCGCCAGATCCATCTCCCCTACCCAT
Seq41-T(SEQ ID NO:0241)3’-TCGCTCCAAGCTCTTCGGGAAGCGAGCCCGCCACAATTGTGCCCGATT
Seq51-T(SEQ ID NO:0251)3’-CTGCAACTTTATCCGCCTCCATCCGCCAGATCCATCTCCCCTACCCAT
Seq32-Fr(SEQ ID NO:0132)5’------TCCGCCATGATTCAAGCGG
Seq32-T(SEQ ID NO:0232)3’-GGATGAGGCGGTACTAAGTTCGCCCGGAAGGGCCGAGCGCAGAAGTGG
Seq42-T(SEQ ID NO:0242)3’-GGATGAGGCGGTACTAAGTTCGCTCGCCATCATCCATCTAGTATTTGA
Seq52-T(SEQ ID NO:0252)3’-CAGTCTATTAATTGTTGCCGGGACCGGAAGGGCCGAGCGCAGAAGTGG
Seq33-Fr(SEQ ID NO:0133)5’------CACGCGTTGTAAAATGAGCTA
Seq33-T(SEQ ID NO:0233)3’-CTAGAGTGCGCAACATTTTACTCGATTTATCAGCAATAAACCAGCCAG
Seq43-T(SEQ ID NO:0243)3’-CTAGAGTGCGCAACATTTTACTCCCCACCCGTCTCGGTGGCGGGTCGT
Seq53-T(SEQ ID NO:0253)3’-AGCTAGAGTAAGTAGTTCGCCAGGATTTATCAGCAATAAACCAGCCAG
Seq34-Fr(SEQ ID NO:0134)5’------CAAGACAAATGTCCCAACCGC
Seq34-T(SEQ ID NO:0234)3’-GGGACGTTCTGTTTACAGGGTTGGCGAGACCCACGCTCACCGGCTCCA
Seq44-T(SEQ ID NO:0244)3’-GGGACGTTCTGTTTACAGGGTTGTATCCTAGTTTTAATGAATCATTAA
Seq54-T(SEQ ID NO:0254)3’-TTAATAGTTTGCGCAACGTTGTTGCGAGACCCACGCTCACCGGCTCCA
Seq35-Fr(SEQ ID NO:0135)5’------CCAGTAGTGCAGAACTGTGA
Seq35-T(SEQ ID NO:0235)3’-CCCGTGGTCATCACGTCTTGACACTGGCCCCAGTGCTGCAATGATACC
Seq45-T(SEQ ID NO:0245)3’-CCCGTGGTCATCACGTCTTGACAATATAAACTTTAAGTCCTCTTGCAA
Seq55-T(SEQ ID NO:0255)3’-GCCATTGCTACAGGCATCGTGGTCTGGCCCCAGTGCTGCAATGATACC
Seq36-Fr(SEQ ID NO:0136)5’------TGTAGGTACCGACTAGGGTG
Seq36-T(SEQ ID NO:0236)3’-GTGGTACATCCATGGCTGATCCCACTACGATACGGGAGGGCTTACCAT
Seq46-T(SEQ ID NO:0246)3’-GTGGTACATCCATGGCTGATCCCTCATATCAATCGTTTAATCAATGAG
Seq56-T(SEQ ID NO:0256)3’-GTCACGCTCGTCGTTTGGTATGGACTACGATACGGGAGGGCTTACCAT
Seq37-Fr(SEQ ID NO:0137)5’------ACGTTCAGATCCTCTGGA
Seq37-T(SEQ ID NO:0237)3’-AGCACTGCAAGTCTAGGAGACCTTGCCTGACTCCCCGTCGTGTAGATA
Seq47-T(SEQ ID NO:0247)3’-AGCACTGCAAGTCTAGGAGACCTGCTGCTAGATATCTCGATTTCACTG
Seq57-T(SEQ ID NO:0257)3’-CTTCATTCAGCTCCGGTTCCCAATGCCTGACTCCCCGTCGTGTAGATA
Seq38-Fr(SEQ ID NO:0138)5’------GAGTGACGTCTCGTTCATA
Seq38-T(SEQ ID NO:0238)3’-AATTGCTCACTGCAGAGCAAGTATCTGTCTATTTCGTTCATCCATAGT
Seq48-T(SEQ ID NO:0248)3’-AATTGCTCACTGCAGAGCAAGTAGAGCTTACGATCCTTCCCGGATCAT
Seq58-T(SEQ ID NO:0258)3’-CGATCAAGGCGAGTTACATGATCTCTGTCTATTTCGTTCATCCATAGT
Seq39-Fr(SEQ ID NO:0139)5’------AGTATGTCCCCGGTGCCC
Seq39-T(SEQ ID NO:0239)3’-AAAACTCATACAGGGGCCACGGGCTCAGTGAGGCACCTATCTCAGCGA
Seq49-T(SEQ ID NO:0249)3’-AAAACTCATACAGGGGCCACGGGCAACATACGAGGCCCCCGAGTCGGT
Seq59-T(SEQ ID NO:0259)3’-CCCCATGTTGTGCAAAAAAGCGGCTCAGTGAGGCACCTATCTCAGCGA
注:带下划线的核苷酸为核糖核苷酸;ddA、ddG、ddT、ddC为具有链终止作用的双脱氧核糖核苷酸;G-C3spacer、C-C3spacer为带有C3 spacer结构的链终止核苷酸;其他为脱氧核糖核苷酸。
针对每个双链底物,进行三次独立的重复实验,实验结果如下:
分析这些实验结果,发现对于具有不同碱基互补配对程度的双链核酸分子,PaRNase H2核酸酶对与融合基因片段结合的单链引导核酸分子的切割效率较高,而对与野生型基因片段结合的单链引导核酸分子则基本不产生有效的切割作用。这表明PaRNase H2核酸酶能对与融合基因片段、野生型基因片段结合的单链引导核酸分子产生差异性切割作用。
实施例四:PaRNase H2对融合基因细胞基因组DNA的检测
为了实现对肿瘤细胞来源的低丰度融合基因的检测,还需要克服海量的来源于野生型细胞基因组DNA对扩增过程的干扰,这是肿瘤早期诊断技术的难度所在。为了解决这个问题,我们以EML4-ALK融合基因片段的选择性扩增为例,开展本实施例研究。
人类棘皮动物微管相关蛋白样4基因(Echinoderm microtubule-associatedprotein like 4,EML4,NM_019063.5)与间变性淋巴瘤激酶基因(Anaplastic lymphomakinase,ALK,NM_004304.5),分别位于人类2号染色体的p21和p23区带,相隔约10Mb的距离。在非小细胞肺癌患者体内,这两个基因常常会发生融合,形成具有致瘤性的EML4-ALK融合基因。通过对临床肿瘤标本的研究,十余年来人类鉴定了一批具有生物学活性的EML4-ALK融合基因,我们选取其中的五个为代表,开展研究。这五个融合基因分别为
1、对融合基因细胞基因组DNA的检测
针对这五个融合基因的基因组序列,分别设计能够适用于PaRNase H2核酸酶差异性切割的单链引导核酸分子、以及能够与之配合实现对融合基因位点进行PCR扩增的下游扩增引物。具体如下:
Seq81-T(SEQ ID NO:0301)5’-aaaactactgtagagccca
Seq81-Fr(SEQ ID NO:0311)5’-CACCTGGGAAAGGACCTAAAGT/rG/TACC-C3spacer
Seq81-Rprimer(SEQ ID NO:0321)5’-GAGTCTGCGGTGCTGTGATA
Seq82-Fr(SEQ ID NO:0312)5’-CTAACTCGGGAGACTATGAAATATTGTACTT/rG/TACC-C3spacer
Seq82-Rprimer(SEQ ID NO:0322)5’-GAGTCTGCGGTGCTGTGATA
Seq83-Fr(SEQ ID NO:0313)5’-CCTGAAAGAGAAATAGAGATATGCTGGATG/rA/GCCC-C3spacer
Seq83-Rprimer(SEQ ID NO:0323)5’-TACATGGCCCTGAATGTCAA
Seq84-Fr(SEQ ID NO:0314)5’-CTCAGTGAAAAAATCAGTCTCAAGTAAAGT/rG/TACC-C3spacer
Seq84-Rprimer(SEQ ID NO:0324)5’-GAGTCTGCGGTGCTGTGATA
Seq85-Fr(SEQ ID NO:0315)5’-TCATGATCTGAATCCTGAAAGAGAAATAGA/rG/CACC-C3spacer
Seq85-Rprimer(SEQ ID NO:0325)5’-GAGTCTGCGGTGCTGTGATA
注:如“/rG/”、“/rA/”所示的核苷酸为核糖核苷酸;C-C3spacer为带有C3 spacer结构的链终止核苷酸;其他为脱氧核糖核苷酸。
以
Seq82-Fr(SEQ ID NO:0312)、Seq82-Rprimer(SEQ ID NO:0322)为针对
获得并培养表达各种EML4-ALK融合基因的非小细胞肺癌细胞株(阳性对照细胞)、以及表达野生型EML4、ALK基因的HEK293T细胞(阴性对照细胞)。对EML4-ALK融合基因表达细胞、以及野生型细胞进行计数,按一定的比例,将两种细胞进行混合,得到含有不同比例EML4-ALK融合基因细胞的混合细胞样本,用以模拟含有不同浓度肿瘤细胞的临床样本。其中,EML4-ALK融合基因细胞/野生型细胞的比例分别为1:10
分别获得表达各种融合基因的阳性对照细胞、阴性对照细胞、以及含有不同浓度融合基因表达细胞的混合细胞样本,提取细胞样本的基因组DNA,作为扩增模板,设置如下扩增反应体系。针对每个扩增反应,进行三次独立的重复实验。
其中,2×Taq Master Mix购自天根生化科技公司。
PCR扩增反应条件为:在95℃条件下孵育1分钟,随后进行50个循环的PCR扩增(95℃,20sec→60℃,20sec→72℃,40sec),最后在72℃条件下,孵育5分钟。每个反应设置3个重复。
利用delta delta Ct法,对扩增产物、以及内参DNA分子的含量进行定量分析。具体方法和步骤可以参见文献:Livak KJ and Schmittgen TD.Analysis of relative geneexpression data usingreal-time quantitative PCR and the 2(-Delta Delta C(T))Method.Methods.2001Dec;25(4):402-408。首先以内参分子的检测值为基础,对扩增产物的检测值进行归一化处理。随后以阳性对照细胞的检测值为参照,计算其他样本中的相对检测值,并进行作图。
图1为
2、扩增片段的长度对检测灵敏度的影响
为了优化扩增条件,我们以
针对这两个融合基因的基因组序列,分别设计能够与单链引导核酸分子Seq81-Fr(SEQ IDNO:0311)、Seq82-Fr(SEQ ID NO:0312)配合,实现对融合基因位点进行PCR扩增的多个下游扩增引物,使PCR扩增反应的产物长度在100-1000个核苷酸之间。
具体的,对
获得并培养表达
按照前面描述的方案,分别提取阳性对照细胞、阴性对照细胞、以及混合细胞样本的基因组DNA。以这些基因组DNA作为扩增模板,利用不同的扩增引物对,进行PCR扩增。针对每个扩增反应,进行三次独立的重复实验。
按照上述实施例描述的方法处理数据,并作图。
图6为对
实施例五:PaRNase H2对融合基因mRNA的检测
为了提高对变异基因检测的灵敏度,我们进一步在mRNA水平检测了融合基因的表达情况,开展了本实施例研究。
按照实施例四描述的方案,以
Seq91-Fr(SEQ ID NO:0411)5’-CACCTGGGAAAGGACCTAAAGT/rG/TACC-C3spacer
Seq91-Rprimer(SEQ ID NO:0421)5’-CACCTGGCCTTCATACACCT
Seq92-Fr(SEQ ID NO:0412)5’-CTAACTCGGGAGACTATGAAATATTGTACTT/rG/TACC-C3spacer
Seq92-Rprimer(SEQ ID NO:0422)5’-CACCTGGCCTTCATACACCT
Seq93-Fr(SEQ ID NO:0413)5’-CCTGAAAGAGAAATAGAGATATGCTGGATG/rA/GCCC-C3spacer
Seq93-Rprimer(SEQ ID NO:0423)5’-ATGATCAGGGCTTCCATGAG
Seq94-Fr(SEQ ID NO:0414)5’-CTCAGTGAAAAAATCAGTCTCAAGTAAAGT/rG/TACC-C3spacer
Seq94-Rprimer(SEQ ID NO:0424)5’-CACCTGGCCTTCATACACCT
Seq95-Fr(SEQ ID NO:0415)5’-TCATGATCTGAATCCTGAAAGAGAAATAGA/rG/CACC-C3spacer
Seq95-Rprimer(SEQ ID NO:0425)5’-CACCTGGCCTTCATACACCT
注:如“/rG/”、“/rA/”所示的核苷酸为核糖核苷酸;C-C3spacer为带有C3 spacer结构的链终止核苷酸;其他为脱氧核糖核苷酸。
Seq91-Fr(SEQ ID NO:0411)、Seq91-Rprimer(SEQ ID NO:0421)为针对
按照实施例四描述的方案,获得并培养表达各种EML4-ALK融合基因的非小细胞肺癌细胞株(阳性对照细胞)、以及表达野生型EML4、ALK基因的HEK293T细胞(阴性对照细胞)。对EML4-ALK融合基因表达细胞、以及野生型细胞进行计数,按一定的比例,将两种细胞进行混合,得到含有不同比例EML4-ALK融合基因细胞的混合细胞样本,用以模拟含有不同浓度肿瘤细胞的临床样本。其中,EML4-ALK融合基因细胞/野生型细胞的比例分别为1:10
分别获得表达各种融合基因的阳性对照细胞、阴性对照细胞、以及含有不同浓度融合基因细胞的混合细胞样本,提取其中的总RNA,将其中的mRNA逆转录成cDNA。
按照实施例四中描述的方案,以获得的cDNA为模板,进行PCR扩增和数据处理。针对每个扩增反应,进行三次独立的重复实验。
图8为
实施例六:PaRNase H2对疾病样本的检测
获得四例具有
按照实施例五描述的方案,以得到的cDNA为被检测样本,检测其中融合基因的含量。以实施例五中的图8、图10为标准曲线,定量分析组织样本中肿瘤细胞的含量。针对每个被检测样本,进行三次独立的重复实验。
实验结果表明,
这些研究结果表明,本申请提供的研究方案能够稳定的检测组织样本中肿瘤细胞的含量。
实施例七:双切割检测方案
为了进一步提高融合基因检测的特异性,本发明还设计了一种双切割检测方案。下面以
针对这两个融合基因的基因组序列,设计靶向融合基因位点、且适应于PaRNaseH2核酸酶差异性切割的单链引导核酸分子、以及能够与之配合实现对融合基因位点进行PCR扩增的“切割型扩增引物”。与上述方案的不同之处在于,切割型扩增引物也被设计成适应于PaRNase H2核酸酶切割的单链引导核酸分子的形式。切割型扩增引物在引导基因扩增之前,需要先与同源的基因组序列互补配对,在经PaRNase H2核酸酶切割后,才能形成有活性的扩增引物,引导PCR扩增。
Seq101-Fr(SEQ ID NO:0511)5’-CTAACTCGGGAGACTATGAAATATTGTACTT/rG/TACC-C3spacer
Seq101-Rprimer(SEQ ID NO:0512)5’-GAGTCTGCGGTGCTGTGATA/rA/CATTCA-C3spacer
Seq102-Fr(SEQ ID NO:0511)5’-TCATGATCTGAATCCTGAAAGAGAAATAGA/rG/CACC-C3spacer
Seq102-Rprimer(SEQ ID NO:0512)5’-GAGTCTGCGGTGCTGTGATA/rA/CATTCAGC-C3spacer
注:如“/rG/”、“/rA/”所示的核苷酸为核糖核苷酸;A-C3spacer、C-C3spacer为带有C3 spacer结构的链终止核苷酸;其他为脱氧核糖核苷酸。
按照实施例四描述的方案,获得并培养表达
分别提取阳性对照细胞、阴性对照细胞、以及混合细胞样本的基因组DNA。以这些基因组DNA作为扩增模板,进行PCR扩增。分别利用扩增引物对Seq101-Fr/Seq101-Rprimer(双切割检测)和实施例四中对应的扩增引物对Seq82-Fr/Seq82-Rprimer(单切割检测)对阳性对照细胞、阴性对照细胞、以及含有
PCR扩增体系,具体如下:
按照上述实施例描述的方法,计算对各样本的扩增情况,并作图。
图13为
序列表
<110> 枣庄康泽生物科技有限公司
<120> 一种基因检测技术和应用
<160> 1
<170> SIPOSequenceListing 1.0
<210> 1
<211> 2663
<212> DNA
<213> 2 Ambystoma laterale x Ambystoma jeffersonianum
<400> 1
gttcgagaag cccttcgcct ctccccgctc caagctcttc gggaagcgga tgaggcggta 60
tcaggagaac gttagtatag atagcaactt taagtcctct tgcaatcata tcaatcgttt 120
aaaggatcaa aattacttag taatttatac gttatcctag ttttaatgaa tcattaaata 180
taaacataaa ctgggtgggc agaggcaccg atctagtatt tgacccaccc gtctcggtgg 240
cgggtggtgt taacacgggc taaagaggta cgcccgccac aattgtgccc gatttcgcca 300
tcatcccatt tttgagtatg tccccgctgc cctagcaagt aaaaactcat acaggggcca 360
cgggcagatt cagatcctct ggattaacaa gtgactgcaa gtctaggaga cctaattgct 420
cactgcagct gtcaccatgt aggtaccggc taggagtctt gacagtggta catccatggc 480
tgatcccagc agacaaatgt cccaacgggc tccagtgacg ttctgtttac agggttgccc 540
gtggtcatca caagcggatc tcacgcgttg ttaaatgcct aagttcgcct agagtgcgca 600
acattttact cgggaggacc acgaaggccg ccatgtgtta gtactacggc ctctttcggt 660
caacaaacga ctaaactata tgtggaaatc caggaaaggg tccacacccg gtgcttccgg 720
cggtacactt aggaggacca cgaaggccgc catgtgaaat ccaggaaagg gtccacaccc 780
ggttcgagaa gcccttcggc cgtctctcgc tccaagctct tcgggaagcc gccagatcca 840
tctcccctac ccattcgctc caagctcttc gggaagcgag cccgccacaa ttgtgcccga 900
ttctgcaact ttatccgcct ccatccgcca gatccatctc ccctacccat tccgccatga 960
ttcaagcggt ccttaggatg aggcggtact aagttcgccc ggaagggccg agcgcagaag 1020
tggggatgag gcggtactaa gttcgctcgc catcatccat ctagtatttg acagtctatt 1080
aattgttgcc gggaccggaa gggccgagcg cagaagtggc acgcgttgta aaatgagcta 1140
tatagctaga gtgcgcaaca ttttactcga tttatcagca ataaaccagc cagctagagt 1200
gcgcaacatt ttactcccca cccgtctcgg tggcgggtcg tagctagagt aagtagttcg 1260
ccaggattta tcagcaataa accagccagc aagacaaatg tcccaaccgc actcgggacg 1320
ttctgtttac agggttggcg agacccacgc tcaccggctc cagggacgtt ctgtttacag 1380
ggttgtatcc tagttttaat gaatcattaa ttaatagttt gcgcaacgtt gttgcgagac 1440
ccacgctcac cggctccacc agtagtgcag aactgtgagc gggggcccgt ggtcatcacg 1500
tcttgacact ggccccagtg ctgcaatgat acccccgtgg tcatcacgtc ttgacaatat 1560
aaactttaag tcctcttgca agccattgct acaggcatcg tggtctggcc ccagtgctgc 1620
aatgatacct gtaggtaccg actagggtgg tgcggtggta catccatggc tgatcccact 1680
acgatacggg agggcttacc atgtggtaca tccatggctg atccctcata tcaatcgttt 1740
aatcaatgag gtcacgctcg tcgtttggta tggactacga tacgggaggg cttaccatac 1800
gttcagatcc tctggatcgg tagcactgca agtctaggag accttgcctg actccccgtc 1860
gtgtagataa gcactgcaag tctaggagac ctgctgctag atatctcgat ttcactgctt 1920
cattcagctc cggttcccaa tgcctgactc cccgtcgtgt agatagagtg acgtctcgtt 1980
catagacaga aattgctcac tgcagagcaa gtatctgtct atttcgttca tccatagtaa 2040
ttgctcactg cagagcaagt agagcttacg atccttcccg gatcatcgat caaggcgagt 2100
tacatgatct ctgtctattt cgttcatcca tagtagtatg tccccggtgc cccagtcaaa 2160
actcatacag gggccacggg ctcagtgagg cacctatctc agcgaaaaac tcatacaggg 2220
gccacgggca acatacgagg cccccgagtc ggtccccatg ttgtgcaaaa aagcggctca 2280
gtgaggcacc tatctcagcg aaaactactg tagagcccac acctgggaaa ggacctaaag 2340
tgtaccgccg gaagcaccag gagctgcaag ccatgcagat ggagctgcag agccctgagt 2400
acaagctgag caagctccgc acctcgacca tcatgaccga ctacaacccc aactactgct 2460
ttgctggcaa gacctcctcc atcagtgacc tgaaggaggt gccgcggaaa aacatcaccc 2520
tcattcggtg agcgccctgc tgccgtcctg ggaggagagg ggtgcagtgt aggggctgaa 2580
tgttatcaca gcaccgcaga ctcctctagc cacaaacacc tgggaaagga cctaaagtgt 2640
accgagtctg cggtgctgtg ata 2663
- 一种基于高通量测序技术检测DMD基因变异的引物及其应用
- 一种基于高通量测序技术用于检测甲状腺癌致病相关基因变异的引物及其应用
- 基于基因编辑技术的双信号输出检测转基因大豆的电化学传感器的制备方法及其应用
- 基于基因编辑技术的双信号输出检测转基因大豆的电化学传感器的制备方法及其应用