一种环磷腺苷葡胺注射液有关物质检测方法
文献发布时间:2024-04-18 19:58:30
技术领域
本发明属于药物分析领域,具体涉及一种环磷腺苷葡胺注射液有关物质检测方法。
背景技术
环磷腺苷为参与调节细胞功能的第二信使物质,其作用非常广泛,注射大剂量的环磷腺苷,能使心肌收缩力增强,引起血压升高,心输出量增高,并能舒张平滑肌,扩张冠状动脉血管,改善肝功能,促进神经再生,抑制皮肤外层上皮细胞分裂及转化异常细胞的功能,促进呼吸链氧化酶的活性及改善心肌缺氧等。
葡甲胺易溶于水,通过增强环磷腺苷脂溶性,提高环磷腺苷纯度,使得环磷腺苷发挥最大程度的药理作用。
环磷腺昔微溶于水,在环磷腺中加入葡甲胺可增加其溶解性。环磷腺苷葡胺注射液为细胞信使,具有调节细胞多种功能,增强心肌收缩力,扩张心脑及外周血管,降低心肌耗氧量,改善心肌代谢等作用,并可促使受损窦房结功能的恢复。用于治疗病窦综合症,急性心肌炎,扩张型心肌病,冠心病心绞痛,各种病因所致的心力衰竭,对洋地黄不敏感或中毒的心衰患者疗效尤佳。
环磷腺苷是以 5′-腺苷酸为原料,在无水条件下通过内酯化反应而制得。在反应液中,若吡啶脱水不完全,有可能产生腺苷[6]。并且环磷腺苷易水解,其产物为 5-磷酸腺苷, 该物质会使得溶液 pH 值降低,杂质含量升高。因此,我们探索条件,建立一种简便、快速、灵敏的高效液相色谱法测定环磷腺苷葡萄糖注射液的含量及有关物质,用于药物生产过程中质量控制。
《中国药典》2020年版仅收载了环磷腺苷原料药的有关物质检测方法,采用高效液相色谱法,我们参考环磷腺苷原料药有关物质检测方法及杂质控制情况,对中国药典中环磷腺苷原料药有关物质检测方法进行优化,得到最终的环磷腺苷葡胺注射液有关物质检测方法
文献:HPLC法测定环磷腺昔葡萄糖注射液含量及有关物质,该方法某些特定杂质不能完全分开,不能对特定杂质进行检测,不能控制其杂质量。本发明方案通过优化后能对5个特定杂质进行很好的分离,从而对特定杂质进行控制,使方法更有特异性。
发明内容
本发明旨在提供一种简单、高效、灵敏度及专属性强的环磷腺苷葡胺注射液有关物质检测方法。
为实现上述发明目的,本发明一种环磷腺苷葡胺注射液有关物质检测方法,具体实施方案为:
本发明所述一种环磷腺苷葡胺注射液有关物质检测方法,该方法采用高效液相色谱法,色谱柱以十八烷基硅烷键合硅胶为填充剂,流动相为磷酸氢二钾和乙腈作为溶液,磷酸氢二钾:乙腈比例为86-96:4-14。
本发明所述一种环磷腺苷葡胺注射液有关物质检测方法,流动相磷酸氢二钾:乙腈比例为90-94:6-10。
本发明所述一种环磷腺苷葡胺注射液有关物质检测方法,流动相磷酸氢二钾:乙腈为比例94:6。
本发明所述一种环磷腺苷葡胺注射液有关物质检测方法,流动相磷酸氢二钾浓度为0.02mol/l。
本发明所述一种环磷腺苷葡胺注射液有关物质检测方法,包括以下步骤:
1)配制供试品溶液:以流动相为溶剂,溶解待测样品,配制得到供试品溶液;
2)取供试品溶液和对照溶液分别进样,用高效液相色谱进行检测,记录色谱图。
本发明所述一种环磷腺苷葡胺注射液有关物质检测方法,步骤2)所述的高效液相色谱条件为:色谱柱:十八烷基硅烷键合硅胶为填充剂;检测波长:242-260nm;柱温:25-30℃;流速:1.0ml/min;流动相:磷酸氢二钾-乙腈。
本发明所述一种环磷腺苷葡胺注射液有关物质检测方法,步骤2)所述的高效液相色谱条件为:色谱柱:十八烷基硅烷键合硅胶为填充剂,SymmetryShield RP18,5μm,4.6mm×250mm;检测波长:258nm;柱温:30℃;流速:1.0ml/min;流动相:磷酸氢二钾-乙腈比例为94:6。
本发明所述一种环磷腺苷葡胺注射液有关物质检测方法,该方法供试品溶液中环磷腺苷及已知杂质与相邻杂质峰之间的分离度不小于1.5。
本发明所述一种环磷腺苷葡胺注射液有关物质检测方法,该方法能有效分离的已知杂质为腺苷(AR)、腺嘌呤(AD)、布达地新杂质5(5’AMP)、腺苷2’3’-单磷酸混合异构体(2’AMP)、腺苷杂质N9(3’AMP)。
本发明所述一种环磷腺苷葡胺注射液有关物质检测方法,该方法能有效分离的杂质结构式为:
。
本发明优势分析:
现有文献方法杂质不能完全分离,本发明方案通过优化后能对5个特定杂质进行很好的分离,从而对特定杂质进行控制,使方法更有特异性,从而提高环磷腺苷葡胺注射液的质量控制。
附图说明
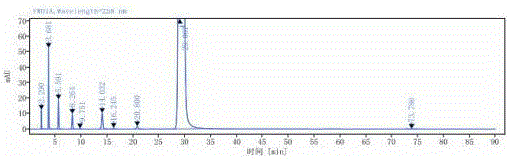
图1:实施例1中系统适用性专属性色谱图
图2:实施例1中空白溶液专属性色谱图
图3:实施例1中空白辅料专属性色谱图
图4:实施例1中供试品专属性色谱图
图5:实施例7中杂质3’AMP、AD、AR、环磷腺苷线性色谱图
图6:实施例7中杂质2’AMP线性色谱图
图7:实施例8中不同流速供试品色谱图
图8:环磷腺苷线性
图9:杂质AD线性
图10:杂质AR线性
图11:杂质5’AMP线性
图12:杂质3’AMP线性
图13:杂质2’AMP线性。
实施方式
下面实施例只为进一步说明本发明技术方案,不以任何形式限制本发明范围。
制剂供试品溶液配制:精密量取本品2ml到20ml量瓶中,用流动相稀释至刻度,摇匀,即得(每1ml含环磷腺苷葡胺1.5mg)。供试品混合溶液:精密量取本品2ml到20ml,精密加入杂质AD、AR、5’AMP、2’(3’)AMP、3’AMP,用流动相稀释至刻度,摇匀,即得(每1ml含环磷腺苷葡胺1.5mg,各杂质2μg/ml)实施例1:专属性
(1)色谱条件:
检测仪器:Agilent1260高效液相色谱仪
色谱柱:SymmetryShield RP18,5μm,4.6mm×250mm
检测波长:258nm
柱温:30℃
流速:1.0ml/min
进样体积:20μl
流动相:0.02mol/l磷酸氢二钾-乙腈(94∶6)
(2)试验方法
取空白溶液(流动相)、空白辅料、对照品溶液、供试品溶液、混合杂质溶液各20µl注入液相色谱仪,记录色谱图。
(3)试验结果
如表1所示,空白溶剂、空白辅料溶液对环磷腺苷葡胺测定无干扰;混合杂质溶液中环磷腺苷峰与相邻杂质峰的分离度符合要求;供试品溶液中环磷腺苷及已知杂质与相邻杂质峰之间的分离度不小于1.5。见附图1-4。
表1-1:专属性试验结果:
表1-2:强制降解试验结果见表:
实施例2:重复性
(1)试验方法
色谱条件同实施例1,平行制备6份含供试品溶液,测定各杂质的含量。
(2)试验结果
如表2所示,6次供试品溶液检测结果的极差最大为0.007,小于0.02。该方法重复性良好。
表2:重复性试验结果
实施例3:中间精密度
(1)试验方法
检测仪器:Agilent1260高效液相色谱仪
其余色谱条件同实施例1,由不同分析人员于不同日期使用不同设备重复测定6份供试品混合溶液各杂质的含量。
(2)试验结果
如表3所示,实验员2的6次供试品溶液检测结果的RSD为0.13%,小于1%。12次检测结果的平均值为100.179%,RSD为0.59%,小于2%。该方法中间精密度符合要求。
表3:中间精密度试验结果
实施例4:准确度
(1)试验方法
色谱条件同实施例1,平行制备6份准确度溶液,测定各杂质的含量。
(2)试验结果
分别按外标法及加校正因子的自身对照法,计算各个杂质的回收率,各浓度点回收率应在80%~120%之间,回收率结果的RSD应不大于15.0%,准确度试验结果见表4-1~4-10。
表4-1 杂质AD外标法回收率试验结果
表4-2 杂质AD加校正因子自身对照法回收率试验结果
表4-3 杂质AR外标法回收率试验结果
表4-4 杂质AR加校正因子自身对照法回收率试验结果
表4-5 杂质5’AMP外标法回收率试验结果
表4-6 杂质5’AMP加校正因子自身对照法回收率试验结果
表4-7 杂质3’AMP外标法回收率试验结果
表4-8 杂质3’AMP加校正因子自身对照法回收率试验结果
表4-9 杂质2’AMP外标法回收率试验结果
表4-10 杂质2’AMP加校正因子自身对照法回收率试验结果
实施例5:溶液稳定性
(1)试验方法
色谱条件同实施例1,将对照溶液及加标供试品溶液置室温条件下,分别于0、2、4、8、12、24、48小时,精密量取20μl注入液相色谱仪,记录色谱图。
(2)试验结果
检测结果见表5-1和表5-2。
表5-1 加标供试品溶液稳定性试验结果
表5-2 自身对照溶液稳定性试验结果
实施例6:定量限检测限
(1)试验方法
色谱条件同实施例1,精密量取环磷腺苷、AD、AR、5'AMP、3'AMP、2'AMP对照品贮备液用流动相逐级稀释制成信噪比(S/N)≥10的溶液作为环磷腺苷、AD、AR、5'AMP、3'AMP、2'AMP定量限溶液,测定6次,各成分峰面积的RSD≤10.0%,环磷腺苷、AD、AR、5’AMP、3’AMP、2’AMP检测限(LOD)溶液:精密量取定量限溶液3ml用流动相稀释至10ml,取该溶液作为检测限溶液。
(2)试验结果
试验结果见表6-1-6-2:
表6-1 定量限试验结果
表6-2 检测限试验结果
实施例7:线性及校正因子
(1)试验方法
色谱条件同实施例1,精密称取AD、AR、5’AMP、3’AMP、2’AMP对照品各2mg,分别置100ml量瓶中,加流动相溶解并稀释至刻度,摇匀,作为对照品贮备液(20μg/ml)。用流动相稀释制成对应限度浓度的线性溶液。分别精密量取20μl注入液相色谱仪,记录色谱图,计算回归方程及相关系数r≥0.990、各浓度点响应因子RSD≤10.0%、Y轴截距占100%浓度响应百分比≤25%,即认为其在该浓度范围内线性关系良好,
(2)试验结果
线性试验结果1见表7-1~7-6及图8~13。
表7-1 环磷腺苷线性试验结果1
/>
环磷腺苷线性见图8。
表7-2 杂质AD线性试验结果1
杂质AD线性见图9。
表7-3 杂质AR线性试验结果1
杂质AR线性见图10
表7-4 杂质5’AMP线性试验结果1
杂质5’AMP线性见图11 。
表7-5 杂质3’AMP线性试验结果1
杂质3’AMP线性见图12。
表7-6 杂质2’AMP线性试验结果1
/>
杂质2’AMP线性见图13。
表7-7 已知杂质校正因子1测定结果
实施例8:耐用性试验
(1)试验方法
色谱条件同实施例1,分别于流速变化±0.1ml/min、柱温变化±2℃、检测波长变化±2nm及不同色谱柱的条件下,精密量取空白溶剂、系统适用性溶液、加标供试品溶液、对照溶液各20μl,注入液相色谱仪,记录色谱图,考察各条件下系统适用性溶液中杂质分离情况是否符合系统适用性要求,同时比较各耐用性条件与正常条件下按加校正因子的自身对照法测得的有关物质结果的差值,是否符合要求
(2)试验结果
耐用性试验结果见表8-1~8-3。
表8-1 耐用性试验系统适用性结果
表8-2 耐用性试验杂质检测结果
表8-3 耐用性条件与原条件杂质含量的差值结果
试验结果表明:经过对环磷腺苷葡胺注射液有关物质测定方法的专属性与系统适用性、溶液稳定性、定量限、线性与范围、校正因子、耐用性、精密度、准确度进行验证,验证结果均符合相关验证要求,本方法可用于环磷腺苷葡胺注射液有关物质的测定。
根据上述实验结果证明:本发明方法操作简便,高效,灵敏度及专属性强,有较高的耐用性,符合中国药典方法验证指导原则要求,能对环磷腺苷葡胺注射液有关物质进行定量分析,进行质量控制。
所属领域的普通技术人员应当理解:以上任何实施例的讨论仅为示例性的,并非旨在暗示本申请的保护范围限于这些例子;在本申请的思路下,以上实施例或者不同实施例中的技术特征之间也可以进行组合,步骤可以以任意顺序实现,并存在如上所述的本申请中一个或多个实施例的不同方面的许多其它变化,为了简明它们没有在细节中提供。
本申请中一个或多个实施例旨在涵盖落入本申请的宽泛范围之内的所有这样的替换、修改和变型。因此,凡在本申请中一个或多个实施例的精神和原则之内,所做的任何省略、修改、等同替换、改进等,均应包含在本申请的保护范围之内。
- 环磷腺苷葡胺大输液制剂及其制备方法和环磷腺苷葡胺含量测定方法
- 环磷腺苷葡胺大输液制剂中环磷腺苷葡胺含量测定方法