一种大尺寸5-羟甲基糠醛氧化电催化剂及其制备方法
文献发布时间:2024-04-18 19:58:30
技术领域
本申请属于氧化电催化剂合成技术领域,具体涉及一种用于5-羟甲基糠醛氧化的大尺寸电催化剂,并具体公开其制备方法与应用。
背景技术
长久以来,由于传统石油基高分子材料的大量使用,给人类的生产生活带来了极为严重“白色污染”等环境问题,再加之化石资源的日渐短缺,迫使人们亟需寻找新型的绿色环保的替代材料。生物基塑料材料因其原料来源绿色广泛、成本低廉、易降解、对环境友好、物理化学性质优异等优势,被认为是传统塑料材料的理想替代品。
聚呋喃二甲酸乙二醇酯(polyethylene furanoate,PEF)是一种新型的生物基高分子材料。其因优异的气阻性能、热稳定性以及机械性能,被认为是最有潜力、最具市场前景的替代石油基聚对苯二甲酸乙二醇酯(Polyethylene terephthalate,PET)的关键材料之一。而2,5-呋喃二甲酸(2,5-Furandicarboxylic acid,FDCA)则是合成PEF的关键单体,其合成工艺对PEF材料的合成具有重要的意义。
目前,工业合成FDCA的主要途径为传统热催化方式,即由5-羟甲基糠醛(5-hydroxymethylfufural,HMF)为原料经氧化制得。该工艺通常采用Pt、Au、Pd等贵金属基固体催化剂以及高压氧化性气体(>5atm)等作为氧化剂,而且该氧化过程需要在高温(100-200℃)的条件下进行。该生产方法由于研究较早且工艺成熟,因而在工业上被广泛采用。但是,由于贵金属成本高且储量稀少,导致该工艺设备成本高昂,难以大规模生产应用;而且,反应条件的苛刻也增大了设备运行的安全风险,并增加了设备安装和维护成本;该工艺在反应过程中会产生大量的能耗以及CO
电催化氧化是使电极及电解质界面上的电荷转移加速反应的一种催化氧化方式。电催化工艺具有反应条件温和、常温常压条件下即可进行的优势,且反应能量效率高;而且,电催化工艺以H
但是,基于电催化方式氧化HMF制备FDCA的工艺目前仍处于实验室研究阶段,尚未实现产业化及商业化。究其原因,制约电催化HMF氧化工艺进一步发展的主要问题包括:已报道的非贵金属基催化剂的制备方法多为溶剂热合成、高温煅烧等,操作复杂繁琐、且反应条件苛刻、不利于合成大尺寸样品,难以进一步放大规模生产;同时,大多数催化剂存在电流密度较低、稳定性较差的问题,其催化HMF氧化的性能并不理想,无法满足工业化的性能要求。
如中国专利CN116377495A公开了一种镍钼双金属电催化剂氧化5-羟甲基糠醛的方法,该催化剂通过溶剂热、煅烧还原等多步反应进行合成,存在工艺步骤多、操作困难、工艺能耗高(最高温度可达600℃)等问题,而且高压和H
因此,本领域期待开发一种高效稳定的用于5-羟甲基糠醛氧化的大尺寸非贵金属基电催化剂,对于电催化氧化HMF制备FDCA工艺的发展具有积极的意义。
发明内容
为此,本发明所要解决的技术问题在于提供一种用于5-羟甲基糠醛氧化的大尺寸非贵金属基电催化剂,具有高效稳定、操作简单、条件温和、安全风险低、合成成本低、重现性好、易于放大化生产的优势;
本发明所要解决的第二个技术问题在于提供上述用于5-羟甲基糠醛氧化的大尺寸非贵金属基电催化剂的制备方法及其应用。
为解决上述技术问题,本发明所述的一种大尺寸5-羟甲基糠醛氧化电催化剂的制备方法,包括如下步骤:
(1)分别配制铜盐溶液和/或镍盐溶液,得到金属盐溶液;
(2)取基底材料加入至所述金属盐溶液中进行浸渍处理,干燥、即得。
具体的,所述步骤(1)中,所述铜盐溶液包括硫酸铜溶液和/或硝酸铜溶液;
优选的,所述铜盐溶液的浓度为25-600mmol/L;更优选为100-300mmol/L;
优选的,所述铜盐溶液的溶剂包括水、醇溶剂等常规溶剂,更优选为水溶液。
具体的,所述步骤(1)中,所述镍盐溶液包括硝酸镍溶液和/或硫酸镍溶液;
优选的,所述镍盐溶液的浓度为25-600mmol/L;更优选为100-300mmol/L;
优选的,所述镍盐溶液的溶剂包括醇溶剂、水等常规溶剂,更优选为乙醇溶液。
具体的,所述步骤(1)中,所述金属盐溶液包括铜盐溶液和镍盐溶液的混合溶液;
所述金属盐溶液中,所述悬浮液中镍离子和铜离子的摩尔比为>0-4:4->0;
优选的,所述金属盐溶液中镍离子和铜离子的摩尔比为1-3:3-1;
优选的,所述金属盐溶液中镍离子和铜离子的摩尔比为2:2。
具体的,所述金属盐溶液的混合步骤包括将所述铜盐溶液滴加至所述镍盐溶液的步骤;
优选的,控制所述滴加时间为5-20min。
具体的,所述步骤(2)中,所述基底材料的尺寸为(1.5-10)cm×(1.5-10)cm,厚度为0.2-1.6mm;
优选的,所述基底材料包括泡沫镍、泡沫铁、泡沫钴、泡沫铜或碳纸基底中的至少一种。
具体的,所述步骤(2)中,所述浸渍处理的时间为10s-10min;
优选的,所述步骤(2)中,还包括将所述基底材料进行预处理的步骤;
优选的,所述预处理步骤包括将所述基底材料进行氧化以及清洗的步骤;
优选的,所述氧化步骤包括采用稀盐酸或硝酸在25-100℃条件下进行氧化处理30-120min的步骤;
优选的,所述清洗步骤包括以乙醇和/或丙酮为溶剂进行超声清理的步骤。
优选的,所述步骤(2)中,还包括将浸渍处理后的电催化剂进行清洗、干燥的步骤。
本发明还公开了由所述方法制备得到的大尺寸5-羟甲基糠醛氧化电催化剂;
优选的,所述5-羟甲基糠醛氧化电催化剂的尺寸为(1.5-10)cm×(1.5-10)cm,厚度为0.2-1.6mm。本发明所述5-羟甲基糠醛氧化电催化剂在常规尺寸下具有较好的性能,即便是大尺寸(10cm×10cm左右)的基底,依然能够实现均匀的活性负载,其性能稳定性较好。
本发明还公开了所述大尺寸5-羟甲基糠醛氧化电催化剂在电催化氧化HMF制备FDCA工艺中的应用。
本发明还公开了一种电催化氧化HMF制备FDCA的工艺,包括在所述催化剂存在下,以HMF为原料进行电催化氧化制备FDCA的步骤。
具体的,所述电催化氧化HMF制备FDCA的工艺中,电化学工作站可选取AutolabPGSTAT302N、Autolabvionic、CHI760或CHI1140等不同型号的直流电化学工作站;
反应容器可选取10mL、50mL等不同规格的H型电解池;
参比电极可选取Hg/HgO或Ag/AgCl等,其中Hg/HgO电极内填充有1M KOH溶液、Ag/AgCl电极内填充有3M KCl或饱和KCl溶液;
对电极可选取铂网、石墨棒等;
工作电极可选取铂夹电极、不锈钢夹电极等;
可选取Nafion117、Nafion212等阳离子交换膜或FAA-3-50、FAB-PK-130等阴离子交换膜将H型电解池2腔室分开;
电解液可选取含有不同浓度HMF的1M KOH或NaOH等;例如,10-100mM HMF+0.5-1.5M KOH/NaOH。
具体的,所述电催化氧化HMF制备FDCA的工艺中,测试方法选取循环扫描伏安法、线性扫描伏安法、恒电压电解法等。
具体的,循环扫描伏安法扫描电压范围为0.2-0.8V(vs Hg/HgO),扫描速率为10-100mV/s,扫描圈数为1-25;
具体的,线性扫描伏安法扫描电压范围为0.2-0.8V(vs.Hg/HgO),扫描速率为2-10mV/s,扫描圈数为1;
具体的,恒电压电解法设置电压范围为0.50-0.65V(vs.Hg/HgO),电解时间为30-60min;
具体的,测试过程中磁子搅拌速度可设置范围0-2000rpm。
本发明所述大尺寸5-羟甲基糠醛氧化电催化剂的制备方法,以铜盐溶液和镍盐溶液为负载活性成分,并通过在常温常压条件下,可以在秒级时间内通过快速浸泡实现活性金属盐的均匀负载,成功合成了一种新型的HMF氧化电催化剂。本发明所述催化剂的制备方法,具有操作简易、快速、合成时间短、安全风险小、重现性好、易于制备大尺寸样品、易于放大规模生产的优势。
本发明所述催化剂的制备方法,尤其适用于大尺寸基底材料的负载制备,可用于合成大尺寸的催化剂,且金属盐活性成分生长均匀、性能相较于小尺寸催化剂没有衰减,可以实现大尺寸氧化电催化剂的有效加工。大尺寸催化剂可以用于组装大尺寸电催化器件,这样可以显著提高HMF氧化效率,提高FDCA产量,并降低工艺成本,更贴近工业实际应用,更具工业化前景和潜力。
本发明所述催化剂的制备方法,相比于传统的溶剂热合成、高温煅烧等合成方法需要高温高压、操作复杂、安全风险大、制备时间长(数小时乃至数天)等问题,该方法仅需要一步浸泡,步骤简单,用时短(数秒、数分钟),且在常温常压下便可实行,安全风险小,条件温和,具备进一步工业化的发展前景。
本发明所述大尺寸5-羟甲基糠醛氧化电催化剂的制备方法,其催化剂合成原液仅为金属盐溶液,可重复循环使用,降低了催化剂的制备成本;同时,该方法同样适用于不同的基底,如泡沫铁、泡沫钴、泡沫铜、碳纤维纸等均适用,具有一定的普适性,应用前景广阔,为HMF氧化电催化剂的设计合成与放大化催化剂制备生产提供了重要参考。
本发明所述大尺寸5-羟甲基糠醛氧化电催化剂,是一种新型的NiCuO
附图说明
为了使本发明的内容更容易被清楚的理解,下面根据本发明的具体实施例并结合附图,对本发明作进一步详细的说明,其中,
图1为实施例1制备催化剂的扫描电子显微镜图;
图2为实施例1制备催化剂的透射电子显微镜图;
图3为实施例1制备催化剂的选区电子衍射图;
图4为实施例5制备催化剂的扫描电子显微镜图;
图5为实施例6制备催化剂的扫描电子显微镜图;
图6为实施例7制备催化剂的扫描电子显微镜图;
图7为实施例8制备催化剂的扫描电子显微镜图;
图8为实施例8制备催化剂的实物照片;
图9为按照实施例1中方法制备的催化剂氧化电催化工艺的HMFOR与OER的LSV曲线(包含5组重复试验);
图10为按照实施例2中方法制备的催化剂氧化电催化工艺的HMFOR与OER的LSV曲线(包含其它不同镍、铜金属离子摩尔比优化样品);
图11为按照实施例3中方法制备的催化剂氧化电催化工艺的HMFOR与OER的LSV曲线(包含其它不同金属离子总浓度优化样品);
图12为按照实施例4中方法制备的催化剂氧化电催化工艺的HMFOR与OER的LSV曲线(包含其它不同浸泡时间优化样品);
图13为按照实施例5-7中方法制备的催化剂与相对应空白基底进行氧化电催化工艺的HMFOR的LSV曲线(即不同基底优化样品);
图14为按照实施例8中方法制备的催化剂氧化电催化工艺的HMFOR与OER的LSV曲线(包含5组平行试验);
图15为实施例1中制备催化剂氧化电催化工艺的HMFOR稳定性测试的FDCA法拉第效率(FE,%)与产率(Yield,%)图;
图16为对比例1制备催化剂的扫描电子显微镜图;
图17为对比例2制备催化剂的扫描电子显微镜图;
图18为对比例3制备催化剂的扫描电子显微镜图;
图19为实施例1与对比例1-3中制备催化剂氧化电催化工艺的HMFOR的LSV曲线。
具体实施方式
实施例1
将尺寸为1.5cm×2cm、厚度为1.0mm的泡沫镍基底在25-50℃下采用稀盐酸进行处理1h,随后采用乙醇/丙酮的混合溶液进行超声清理,以去除基底表面的氧化物以及有机杂质,并将泡沫镍基底干燥,备用。
分别配置300mmol/L的Ni(NO
将C溶液转移到玻璃容器中,经超声、静置;将干燥后的泡沫镍基底浸入C溶液中,浸泡时间为10s。而后取出所得催化剂,经清洗、干燥,即得。
本实施例制备催化剂的扫描电子显微镜图、透射电子显微镜图以及选区电子衍射图分别见附图1-3。可见,本实施例制备的催化剂展现了均一的纳米针形貌,且该催化剂物相主要为无定型相。
采用3电极体系和H型电解池测试所得催化剂的性能。以50mM HMF+1M KOH为电解液,以Hg/HgO和铂网分别为参比电极和对电极,以阳离子交换膜Nafion 212将H型电解池2腔室分开。使用线性扫描伏安法测试催化剂性能,扫速为10mV s
实施例2
将尺寸为1.5cm×2cm、厚度为1.0mm的泡沫镍基底在25-50℃下采用稀盐酸处理1h,而后采用乙醇/丙酮混合溶液超声清理,以去除基底表面的氧化物以及有机杂质,最后将基底干燥,备用。
分别配置200mmol/L的Ni(NO
将C溶液转移到玻璃容器中,经超声、静置;将干燥后的泡沫镍基底浸入溶液中,浸泡时间为10s,而后取出催化剂,清洗干燥,备用。
采用3电极体系和H型电解池测试催化剂性能。以50mM HMF+1MKOH为电解液,以Hg/HgO和铂网分别为参比电极和对电极,以阳离子交换膜Nafion 212将H型电解池2腔室分开,使用线性扫描伏安法测试催化剂性能,扫速为10mV s
实施例3
将尺寸为1.5cm×2cm、厚度为1.0mm的泡沫镍基底在25-50℃下采用稀盐酸处理1h,而后采用乙醇/丙酮混合溶液超声清理,以去除基底表面的氧化物以及有机杂质,最后将基底干燥备用。
分别配置75mmol/L的Ni(NO
然后,将C溶液转移到玻璃容器中,超声,静置;将干燥后的泡沫镍基底浸入溶液中,浸泡时间为10s,而后取出催化剂,清洗干燥,备用。
采用3电极体系和H型电解池测试催化剂性能。以50mM HMF+1MKOH为电解液,以Hg/HgO和铂网分别为参比电极和对电极,以阳离子交换膜Nafion 212将H型电解池2腔室分开,使用线性扫描伏安法测试催化剂性能,扫速为10mV s
实施例4
将尺寸为1.5cm×2cm、厚度为1.0mm的泡沫镍基底在25-50℃下采用稀盐酸处理1h,而后采用乙醇/丙酮混合溶液超声清理,以去除基底表面的氧化物以及有机杂质,最后将基底干燥备用。
分别配置300mmol/L的Ni(NO
然后,将C溶液转移到玻璃容器中,超声,静置;将干燥后的泡沫镍基底浸入溶液中,浸泡时间为10min,而后取出催化剂,清洗干燥,备用。
采用3电极体系和H型电解池测试催化剂性能,以50mM HMF+1MKOH为电解液,以Hg/HgO和铂网分别为参比电极和对电极,以阳离子交换膜Nafion 212将H型电解池2腔室分开,使用线性扫描伏安法测试催化剂性能,扫速为10mV s
实施例5
将尺寸为1.5cm×2cm、厚度为0.75mm的泡沫铁基底在25℃下采用稀盐酸处理30s,而后采用乙醇/丙酮混合溶液超声清理,以去除基底表面的氧化物以及有机杂质,最后将基底干燥备用。
分别配置300mmol/L的Ni(NO
然后,将C溶液转移到玻璃容器中,超声,静置;将干燥后的泡沫镍基底浸入溶液中,浸泡时间为10s,而后取出催化剂,清洗干燥,备用。
本实施例制备催化剂的扫描电子显微镜图见附图4。可见,本实施例制备的催化剂展现了均一的纳米片形貌。
采用3电极体系和H型电解池测试催化剂性能,以50mM HMF+1MKOH为电解液,以Hg/HgO和铂网分别为参比电极和对电极,以阳离子交换膜Nafion 212将H型电解池2腔室分开,使用线性扫描伏安法测试催化剂性能,扫速为10mV s
实施例6
将尺寸为1.5cm×2cm、厚度为0.5mm的泡沫铜基底在25-50℃下采用稀盐酸处理2h,而后采用乙醇/丙酮混合溶液超声清理,以去除基底表面的氧化物以及有机杂质,最后将基底干燥备用。
分别配置300mmol/L的Ni(NO
然后,将C溶液转移到玻璃容器中,超声,静置;将干燥后的泡沫镍基底浸入溶液中,浸泡时间为10s,而后取出催化剂,清洗干燥,备用。
本实施例制备催化剂的扫描电子显微镜图见附图5。可见,本实施例制备的催化剂以纳米块的形式均匀分散在基底表面。
采用3电极体系和H型电解池测试催化剂性能,以50mM HMF+1MKOH为电解液,以Hg/HgO和铂网分别为参比电极和对电极,以阳离子交换膜Nafion 212将H型电解池2腔室分开,使用线性扫描伏安法测试催化剂性能,扫速为10mV s
实施例7
将尺寸为1.5cm×2cm、厚度为1.6mm的泡沫钴基底在25-50℃下采用稀盐酸处理2h,而后采用乙醇/丙酮混合溶液超声清理,以去除基底表面的氧化物以及有机杂质,最后将基底干燥备用。
分别配置300mmol/L的Ni(NO
然后,将C溶液转移到玻璃容器中,超声,静置;将干燥后的泡沫镍基底浸入溶液中,浸泡时间为10s,而后取出催化剂,清洗干燥,备用。
本实施例制备催化剂的扫描电子显微镜图见附图6。可见,本实施例制备的催化剂展现了均一的纳米片形貌。
采用3电极体系和H型电解池测试催化剂性能,以50mM HMF+1MKOH为电解液,以Hg/HgO和铂网分别为参比电极和对电极,以阳离子交换膜Nafion 212将H型电解池2腔室分开,使用线性扫描伏安法测试催化剂性能,扫速为10mV s
实施例8
将尺寸为10cm×10cm、厚度为1.0mm的泡沫镍基底在25-50℃下采用稀盐酸处理2h,而后采用乙醇/丙酮混合溶液超声清理,以去除基底表面的氧化物以及有机杂质,最后将基底干燥备用。
分别配置300mmol/L的Ni(NO
然后,将C溶液转移到玻璃容器中,超声,静置;将干燥后的泡沫镍基底浸入溶液中,浸泡时间为1min,而后取出催化剂,清洗干燥,备用。本实施例制备催化剂的扫描电子显微镜图见附图7,实物照片见附图8。可见,本实施例制备的催化剂在不同尺度下均展现了其均一性。
采用3电极体系和H型电解池测试催化剂性能,以50mM HMF+1MKOH为电解液,以Hg/HgO和铂网分别为参比电极和对电极,以阳离子交换膜Nafion 212将H型电解池2腔室分开,使用线性扫描伏安法测试催化剂性能,扫速为10mV s
对比例1
本对比例所述催化剂的制备方法同实施例1,其区别仅在于,所述基底尺寸选择为10cm×10cm、厚度为1.0mm,且铜盐溶液和镍盐溶液各500mL,还添加100mL质量分数为30%的H
然后,将C溶液转移到玻璃容器中,短暂超声后,静置C液中出现大量由H
本对比例制备催化剂的扫描电子显微镜图见附图16。可见,该催化剂的基底大面积暴露,未见催化剂成功负载,均一性较差。
本对比例所述催化剂的性能测试方法同实施例1。
对比例2
本对比例所述催化剂的制备方法同实施例1,其区别仅在于,所述基底尺寸选择10cm×10cm、厚度为1.0mm,且金属盐溶液仅选用硝酸镍溶液,其用量为1000mL,还添加100mL质量分数为30%的H
然后,将C溶液转移到玻璃容器中,短暂超声,将干燥后的泡沫镍基底浸入溶液中,浸泡时间为1min,而后取出催化剂,清洗干燥,备用。
本对比例制备催化剂的扫描电子显微镜图见附图17。可见,该催化剂未成功大量负载,基底暴露明显。
本对比例所述催化剂的性能测试方法同实施例1。
对比例3
本对比例所述催化剂的制备方法同实施例1,其区别仅在于,所述基底尺寸选择10cm×10cm、厚度为1.0mm,且金属盐溶液仅选用硫酸铜溶液,其用量为1000mL,还添加100mL质量分数为30%的H
然后,将C溶液转移到玻璃容器中,短暂超声,将干燥后的泡沫镍基底浸入溶液中,浸泡时间为1min,而后取出催化剂,清洗干燥,备用。
本对比例制备催化剂的扫描电子显微镜图见附图18。可见,该催化剂的基底暴露明显,催化剂负载少,无特定形貌。
本对比例所述催化剂的性能测试方法同实施例1。
实验例
1、电氧化催化工艺性能
本实验例中,采用3电极体系分别对上述实施例1-8及对比例1-3记载方法制备的催化剂进行氧化电催化工艺测试。
本实验例中,参比电极选取Hg/HgO电极,其中,Hg/HgO电极内填充有1M KOH溶液,对电极选取铂网,工作电极选取铂夹电极;选取Nafion212阳离子交换膜将H型电解池2腔室分开;电解液选取含有50mM HMF+1MKOH的电解液。
本实验例中,测试方法选取线性扫描伏安法,线性扫描伏安法扫描电压范围为0.2-0.8V(vs.Hg/HgO),扫描速率为2-10mV/s,扫描圈数为1。
按照实施例1中方法及参数进行催化剂的制备,并进行5组重复试验,将制备催化剂进行氧化电催化工艺的HMFOR与OER的LSV曲线见附图9。如图所示结果,该催化剂制备方法展现了优异的重现性,5组重复试验的催化剂性能差异很小。
按照实施例2中方法及参数制备所需催化剂,并分别制备其它不同镍、铜金属离子摩尔比优化样品,将制备的各组催化剂进行氧化电催化工艺的HMFOR与OER的LSV曲线见附图10。如图所示结果,镍与铜金属离子摩尔比为3:1的催化剂,展现了最为优异的催化活性。
按照实施例3中方法及参数制备所需催化剂,并分别制备其他不同金属离子总浓度优化样品,将制备的各组催化剂进行氧化电催化工艺的HMFOR与OER的LSV曲线见附图11。如图所示结果,金属离子总浓度为400mM的催化剂,展现了最为优异的催化活性,且随着浓度从100mM到600mM的不断增大,其催化剂性能呈现了先升高后下降的“火山型”规律。
按照实施例4中方法及参数制备所需催化剂,并分别制备其他不同浸渍时间下的样品,将各组催化剂进行氧化电催化工艺的HMFOR与OER的LSV曲线见附图12。如图所示结果,随着浸渍时间的延长,其催化剂活性逐渐下降,浸渍时间最短的催化剂展现了最为优异的催化活性。
分别将实施例5-7中制备的不同基底(泡沫铁FF、泡沫钴CoF、泡沫铜CuF、碳纤维纸CP)的催化剂与相对应空白基底进行氧化电催化工艺的HMFOR的LSV曲线见附图13。如图所示结果,通过浸渍处理后,不同基底在负载活性组分后其对催化HMF氧化的活性均显著提高,体现了该催化剂制备方法的普适性高。
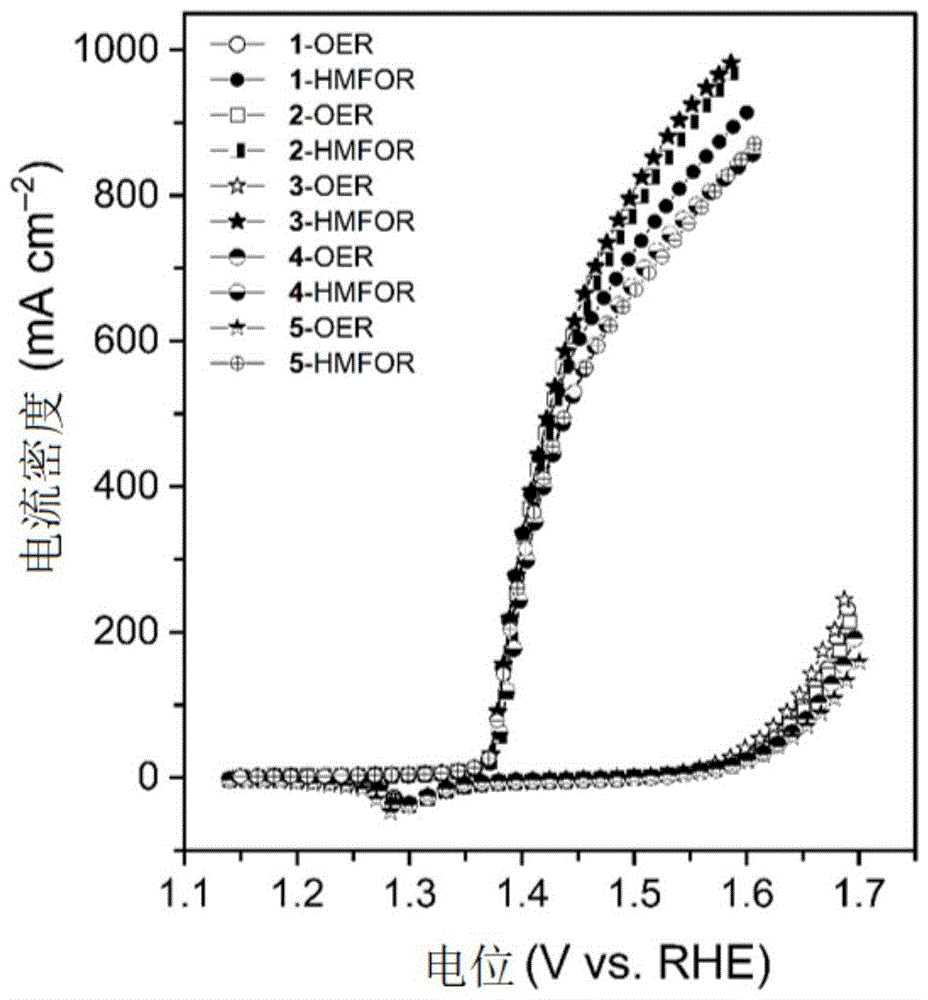
按照实施例8中方法及参数制备所需催化剂,并进行5组平行试验,将各组催化剂进行氧化电催化工艺的HMFOR与OER的LSV曲线见附图14。如图所示结果,该催化剂制备方法在制备大尺寸催化剂方面,展现了优异的重现性,5组重复试验的催化性能差异很小。
取实施例1中催化剂氧化电催化工艺的HMFOR稳定性测试的FDCA法拉第效率(FE,%)与产率图(Yield,%)见附图15。如图所示结果,该浸渍方法处理的催化剂在长达18个循环的电解中,其FDCA法拉第效率均维持在95%以上,FDCA产率均维持在91%以上,展现了优异的稳定性。
对比例1-3与实施例1中催化剂氧化电催化工艺的HMFOR的LSV曲线见附图19。如图所示结果,对比例1-3所述催化剂催化HMF氧化的性能显著低于实施例1。这表明在前驱体悬浮液中额外加入H
综上,通过该催化剂制备方法,合成的催化剂在催化HMF氧化方面展现了优异的催化活性与稳定性,且该方法适用于多种不同基底材料,方法普适性高,同时,该法制备的大尺寸催化剂展现了良好的重现性,易于放大规模生产。
2、与已知催化剂的制备方法及性能对比
本发明实施例1中制备催化剂的特征与部分已报道HMF氧化电催化剂合成过程的对比特征见下表1。
表1催化剂合成过程对比特征
本发明实施例1中制备催化剂的特征与部分已报道HMF氧化电催化剂性能的对比特征见下表2,测试方法参见实验例1。
表2催化剂性能对比特征
注:a.在1.45V(vs.RHE)下的电流密度;b.在1.40V(vs.RHE)下的电流密度;
综上,相比于已知催化剂的制备方法及性能对比,本发明实施例1制备催化剂的工艺所需总时间显著缩短,由数小时、数十小时缩短至数十秒;工艺所需最高温度从普遍的数百摄氏度降至室温;尤其是催化剂尺寸从数平方厘米增大至数百平方厘米,体现了该制备方法操作简易、快速、合成时间短、易于制备大尺寸催化剂的优势。同时,该催化剂展现了优异的催化HMF氧化性能,尤其是稳定性显著优于部分已报道催化剂,具备进一步工业化的潜力。
显然,上述实施例仅仅是为清楚地说明所作的举例,而并非对实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。而由此所引伸出的显而易见的变化或变动仍处于本发明创造的保护范围之中。
- 一种选择性氧化5-羟甲基糠醛制备2,5-二甲酰基呋喃的方法
- 氮掺杂碳材料催化剂及其在催化5-羟甲基糠醛氧化制备2,5-二甲酰基呋喃中的应用
- 大电流密度电催化5-羟甲基糠醛为2,5-呋喃二甲酸的催化剂及其制备方法
- 一种非均相催化剂催化5-羟甲基糠醛氧化制备2,5-呋喃二甲酸的方法