一种1-NEA对映异构体过量率的测定方法
文献发布时间:2023-06-19 12:27:31
技术领域
本发明属于药物分析技术领域,具体涉及一种1-NEA对映异构体过量率的测定方法。
背景技术
1-萘乙胺(1-NEA)是新型口服拟钙剂盐酸西那卡塞重要的手性中间体,而(R)-1-NEA是(R)-盐酸西那卡塞唯一的手性中心引入源,因此,1-NEA的对映异构体过量率的测定对于制备高光学纯度盐酸西那卡塞具有重要的理论意义与实际意义。
手性色谱柱分析法是测定1-NEA对映异构体过量率的主要方法,此方法结果准确、可靠。张峰等(华西药学杂志,2014,29(01):75-76)采用Daicel Chirlcel OJ-H(250mm×4.6mm,5μm)手性色谱柱,流动相为95%正己烷+5%乙醇+0.1%乙醇胺,流速为1.0mL/min,紫外吸收波长设定在222nm,柱温为25℃,获得1-NEA的对映异构体过量率;Pan等(TheJournal of Organic Chemistry,2018,83(19):11502-11509)采用Daicel Chiralcel OD-H column(250mm×4.6mm,5μm)手性柱测定1-NEA的手性纯度,使用Agilent 1100HPLC仪器,流动相为90%己烷+10%异丙醇,流速为0.8mL/min,紫外吸收波长设定在285nm,获得对映体过量测量值。
手性色谱柱价格约1.5-2万元左右,价格昂贵,同时手性色谱柱使用条件苛刻、寿命短等,相比之下采用非手性色谱柱测定操作简便且成本低廉。在以往的报道中,Calmes等(Tetrahedron Asymmetry,1993,4(12):2437-2440)采用Marfey试剂作为手性衍生试剂与1-NEA进行衍生化反应,乙腈和水作为流动相,Jun等采用DBD-顺式-4-羟基-L-脯氨酸、DBD-反式-4-羟基-L-脯氨酸等作为手性衍生试剂与1-NEA进行衍生化反应,甲醇和水作为流动相,在ULTRON VX-ODS(150mm×4.6mm,5μm)色谱柱进行测定,前者所测得衍生产物的保留时间为20min左右,后者为40min左右。以上两种方法经过衍生化反应后,使用非手性色谱柱进行对映异构体过量率的测定,虽然节约了检测成本,但衍生产物的保留时间过长。Mochizuki等(Analytica Chimica Acta,2013,773:76-82)采用L-PGA作为手性衍生试剂,在UPLC上使用乙腈和水作为流动相,衍生产物的保留时间分别为8.58min和9.03min,虽然缩短了衍生产物的保留时间,但是衍生化过程使用EDC和HOBt作为缩合剂,存在原子经济性较差的问题。
现有专利技术中也有披露过衍生化过程,比如专利号ZL201610043272.9、ZL201610043273.3、ZL201610043277.1和ZL201610043270.X中披露了3-氨基哌啶和2-氨基丁醇的对映异构体的HPLC分离检测方法,但是由于目标底物不同,相应的特定的手性衍生试剂是技术关键,手性试剂的选择并不是显而易见或者推导出来的,原因有以下几点:(1)一些手性试剂不易购买,难以合成,如在先专利用到的手性衍生试剂(R)-(+)-1-苯基乙磺酰氯不易购买,若先自制合成(R)-(+)-1-苯基乙磺酸,再经过氯代反应制得手性衍生试剂(R)-(+)-1-苯基乙磺酰氯,不仅增加了检测成本,而且大大增加了检测时间,过程不再具有便捷性。
(2)一些化合物如(S)-COXA-Osu、OTPTHE等虽然可直接用作手性衍生试剂,但其价格较为昂贵,增加了检测成本。
(3)另外常见的手性氨基酸,如(D)-苯甘氨酸、(D)-对羟基苯甘氨酸,在经过常规的氯代反应过程中,会有副产物生成,所以不适用于作为手性衍生试剂;如果选用缩合剂直接与1-NEA进行缩合,不仅大大增加了反应时间,而且不利于原子经济化。
(4)采用手性酸(S)-扁桃酸,经取代反应合成(S)-扁桃酸酰氯作为手性衍生试剂,TLC结果显示有副产物生成,考虑(S)-扁桃酸在酸性条件下自身进行酯化反应。不适合作为手性衍生试剂。
针对这一现状,需要研发一种操作简单、灵敏度高、衍生产物保留时间短的方法,为新型口服拟钙剂盐酸西那卡塞的研发创造了不可或缺的条件。
发明内容
本发明所要解决的技术问题是克服上述现有技术的不足,提供一种1-NEA对映异构体过量率的测定方法,该方法具有操作简单、灵敏度高以及衍生产物保留时间短的优点。
本发明解决其技术问题所采取的技术方案为:其包括以下步骤,
一种1-NEA对映异构体过量率的测定方法,其特征在于:其包括以下步骤,
(1)衍生化
将1-NEA溶于有机溶剂中,在一定的温度条件下,以(S)-萘普生酰氯为衍生化试剂,使用三乙胺作为缚酸剂,控制1-NEA和(S)-萘普生酰氯的的摩尔比,进行衍生化反应,得到衍生化后的1-NEA;反应式见式I;
(2)分离检测
采用反相高效液相色谱-紫外检测器对衍生化后的1-NEA进行定性、定量和对映异构体过量率测定;
所述的反相高效液相色谱使用的色谱柱是普通反相柱,优选色谱柱为反相C18柱。
进一步地,步骤(1)中,所述有机溶剂选自二氯甲烷、氯仿和四氯化碳中的一种。
进一步地,步骤(1)中,所述1-NEA与有机溶剂的固液质量比为1:5~20,优选的体积比为1:5。
进一步地,步骤(1)中,所述一定的温度条件指从15~45℃,优选为25℃。
进一步地,:所述1-NEA和(S)-萘普生酰氯的摩尔比为1:0.8~1.2,优选的摩尔比是1:0.8;
所述1-NEA与缚酸剂三乙胺的摩尔比为1:1~5,优选的摩尔比是1:3。
进一步地,所述紫外检测器的检测波长为224nm。
进一步地,所述的反相高效液相色谱的流动相由水和有机溶剂组成,该有机溶剂选自甲醇和乙腈中的一种,优选的为乙腈。
进一步地,所述反相高效液相色谱的流动相中,有机溶剂占流动相的体积分数为65%~80%,优选的为75%。
进一步地,所述流动相流速在0.5~1.5mL/min,优选的流速为1.0mL/min。
进一步地,步骤(2)中,反相高效液相色谱的流动相条件为:采用Agilent XDB-C18色谱柱,流动相由水和乙腈组成,其中水和乙腈的体积比为25:75,紫外检测波长为224nm,流速为1.0mL/min,柱温为25℃,进样量为20μL。
本发明的有益效果:
本发明给出1-NEA的衍生化方法,以(S)-萘普生酰氯为手性衍生试剂,通过控制反应条件,(S)-萘普生酰氯与1-NEA发生衍生反应,使1-NEA的对映异构体转化为非对映异构体,使用非手性固定相色谱即可测定1-NEA的非对映异构体过量值(d.e.%)。1-NEA的对映异构体过量值(e.e.%)等于两种非对映异构体的非对映异构体过量值。该方法操作简单、重现性好、灵敏度高、衍生产物保留时间短。通过对衍生化后的1-NEA进行定性、定量和对映体过量率分析,从而实现对1-NEA的定性、定量和对映体过量率分析。
本发明对手性衍生试剂进行筛选,以(S)-萘普生酰氯为手性衍生试剂,以二氯甲烷为溶剂,以三乙胺为缚酸剂,经取代反应得到产物(S)-萘普生酰基·(R)-1-萘乙胺和(S)-萘普生酰基·(S)-1-萘乙胺,采用非手性固定相HPLC即可快速测定1-NEA的对映异构体过量率。本发明公开的工艺路线以(S)-萘普生酰氯为手性衍生试剂,采用酰氯法合成酰胺,以三乙胺为缚酸剂,促进反应正向进行,使反应更彻底。反应时间短、效率高。
附图说明
图1:实施例1-1中,(S)-萘普生酰氯与(R)-1-NEA衍生化后色谱图;
图2:实施例1-2中,(S)-萘普生酰氯与(S)-1-NEA衍生化后色谱图;
图3:实施例1-3中,(S)-萘普生酰氯与(RS)-1-NEA衍生化后色谱图;
图4:对比例1中合成(R)-(+)-1-苯基乙磺酰氯反应的薄层色谱图;
在附图中:1为(S)-萘普生酰氯与(R)-1-NEA的衍生物;2为(S)-萘普生酰氯与(S)-1-NEA的衍生物。
具体实施方式
以下结合附图对本发明进行进一步详细的叙述。
实施例1:(S)-萘普生酰氯与1-NEA衍生物的液相色谱分析
1-1:(S)-萘普生酰氯与(R)-1-NEA衍生物的液相色谱分析
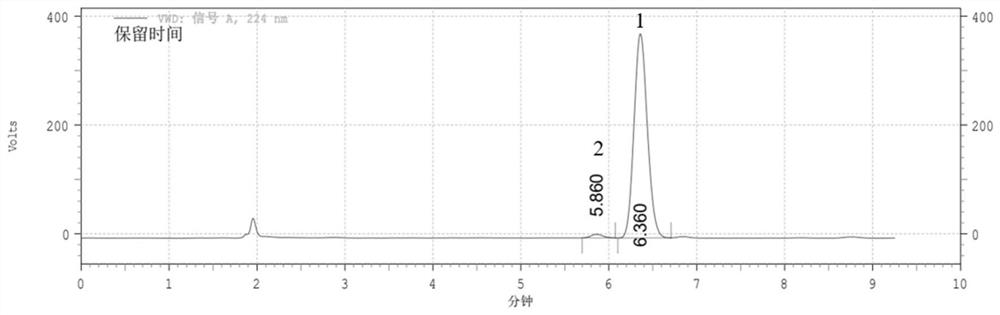
取(R)-1-NEA 5g(0.029mol),三乙胺12mL(0.087mol)溶于15g(11.32mL)二氯甲烷中,25℃下搅拌,将(S)-萘普生酰氯5.97g(0.024mol)溶于10g(7.55mL)二氯甲烷中,搅拌均匀后转移至恒压滴液漏斗,将其缓慢滴加到体系中,TLC监测反应,反应结束后蒸干溶剂得到(S)-萘普生酰基·(R)-1-萘乙胺衍生物,为了能更好的提取固体产物,进一步地,使用正庚烷热洗涤进行纯化。
将(S)-萘普生酰基·(R)-1-萘乙胺衍生物用流动相溶解,浓度为13μg/mL,采用高效液相色谱进行检测分析。液相色谱条件:Agilent C18色谱柱,流动相为乙腈-水(体积比为75:25),紫外检测波长为224nm,流速为1.0mL/min,柱温为25℃,进样体积20μL,保留时间和手性纯度分析见表1,其图谱见图1。
1-2:(S)-萘普生酰氯与(R)-1-NEA衍生物的液相色谱分析
取(S)-1-NEA 5g(0.029mol),三乙胺12mL(0.087mol)溶于15g(11.32mL)二氯甲烷中,25℃下搅拌,将(S)-萘普生酰氯5.97g(0.024mol)溶于10g(7.55mL)二氯甲烷中,搅拌均匀后转移至恒压滴液漏斗,将其缓慢滴加到体系中,TLC监测反应,反应结束后蒸干溶剂得到(S)-萘普生酰基·(S)-1-萘乙胺衍生物,使用正庚烷热洗涤进行纯化。
将(S)-萘普生酰基·(S)-1-萘乙胺衍生物用流动相溶解,浓度为13μg/mL,采用高效液相色谱进行检测分析。液相色谱条件:Agilent C18色谱柱,流动相为乙腈-水(体积比为75:25),紫外检测波长为224nm,流速为1.0mL/min,柱温为25℃,进样体积20μL,保留时间和手性纯度分析见表1,其图谱见图2。
1-3:(S)-萘普生酰氯与(RS)-1-NEA衍生物的液相色谱分析
取(RS)-1-NEA 5g(0.029mol),三乙胺12mL(0.087mol)溶于15g(11.32mL)二氯甲烷中,25℃下搅拌,将(S)-萘普生酰氯5.97g(0.024mol)溶于10g(7.55mL)二氯甲烷中,搅拌均匀后转移至恒压滴液漏斗,将其缓慢滴加到体系中,TLC监测反应,反应结束后蒸干溶剂得到衍生物(S)-萘普生酰基·(R)-1-萘乙胺和(S)-萘普生酰基·(S)-1-萘乙胺,使用正庚烷热洗涤进行纯化。
将(S)-萘普生酰基·(RS)-1-萘乙胺衍生物用流动相溶解,浓度为26μg/mL,采用高效液相色谱进行检测分析。液相色谱条件:Agilent C18色谱柱,流动相为乙腈-水(体积比为75:25),紫外检测波长为224nm,流速为1.0mL/min,柱温为25℃,进样体积20μL,两个非对映异构体衍生产物分离度为1.93,保留时间和手性纯度分析见表1,其图谱见图3。
实施例2:1-NEA与(S)-萘普生酰氯的衍生化反应条件考察试验
2-1:取(RS)-1-NEA 5g(0.029mol),三乙胺4mL(0.029mol)溶于25g(16.89mL)氯仿中,25℃下搅拌,将(S)-萘普生酰氯7.21g(0.029mol)溶于25g(16.89mL)氯仿中,搅拌均匀后转移至恒压滴液漏斗,将其缓慢滴加到体系中,TLC监测反应,反应结束后蒸干溶剂得到(S)-萘普生酰基·(RS)-1-萘乙胺衍生物,使用正庚烷热洗涤进行纯化。
将(S)-萘普生酰基·(RS)-1-萘乙胺衍生物用流动相溶解,浓度为26μg/mL,采用高效液相色谱进行检测分析。液相色谱条件:Agilent C18色谱柱,流动相为乙腈-水(体积比为75:25),紫外检测波长为224nm,流速为1.0mL/min,柱温为25℃,进样体积20μL,保留时间和手性e.e.%值同实施例1-3。
2-2:取(RS)-1-NEA 5g(0.029mol),三乙胺20mL(0.145mol)溶于45g(28.21mL)四氯化碳中,15℃下搅拌,将(S)-萘普生酰氯8.70g(0.035mol)溶于30g(18.81mL)四氯化碳中,搅拌均匀后转移至恒压滴液漏斗,将其缓慢滴加到体系中,TLC监测反应,反应结束后蒸干溶剂得到(S)-萘普生酰基·(RS)-1-萘乙胺衍生物,使用正庚烷热洗涤进行纯化。
将(S)-萘普生酰基·(RS)-1-萘乙胺衍生物用流动相溶解,浓度为25μg/mL,采用高效液相色谱进行检测分析。液相色谱条件:Agilent C18色谱柱,流动相为乙腈-水(体积比为75:25),紫外检测波长为224nm,流速为1.0mL/min,柱温为25℃,进样体积20μL,保留时间和手性e.e.%值同实施例1-3。
2-3:取(RS)-1-NEA 5g(0.029mol),三乙胺12mL(0.087mol)溶于50g(37.74mL)二氯甲烷中,25℃下搅拌,将(S)-萘普生酰氯5.97g(0.024mol)溶于50g(37.74mL)二氯甲烷中,搅拌均匀后转移至恒压滴液漏斗,将其缓慢滴加到体系中,TLC监测反应,反应结束后蒸干溶剂得到(S)-萘普生酰基·(RS)-1-萘乙胺衍生物,使用正庚烷热洗涤进行纯化。
将(S)-萘普生酰基·(RS)-1-萘乙胺衍生物用流动相溶解,浓度为25μg/mL,采用高效液相色谱进行检测分析。液相色谱条件:Agilent C18色谱柱,流动相为乙腈-水(体积比为75:25),紫外检测波长为224nm,流速为1.0mL/min,柱温为25℃,进样体积20μL,保留时间和手性e.e.%值同实施例1-3。
2-4:取(R)-1-NEA 5g(0.029mol),三乙胺12mL(0.087mol)溶于15g(11.32mL)二氯甲烷中,35℃下搅拌,将(S)-萘普生酰氯5.97g(0.024mol)溶于10g(7.55mL)二氯甲烷中,,搅拌均匀后转移至恒压滴液漏斗,将其缓慢滴加到体系中,TLC监测反应,反应结束后蒸干溶剂得到(S)-萘普生酰基·(R)-1-萘乙胺衍生物,使用正庚烷热洗涤进行纯化。
将(S)-萘普生酰基·(R)-1-萘乙胺衍生物用流动相溶解,浓度为13.3μg/mL,采用高效液相色谱进行检测分析。液相色谱条件:Agilent C18色谱柱,流动相为乙腈-水(体积比为75:25),紫外检测波长为224nm,流速为1.0mL/min,柱温为25℃,进样体积20μL,保留时间和手性e.e.%值同实施例1-1。
2-5:取(S)-1-NEA 5g(0.029mol),三乙胺12mL(0.087mol)溶于15g(11.32mL)二氯甲烷中,45℃回流状态下搅拌,将(S)-萘普生酰氯5.97g(0.024mol)溶于10g(7.55mL)二氯甲烷中,搅拌均匀后转移至恒压滴液漏斗,将其缓慢滴加到体系中,体系加热至回流状态,TLC监测反应,反应结束后蒸干溶剂得到(S)-萘普生酰基·(R)-1-萘乙胺衍生物,使用正庚烷热洗涤进行纯化。
将(S)-萘普生酰基·(R)-1-萘乙胺衍生物用流动相溶解,浓度为13.3μg/mL,采用高效液相色谱进行检测分析。液相色谱条件:Agilent C18色谱柱,流动相为乙腈-水(体积比为75:25),紫外检测波长为224nm,流速为1.0mL/min,柱温为25℃,进样体积20μL,保留时间和手性e.e.%值同实施例1-2。
实施例3:色谱条件考察试验
(1)衍生化
取(RS)-1-NEA 5g(0.029mol),三乙胺12mL(0.09mol)溶于15mL二氯甲烷中,25℃下搅拌,将(S)-萘普生酰氯5.7g(0.023mol)溶于10mL二氯甲烷中,搅拌均匀后转移至恒压滴液漏斗,将其缓慢滴加到体系中,TLC监测反应,反应结束后蒸干溶剂得到(S)-萘普生酰基·(RS)-1-萘乙胺衍生物,使用正庚烷热洗涤进行纯化。
(2)对(S)-萘普生酰氯与(RS)-1-NEA衍生物采用以下不同色谱条件进行分离检测
3-1:将(S)-萘普生酰基·(RS)-1-萘乙胺衍生物用流动相溶解,浓度为25μg/mL,采用高效液相色谱进行检测分析。液相色谱条件:Agilent C18色谱柱,流动相为甲醇-水(体积比为75:25),紫外检测波长为224nm,流速为1.0mL/min,柱温为25℃,进样体积20μL,手性e.e.%值同实施例1-3,但出峰时间延后,(S)-萘普生酰基·(R)-1-萘乙胺衍生物出峰时间在8.63min,(S)-萘普生酰基·(S)-1-萘乙胺衍生物出峰时间在8.05min,分离度为1.71。
3-3:将(S)-萘普生酰基·(RS)-1-萘乙胺衍生物用流动相溶解,浓度为25μg/mL,采用高效液相色谱进行检测分析。液相色谱条件:Agilent C18色谱柱,流动相为乙腈-水(体积比为80:20),紫外检测波长为224nm,流速为1.0mL/min,柱温为25℃,进样体积20μL,手性e.e.%值同实施例1-3,(S)-萘普生酰基·(R)-1-萘乙胺衍生物出峰时间提前至4.56min,(S)-萘普生酰基·(S)-1-萘乙胺衍生物出峰时间提前至4.19min,但两个非对映异构体衍生产物分离度降低,分离度为1.52。
3-4:将(S)-萘普生酰基·(RS)-1-萘乙胺衍生物用流动相溶解,浓度为25μg/mL,采用高效液相色谱进行检测分析。液相色谱条件:Agilent C18色谱柱,流动相为乙腈-水(体积比为70:30),紫外检测波长为224nm,流速为1.0mL/min,柱温为25℃,进样体积20μL,手性e.e.%值同实施例1-3,但出峰时间推迟,(S)-萘普生酰基·(R)-1-萘乙胺衍生物出峰时间在7.6min,(S)-萘普生酰基·(S)-1-萘乙胺衍生物出峰时间在6.95min,分离度为2.09。
3-5:将(S)-萘普生酰基·(RS)-1-萘乙胺衍生物用流动相溶解,浓度为25μg/mL,采用高效液相色谱进行检测分析。液相色谱条件:Agilent C18色谱柱,流动相为乙腈-水(体积比为65:35),紫外检测波长为224nm,流速为1.0mL/min,柱温为25℃,进样体积20μL,手性e.e.%值同实施例1-3,但出峰时间推迟,(S)-萘普生酰基·(R)-1-萘乙胺衍生物出峰时间在7.98min,(S)-萘普生酰基·(S)-1-萘乙胺衍生物出峰时间在7.21min,分离度为2.31。
3-6:将(S)-萘普生酰基·(RS)-1-萘乙胺衍生物用流动相溶解,浓度为25μg/mL,采用高效液相色谱进行检测分析。液相色谱条件:Agilent C18色谱柱,流动相为乙腈-水(体积比为75:25),紫外检测波长为224nm,流速为0.5mL/min,柱温为25℃,进样体积20μL,手性e.e.%值同实施例1-3,但出峰时间推迟,(S)-萘普生酰基·(R)-1-萘乙胺衍生物出峰时间在15.87min,(S)-萘普生酰基·(S)-1-萘乙胺衍生物出峰时间在14.19min,分离度为2.77。
3-7:将(S)-萘普生酰基·(RS)-1-萘乙胺衍生物用流动相溶解,浓度为25μg/mL,采用高效液相色谱进行检测分析。液相色谱条件:Agilent C18色谱柱,流动相为乙腈-水(体积比为75:25),紫外检测波长为224nm,流速为1.5mL/min,柱温为25℃,进样体积20μL,手性e.e.%值同实施例1-3,(S)-萘普生酰基·(R)-1-萘乙胺衍生物出峰时间提前至5.76min,(S)-萘普生酰基·(S)-1-萘乙胺衍生物出峰时间提前至5.33min,但两个非对映异构体衍生产物分离度降低,分离度为1.51。
3-8:将(S)-萘普生酰基·(RS)-1-萘乙胺衍生物用流动相溶解,浓度为25μg/mL,采用高效液相色谱进行检测分析。液相色谱条件:Agilent C18色谱柱,流动相为乙腈-水(体积比为85:15),紫外检测波长为224nm,流速为1.0mL/min,柱温为25℃,进样体积20μL,手性e.e.%值同实施例1-3,虽然出峰时间提前,(S)-萘普生酰基·(R)-1-萘乙胺衍生物出峰时间在4.16min,(S)-萘普生酰基·(S)-1-萘乙胺衍生物出峰时间在3.79min,但分离度为1.44,未达到完全分离。
3-9:将(S)-萘普生酰基·(RS)-1-萘乙胺衍生物用流动相溶解,浓度为25μg/mL,采用高效液相色谱进行检测分析。液相色谱条件:Agilent C18色谱柱,流动相为乙腈-水(体积比为75:25),紫外检测波长为224nm,流速为2.0mL/min,柱温为25℃,进样体积20μL,手性e.e.%值同实施例1-3,虽然出峰时间提前,(S)-萘普生酰基·(R)-1-萘乙胺衍生物出峰时间在2.09min,(S)-萘普生酰基·(S)-1-萘乙胺衍生物出峰时间在1.96min,但分离度为1.22,未达到完全分离。
本发明的研究思路涉及一种衍生化技术,就是通过化学反应将样品中难于分析检测的目标化合物定量的转化成另一易于分析检测的化合物,通过后者的分析检测可以对目标化合物进行定性和定量分析,该技术在色谱分析中得到广泛应用。采用手性衍生试剂将手性化合物的一对对映体转化为一对非对映异构体,使用非手性固定相色谱即可测定非对映异构体过量值(%d.e.)。对映异构体过量值(%e.e.)等于两种非对映异构体的非对映异构体过量值,间接表征了手性纯度。
采用非手性固定相色谱测定手性纯度成功的关键是选择合适的手性衍生试剂和HPLC检测条件。合适的手性衍生试剂应该易于使对映体生成非对映异构体,且非对映异构体理化性质差别较大;合适的HPLC检测条件应能够使非对映异构体完全分离,并且尽量缩短检测时间,提高检测效率。因此筛选合适的手性衍生试剂和HPLC检测条件的过程之间几乎没有参考价值和技术启示,筛选手性衍生试剂和HPLC检测条件的过程都属于探索创新的过程。非对映异构体的分离度高低是衍生过程与检测过程是否成功的重要指标,是衍生方法能否作为工业化方法的关键。
本发明中,衍生化过程中的衍生化试剂的选择是本发明成功分离的技术关键点之一,为了说明本发明的衍生化试剂的创造性,结合一下两个对比例进行说明。
对比例1
以选用(R)-(+)-1-苯基乙磺酰氯为衍生化试剂进行说明:
(1)衍生化
由于手性衍生试剂(R)-(+)-1-苯基乙磺酰氯不易购买,故采用实验室制备(R)-(+)-1-苯基乙磺酸时,合成的(R)-(+)-1-苯基乙磺酸与(D)-对羟基苯甘氨酸形成的复盐((+)-PES·D-HPG)进行衍生试剂的合成。
将复盐(+)-PES·D-HPG(15g)溶到50mL水中,用5%的NaOH调溶液的pH值在5.0-5.5,搅拌1h,静置析晶,过滤,滤饼用冰水淋洗,烘干滤饼,得白色固体D-HPG。将滤液旋干得到(+)-PES·Na,通过离子交换得到(+)-PES,即(R)-(+)-1-苯基乙磺酸。将(R)-(+)-1-苯基乙磺酸溶于二氯甲烷中,冰浴条件下滴加过量的氯化亚砜,滴加完毕后室温下搅拌过夜。
TLC结果显示,原料有较多剩余,得到的手性衍生试剂(R)-(+)-1-苯基乙磺酰氯纯度较低,若再经过纯化过程,将大大增加反应的时间,不利于后续的衍生化反应。综合以上原因,1-NEA的衍生不可以选择(R)-(+)-1-苯基乙磺酰氯作为手性衍生试剂。
图4为合成(R)-(+)-1-苯基乙磺酰氯反应的薄层色谱图,展开剂甲醇:二氯甲烷=1:1。从图4中可以看出,有较多原料剩余,反应不完全。
对比例2
以选用(R)-α-甲基-2-萘乙酰氯为衍生化试剂进行说明:
(1)衍生化
(R)-α-甲基-2-萘乙酰氯不易购买,若以此作为手性衍生试剂,需先合成(R)-α-甲基-2-萘乙酸,再经过氯代反应才能合成(R)-α-甲基-2-萘乙酰氯,增加了整个衍生、检测过程反应的难度。综合分析,1-NEA的衍生不能选择(R)-α-甲基-2-萘乙酰氯作为手性衍生试剂。
对于本发明的研究对象1-萘乙胺,本发明的发明人发现仅具有单一羧酸或磺酸官能团、结构中无易与羧基反应的其他官能团的(D)-樟脑磺酸和(S)-萘普生,不仅价格实惠、性质稳定、无毒害,且经过氯代反应,无其他副产物生成,即可合成相应的酰氯作为手性衍生试剂,可以长期存放。采用(D)-樟脑磺酰氯和(S)-萘普生酰氯作为手性衍生试剂尚未见报道。
在衍生化过程中,1-NEA与(S)-萘普生酰氯的优选的摩尔比是1:0.8,选择三乙胺作为缚酸剂促进反应正向进行,反应结束后以盐酸水溶液洗涤除去过量的1-NEA,提升衍生产物纯度。因此筛选合适的手性衍生试剂及衍生化反应过程中没有任何参考价值和技术启示,没有规律可以遵循,对所属领域技术人员而言也不是显而易见的,这些筛选手性衍生试剂的过程都属于探索创新的过程。
以上所述实施方式仅为本发明的优选实施例,而并非本发明可行实施的穷举。对于本领域一般技术人员而言,在不背离本发明原理和精神的前提下对其所作出的任何显而易见的改动,都应当被认为包含在本发明的权利要求保护范围之内。
- 一种1-NEA对映异构体过量率的测定方法
- 一种3-氨基哌啶对映异构体过量率的测定方法