一种生产四乙酰基六氮杂异伍兹烷的方法
文献发布时间:2024-01-17 01:23:17
技术领域
本发明涉及一种生产四乙酰基六氮杂异伍兹烷的方法,属于精细化工技术领域。
背景技术
四乙酰基六氮杂异伍兹烷,全名2,6,8,12-四乙酰基-2,4,6,8,10,12-六氮杂异伍兹烷,英文名:2,6,8,12-tetraacetyl-2,4,6,8,10,12-hexaazaisowurtzitane(TAIW),CASNo.:181940-38-5,是一种白色粉状固体,其分子结构式如下:
四乙酰基六氮杂异伍兹烷是用于制造新型高密度含能材料六硝基六氮杂异伍兹烷的关键原材料。六硝基六氮杂异伍兹烷(HNIW)俗称CL-20,它的最大爆速、爆压、密度等几个材料参数都优于奥克托今,同等重量能量输出比奥克托今高10-15%,是目前已知能够实际应用的能量最高、威力最强大的非核单质炸药,在先进武器装备的制造中有广泛的运用需求。CL-20的合成路线有很多,目前经过验证较为安全可行的方法就是以四乙酰基六氮杂异伍兹烷为原料经简单硝化一步来制备。因此四乙酰基六氮杂异伍兹烷的价格与产量决定了后续CL-20的使用场景,开发一种规模化低成本的四乙酰基六氮杂异伍兹烷生产方法显得非常必要。
六硝基六氮杂异伍兹烷(HNIW)的合成方法有很多,可以通过硝化以下五种原料来合成得到:四乙酰基二甲酰基六氮杂异伍兹烷(TADFIW)、四乙酰基二乙基六氮杂异伍兹烷(TADEIW)、六乙酰基六氮杂异伍兹烷(HAIW)、四乙酰基六氮杂异伍兹烷(TAIW)及四乙酰基二苄基六氮杂异伍兹烷(TADBIW)。前四种原料都需要由六苄基六氮杂异伍兹烷(HBIW)经过两次加氢才能制备,TADBIW仅需HBIW经过一次加氢就可制得。TADBIW为原料时虽然可省去第二步氢解,但是在硝化时实际上是亚硝解——硝解工艺,即TADBIW中六元环上的两个苄基是通过亚硝解脱除的,其反应过程是先由TADBIW亚硝解为四乙酰基二亚硝基六氮杂异伍兹烷(TADNSIW),再由TADNSIW硝解为HNIW。但是以TADBIW为原料的亚硝解——硝解反应所用的硝化试剂价格昂贵,工艺较普通硝酸/硫酸的硝化工艺更为危险,而且该方法制备的HNIW产品以为含有难分离的硝化不完全的中间体,产品纯度最高只能做到95%~98%,这些难分离的杂质不仅会降低HNIW的性能,还会大大增加HNIW在后续的转晶、分离、干燥机运输途中发生不可控爆炸的危险。
现阶段HNIW制备成本居高不下,主要原因是反应两步氢解过程需要使用大量的Pd催化剂。又因为氢解过程溶剂体系复杂,Pd颗粒极易从载体脱落和发生自身团聚,从而导致催化剂失活、循环性能差。因此,探索高效、稳定、高原子利用率的工业催化剂是降低HNIW成本的重要途径之一。
为了解决催化剂成本占比过高的问题,有很多文献和专利对负载型Pd催化剂的载体的性质对其催化性能的影响做了报道。Nielsen等用活性炭负载的20% Pd(OH)
随着国防工业对HNIW的需求量越来越大,而以TAIW为原料一步法硝化制备HNIW是目前经过实际论证唯一能规模化工业生产的方法,因此迫切需要一种工艺简单、原料易得、产品收率高、质量好,能够规模化生产TAIW的方法。选择一种合适的催化剂及合理的反应体系,是解决这一问题的关键。
发明内容
针对现有技术中的缺陷,本发明的目的是提供一种生产四乙酰基六氮杂异伍兹烷的方法。
本发明是通过以下技术方案实现的:
<第一方面>
本发明提供一种六苄基六氮杂异伍兹烷提纯精制的方法,包括如下步骤:
将六苄基六氮杂异伍兹烷用混合溶剂进行提纯精制,所述混合溶剂包括易溶解六苄基六氮杂异伍兹烷的溶剂和微溶或不溶解六苄基六氮杂异伍兹烷的溶剂的组合。
所述的六苄基六氮杂异伍兹烷提纯精制的方法具体为:将含量在95~98%的六苄基六氮杂异伍兹烷、混合溶剂、活性炭投入反应釜中,加热至35~75℃保温15min~2h,压滤除去活性炭及不溶物,滤液搅拌冷却降温至-5℃-5℃保持30min~2h;析出固体,过滤,固体再用溶剂洗涤、干燥,制得粉状固体,即为精制的六苄基六氮杂异伍兹烷。
所述易溶解六苄基六氮杂异伍兹烷的溶剂包括乙醚、甲基叔丁基迷、乙基丁基醚、四氢呋喃、1,4-二氧六环、2-甲基四氢呋喃、3-甲基四氢呋喃、乙二醇二甲醚中的一种或几种;
所述微溶或不溶解六苄基六氮杂异伍兹烷的溶剂的溶剂包括甲醇、乙醇、异丙醇、乙腈、丙腈、甲苯、二甲苯、乙酸乙酯、二氯甲烷、1,2-二氯乙烷中的一种或几种。
<第二方面>
本发明还提供如上所述的制备方法制备获得的六苄基六氮杂异伍兹烷
<第三方面>
本发明提供一种生产四乙酰基六氮杂异伍兹烷的方法,包括如下步骤:
S1、六苄基六氮杂异伍兹烷用混合溶剂进行提纯精制;
S2、将步骤S1提纯精制的六苄基六氮杂异伍兹烷在催化剂A的催化下进行第一次加氢反应,制得四乙酰基二苄基六氮杂异伍兹烷;
S3、将四乙酰基二苄基六氮杂异伍兹烷在催化剂B的催化下通过第二次加氢反应,制得所述四乙酰基六氮杂异伍兹烷。
步骤S1具体为:将含量在95~98%的六苄基六氮杂异伍兹烷、混合溶剂、活性炭投入反应釜中,加热至35~75℃保温15min~2h,压滤除去活性炭及不溶物,滤液搅拌冷却降温至-5℃-5℃保持30min~2h;析出固体,过滤,固体再用溶剂洗涤、干燥,制得粉状固体,即为精制的六苄基六氮杂异伍兹烷。
所述混合溶剂包括易溶解六苄基六氮杂异伍兹烷的溶剂和微溶或不溶解六苄基六氮杂异伍兹烷的溶剂的组合。
所述易溶解六苄基六氮杂异伍兹烷的溶剂包括乙醚、甲基叔丁基迷、乙基丁基醚、四氢呋喃、1,4-二氧六环、2-甲基四氢呋喃、3-甲基四氢呋喃、乙二醇二甲醚中的一种或几种。优选四氢呋喃。
所述微溶或不溶解六苄基六氮杂异伍兹烷的溶剂的溶剂包括甲醇、乙醇、异丙醇、乙腈、丙腈、甲苯、二甲苯、乙酸乙酯、二氯甲烷、1,2-二氯乙烷中的一种或几种。优选乙腈。
做为优选方案,用于提纯精制的活性炭可以是煤质活性炭、木质活性炭或合成材料活性炭,优选为氢氧化钾活化处理法制备的中性椰壳活性炭。
做为优选方案,易溶解溶剂:微溶或难溶溶剂:活性炭:六苄基六氮杂异伍兹烷粗品的重量比是:0.5~2:0.5~5:0.01~0.05:1.0,优选为0.7~1.2:2.5~3.5:0.01~0.02:1.0。
做为优选方案,步骤S1中,加热温度控制在35~68℃,优选45~60℃;冷却温度控制在-5~5℃,优选0~5℃。
步骤S2具体包括如下步骤:
S2.1、将步骤S1提纯精制的六苄基六氮杂异伍兹烷、溶剂A、催化活化剂A混合搅拌均匀,制得浆状物料;
S2.2、将溶剂B、催化活化剂B、乙酸酐、催化剂A在反应釜A中混合均匀,通入氢气,控制反应氢气压力为0.25~1.5MPa,降温至-10~30℃;然后将步骤S2.1的浆状物料缓慢压入压力反应釜中,压料时间控制在2-8h,压料结束后,继续在-10~30℃、0.25~1.5MPa保温反应2-8h;然后升温至30~60℃,继续在0.25~1.5MPa压力下反应2-8h;降温到15-30℃,过滤,洗涤,制得四乙酰基二苄基六氮杂异伍兹烷。
步骤S2.2中,所述催化剂A为活性炭附载型钯或氢氧化钯催化剂;钯或氢氧化钯在催化剂中的负载重量比为3%~7%;催化剂A的用量按催化剂中纯钯与精制的六苄基六氮杂异伍兹烷的重量比折算为0.01%~0.20%。优选的,所钯或氢氧化钯在催化剂中的负载重量比为3%~5%。
所述溶剂A+溶剂B、乙酸酐、催化活化剂A+催化活化剂B、精制的六苄基六氮杂异伍兹烷的重量比为1~5:(0.6~2):(0.01~0.1):1.0。优选为2~4:(0.8~1.5):(0.02~0.05):1.0。
所述催化活化剂A和催化活化剂B为在催化加氢条件下能分解出溴离子的化合物;可以是液溴、溴化氢、溴苯、苄溴、乙酰溴、三溴乙酸、NBS。优选催化活化剂为苄溴;
所述溶剂A和溶剂B均为DMF。
其中所述压力反应釜A可以进行先加压,再搅拌;再加压,再搅拌。其所述压力反应釜A可以为多层推进自吸式搅拌反应釜,反应釜的材质包括酸性条件下对溴离子耐腐蚀的金属合金材质(哈氏合金、钛合金);或,反应釜内衬高分子聚合物(PTEE、FEP、PFA、ECTFE)或耐酸无机非金属材质(搪玻璃材质、陶瓷材质、碳化硅材质);优选反应釜为碳化硅改性聚四氟乙烯材料内衬的高压反应釜。
步骤S3具体为:将溶剂C、水、钯或氢氧化钯催化剂B、四乙酰基二苄基六氮杂异伍兹烷按照一定比例加入到反应釜B中,通入氢气,控制反应压力为0.25~1.5MPa,温度25~80℃反应12~48h;过滤,收集滤液,减压浓缩,干燥,制的所述四乙酰基六氮杂异伍兹烷。
催化剂B为活性炭附载型钯或氢氧化钯催化剂;其中,钯或氢氧化钯在催化剂中的负载重量比为5%~10%;催化剂B的用量按催化剂中纯钯与TADBIW(四乙酰基二苄基六氮杂异伍兹烷)的重量比折算为0.02%~0.20%。
步骤S3中,所述溶剂C、水、TADBIW的重量比为1~5:(0.6~2):1.0;所述溶剂C包括乙酸。
反应釜B材质可以为316L不锈钢、904不锈及T2型钛材。
作为本发明的又一实施方式,所述的生产四乙酰基六氮杂异伍兹烷的方法,包括如下步骤:
(1)、六苄基六氮杂异伍兹烷用混合溶剂进行提纯精制;
(2)、一次加氢反应:将步骤(1)提纯精制的六苄基六氮杂异伍兹烷、溶剂A、催化活化剂A混合搅拌均匀,制得浆状物料;将溶剂B、催化活化剂B、乙酸酐、催化剂A在反应釜A中混合均匀,通入氢气,控制反应氢气压力为0.25~1.5MPa,降温至-10~30℃;然后将浆状物料缓慢压入反应釜中,压料时间控制在2-8h,压料结束后,继续在-10~30℃、0.25~1.5MPa保温反应2-8h;然后升温至30~60℃,继续在0.25~1.5MPa压力下反应2-8h;降温到15-30℃,过滤,洗涤,制得四乙酰基二苄基六氮杂异伍兹烷;
(3)、二次加氢反应:将溶剂C、水、催化剂B、四乙酰基二苄基六氮杂异伍兹烷按照一定比例加入到反应釜B中,通入氢气,控制反应压力为0.25~1.5MPa,温度25~80℃反应12~48h;过滤,收集滤液,减压浓缩,干燥,制的所述四乙酰基六氮杂异伍兹烷。
所述的生产四乙酰基六氮杂异伍兹烷的方法还包括步骤(4)、将步骤(3)的产物进行纯化的过程,具体如下:
步骤(3)的产物加入无水醋酸,升温到60~110℃,搅拌30min~2h,搅拌下加入无水乙醇,继续回流搅拌30min~2h,搅拌,降温到25~38℃,过滤,固体再用无水乙醇洗涤,烘干。
作为本发明的又一实施方式,生产四乙酰基六氮杂异伍兹烷的方法,其包括如下步骤:
1、首先是对原料六苄基六氮杂异伍兹烷粗品进行提纯精制,将含量在95~98%的六苄基六氮杂异伍兹烷粗品用混合溶剂进行重结晶:混合溶剂是两种或两种以上溶剂按一定比例组成的混合物,将混合溶剂、六苄基六氮杂异伍兹烷、活性炭按照一定比列投入反应釜中,加热至35~75℃保温15min~2h使六苄基六氮杂异伍兹溶解完全,保温结束趁热压滤除去活性炭及不溶物,滤液搅拌降温至-5℃-5℃保持30min~2h析出固体,过滤,固体再用溶剂洗涤、干燥,得雪白的粉状固体,取样HPLC检测主含量>99.80%,乙二酰二苄胺类杂质含量合计<0.001%,即可用做下一步加氢原料;
2、将一部分DMF及催化活化剂,乙酸酐及催化剂在特殊材质压力反应釜中混匀后,通入氢气,控制反应压力为0.25~1.5MPa,降温至-10~30℃后,用计量隔膜泵以一定速度压入六苄基六氮杂异伍兹烷与DMF和催化活化剂的浆状混合物,压料时间控制在2-8h,待物料全部压入后全部压入后,继续在-10~30℃、0.25~1.5MPa保温反应2-8h,然后升温至30~60℃,继续在0.25~1.5MPa压力下反应2-8h至反应完全,取样HPLC检测原料及中间体含量合计<0.5%即可降温到15-30℃,过滤,产品依次用DMF洗涤,再用去离子水洗涤,不用干燥直接用于下步加氢反应原料;
3、将加氢反应溶剂乙酸、水、催化剂和2步骤制备的四乙酰基二苄基六氮杂异伍兹烷按照一定比例加入到氢化压力反应釜中,通入氢气,控制反应压力为0.25~1.5MPa,温度25~80℃反应12~48h至反应完毕,取样HPLC检测原料及中间体含量合计<0.5%,即可趁热过滤回收催化剂,催化剂用水洗涤,合并滤液,浓缩至干后,重新加入一定量无水醋酸,升温到60~110℃,搅拌30min~2h,搅拌下加入一定量无水乙醇,继续回流搅拌30min~2h,搅拌,降温到25~38℃,过滤,固体再用无水乙醇洗涤,烘干,得到含量>99.5%的白色粉状四乙酰基六氮杂异伍兹烷产品。
通过所述的生产四乙酰基六氮杂异伍兹烷的方法制备得到的四乙酰基六氮杂异伍兹烷也属于本发明的保护范围。
在本发明提出物料浓度配比及反应温度、压力等条件下,采用将原料经过重结晶精制后将其缓慢压入特殊材质反应釜进行催化加氢的方法,基本解决了现有工艺技术会遇到上面的问题。由于该反应方式在氢化反应过程中不容易生成能对钯炭催化剂产生毒性的N-乙酰基苄胺、以及其因溴离子对设备腐蚀产生的重金属离子,使得反应在使用及少量的催化剂条件下即可完成催化脱苄基反应,而且催化剂反应完后还能保持较好活性,在保证反应收率不降低的能进行一定次数的套用。
本发明中,第一步加氢反应中反应釜材质的选择,对实现本发明至关重要,第一步加氢生成的产物是不溶于体系的固体,固体析出过程中对催化剂表面很不友好,这就需要添加原位生成的活性源溴离子来保持催化剂活性,再加上反应生成醋酸,酸性条件下溴离子对普通的金属合金材质都有很强的腐蚀性,随着设备腐蚀,重金属离子溶解于反应体系,在氢气还原性催化剂表面形成沉积,会导致催化剂活性减低,导致催化剂用量减少后(催化剂中金属钯的重量与HBIW的重量比值<0.2%)反应收率降低很多,这也是现有技术中不能减低催化剂用量的原因。而催化剂是该产品生产工艺的主要成本,所以第一步反应釜的材质对降低催化剂用量及稳定产品收率有关键作用。
第二步加氢反应中,因为已经通过溶剂及去离子水洗涤除去了TADBIW中间体的溴离子,反应的醋酸/水体系在该反应条件下对316L材质反应釜几乎无腐蚀,反应釜材质对该反应没有明显影响,所以这一步可以不使用成本昂贵的内衬材质的反应釜。
本发明通过研究发现压料时间过快与过慢都会影响到反应的收率。我们研究发现HBIW在酸性受热的条件下会分解,分解产物会与醋酐反应生成对催化剂有毒害的产物,所以将反应体系降低到安全温度后再缓慢将HBIW压入反应体系可以有效避免这种情况。在HBIW加氢制备TADBIW过程中是分多步骤进行的,首先是15~23℃条件下脱出1~2个苄基,在笼状结构上接上1~2个乙酰基后笼状结构电负性降低,中间体酸性条件下稳定性得到了很大的提升,且此时中间体是溶于反应体系的;然后再升温反应,继续脱出苄基,得到不溶于反应体系的TADBIW。将HBIW缓慢压入,可以有效控制反应体系内HBIW的浓度,抑制HBIW的分解,提高反应收率,降低催化剂用量。
本研究也进行过大量的HBIW精制实验,发现只有THF类结构的醚对其有很好的溶解性,但是仅使用这一种溶剂的话重结晶的分离收率会很低。使用本发明申请的混合催化剂能很好的兼顾重结晶产品含量及收率,我们也实验论证了引用丙酮作为重结晶溶剂,但是发现需要使用超过16倍重量溶剂且长时间加入回流才能溶解HBIW粗品,这不仅使得重结晶产品收率降低,所得产品含量最高仅为99.3%。我们通过实验发现,当HBIW的含量小于99.5%时,钯催化剂的用量必须要超过0.3%(催化剂的用量按催化剂中纯钯与HBIW的重量比折算),否则TADBIW的收率会很低甚至得不到产品,而本发明通过提纯精制的步骤,HBIW的含量大于99.9%,因此可以减少催化剂用量。
为与现有技术相比,本发明具有如下的有益效果:
1、使用混合溶剂与活性炭吸附对HBIW粗品进行重结晶精制,以较少的溶剂用量及较高的分离收率得到了高纯度HBIW精品,完全除去了对钯炭催化剂有毒害作用的杂质,使得在催化加氢的时候降低催化剂的用量及提高催化剂使用次数得以实现;
2、在HBIW催化加氢制备TADBIW(四乙酰基二苄基六氮杂异伍兹烷)过程中,采用缓慢将原料和催化活化剂压入反应体系的方法,使得在非还原性条件下HBIW不会与醋酐接触,避免其酸性条件下分解及与醋酐生成会使钯催化剂毒性的N-乙酰基苄胺类物质,催化活化剂和原料一同缓慢压入,保证在第一反应阶段催化剂能保持长时间的较高活性,减少开环副产物,提高反应收率;
3、选择对酸性条件下对溴离子有极好耐腐蚀性能的无机非金属碳化硅材料制作的特殊自吸式多层推进搅拌反应釜,不仅可以避免因使用金属合金材料制作的反应釜由于压力设备腐蚀原因导致的安全风险,还可以杜绝因设备腐蚀产生的重金属离子在还原体系里面对钯催化剂的毒化,极大的提高了生产的安全性、降低了催化剂在生产中的成本占比。
附图说明
通过阅读参照以下附图对非限制性实施例所作的详细描述,本发明的其它特征、目的和优点将会变得更明显:
图1为本发明实施例8中TAIW液相色谱图;
图2为本发明实施例8中TAIW核磁氢谱图。
具体实施方式
下面结合具体实施例对本发明进行详细说明。以下实施例将有助于本领域的技术人员进一步理解本发明,但不以任何形式限制本发明。应当指出的是,对本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进。这些都属于本发明的保护范围。
本发明提供一种生产四乙酰基六氮杂异伍兹烷的方法,反应式如下:
六苄基六氮杂异伍兹烷提纯精制
1000L搪瓷反应釜(普通锚式搅拌,转速45转/分钟),洗净干燥,氮气保护下通过高位槽加入150.0Kg THF(四氢呋喃),450.0Kg乙腈,然后加入150.0Kg含量约为98%左右的HBIW(六苄基六氮杂异伍兹烷)粗品,2.0Kg活性炭,搅拌升温到55℃,保持30分钟使得粗品溶解完全,然后趁热将其压滤到1000L的结晶釜中,搅拌降温到0~5℃保温2小时使产品析出完全,保持低温过滤,固体再用冷的乙腈洗涤,真空干燥,得142.0Kg白色固体,取样HPLC分析,HBIW含量99.90%,未检出乙二酰二苄胺类杂质,可用于后续氢化反应。
1000L搪瓷反应釜(普通锚式搅拌,转速45转/分钟),洗净干燥,氮气保护下通过高位槽加入丙酮800Kg,然后从人孔再投入50.0Kg含量约为98%左右的HBIW(六苄基六氮杂异伍兹烷)粗品,0.7Kg活性炭,搅拌升温到55℃,保持30分钟粗品未能溶解完全,继续升温到57℃回流搅拌30分钟,粗品基本溶解完全。然后趁热将其压滤到1000L的结晶釜中,搅拌降温到0~5℃保温2小时使产品析出完全,保持低温过滤,固体再用冷的丙酮洗涤,真空干燥,得41.0Kg稍微带浅黄色的固体,取样HPLC分析,HBIW含量99.2%,乙二酰二苄胺类杂质含量为0.08%。
结果表明:因为HBIW在丙酮中的溶解度低,需要16倍重量的溶剂在长时间加热回流的条件下才能使粗品溶解完全,这不仅提高了重结晶的处理成本,也使得粗品因加热时间过长分解,使用丙酮作为重结晶溶剂不仅分离收率及效率低,而且也无法完全除去对加氢敏感的杂质。
第一次加氢反应制备TADBIW(四乙酰基二苄基六氮杂异伍兹烷)
1、在10L带机械搅拌玻璃反应瓶中,依次加入2.80Kg经过制备例1重结晶精制过的HBIW,3.60Kg DMF,60.0g苄溴,搅拌均匀,大部分HBIW不溶,得到一白色浆状物料;
2、20升碳化硅改性聚四氟乙烯材质内衬加氢反应釜(双层推进自吸式反应釜),向反应釜引入溶剂DMF 5.60Kg、乙酸酐2.30Kg、苄溴20.0g,以及40.0g 3.5%(钯在催化剂中的负载重量比)钯/C催化剂(净重,事先用DMF洗涤置换进行无水处理),反应釜经氮气和氢气置换后,氢气加压到0.5MPa,反应釜冷冻水,开搅拌800转/分钟,降温到15℃。
3、玻璃反应瓶中配制的原料通过精密高压活塞泵打入加氢压力反应釜中,打料速率控制为1.40~1.60Kg/小时,压料过程中控制氢气压力在0.4~0.6MPa、温度控制在14~17℃,4~5小时原料打入结束后,缓慢升温至17~21℃、继续在0.4~0.6MPa压力下反应6小时,然后缓慢升温至50~53℃,继续在0.4~0.6MPa压力下反应8小时至反应基本不耗氢,此时反应液取样HPLC分析,原料HBIW剩余基本检测不出,中间体剩余<0.5%,;降温到15~20℃,氮气置换,抽滤分离,固体产物再用DMF洗涤三次除去杂质,去离子水洗涤两次除去DMF,抽干,真空干燥至恒重,得1.80Kg灰白色四乙酰基二苄基六氮杂异伍兹烷产品。产品取样HPLC分析,含量为98.6%,扣除水和钯炭催化剂的重量,此步加氢的分离收率为85.3%。
实施例2仅HBIW的压料时间与实施例1不同,实施例2验证了HBIW加氢制备TWDBIW过程中,HBIW的压料时间的不同对反应结果的影响。
表1
*、TADBIW的收率为分离产品真空干燥后扣除催化剂再经过含量折算后的对比原料HBIW的摩尔收率。
实施例3与实施例1区别在于:使用的是不同附载量的钯炭催化剂,其中催化剂中金属钯的总重量与实施例1相同,均为1.40g,金属钯与原料HBIW的重量比均为0.05%。实施例3验证了催化剂中金属钯与原料HBIW的重量比均为0.05%时,催化剂中金属钯在催化剂中的不同负载重量比值对反应结果的影响。
表2
*、TADBIW的收率为分离产品真空干燥后扣除催化剂再经过含量折算后的对比原料HBIW的摩尔收率。
实施例4使用的除反应釜内衬材质有区别外,其它操作与实施例1相同,实施例4验证了反应釜物料接触部分为316L不锈钢、904不锈及T2型钛材时对反应收率的影响。
表3
*、TADBIW的收率为分离产品真空干燥后扣除催化剂再经过含量折算后的对比原料HBIW的摩尔收率。
和实施例1相比,在催化剂相同的条件下,反应设备的材质对反应收率及产品含量影响较大。
对比例1和实施例1的区别在于,对比例1所用原料为对比制备例1中用丙酮重结晶制备的HBIW原料。
HBIW原料打入结束后,缓慢升温至17~21℃、继续在0.4~0.6MPa压力下反应6小时,然后缓慢升温至50~53℃,继续在0.4~0.6MPa压力下反应,2小时后反应耗氢很慢,此时反应液取样HPLC分析,大量HBIW原料和中间体剩余,继续保温反应10小时基本不消耗氢气,取样HPLC分析,HBIW分解,TADIW含量小于15%,降温到15~20℃,氮气置换,抽滤分离,固体产物再用DMF洗涤三次除去杂质,去离子水洗涤两次除去DMF,抽干,真空干燥,得0.54Kg深灰黑色粘稠固体,取样HPLC分析,TADBIW含量为47.3%,此方法加氢反应TADBIW的分离收率为11.5%。
对比例2和实施例1的区别在于HBIW的加料方式采用直接投料,具体如下:
20升碳化硅改性聚四氟乙烯材质内衬加氢反应釜,向反应釜引入溶剂DMF9.20Kg、乙酸酐2.30Kg、苄溴80.0g,反应釜经氮气置换后搅拌降温到10~15℃,再加入2.80Kg经过重结晶精制的HBIW,以及40.0g净重的3.5%(金属钯在催化剂中的重量比)钯/C催化剂(净重,事先用DMF洗涤置换进行无水处理),氮气和氢气置换后加压到0.5MPa,反应釜继续通冷冻水,开搅拌800转/分钟,降温到15℃,温度控制在14~17℃,0.4~0.6MPa压力下反应4~5小时,然后缓慢升温至17~21℃、继续在0.4~0.6MPa压力下反应6小时,然后缓慢升温至50~53℃,继续在0.4~0.6MPa压力下反应8小时至反应基本不耗氢;降温到15~20℃,氮气置换,抽滤分离,固体产物再用DMF洗涤三次除去杂质,去离子水洗涤两次除去DMF,抽干,真空干燥,得1.31Kg灰白色四乙酰基二苄基六氮杂异伍兹烷产品,产品取样HPLC分析,含量为90.1%,扣除钯炭催化剂的重量,此方法加氢反应TADBIW的分离收率为55.4%。
第二次加氢反应制备TAIW(四乙酰基六氮杂异伍兹烷)
20升碳316L材质氢反应釜(双层推进自吸式反应釜,转速800转/分钟),用去离子水清洗干净,依次加入1.80Kg去离子水,9.00Kg醋酸,26.0g 7.0%(金属钯在催化剂中的负载重量比)钯/C催化剂(净重)以及实施例1制备得到的1.80Kg TDABIW,反应釜经氮气和氢气置换后,氢气加压到0.5MPa,搅拌下缓慢升温至50~55℃,继续在此温度下继续在0.4~0.6Mpa反应16小时至基本不耗氢,反应液取样HPLC分析,原料及中间体含量合计为0.2%,氮气置换,趁热过滤分离催化剂,催化剂再用去离子水洗涤两次,合并滤液,转移到20升玻璃反应瓶中;滤液减压浓缩至干,加入1.20Kg醋酸,搅拌升温至100~105℃打浆30分钟,加入4.80Kg无水乙醇,继续回流打浆搅拌2小时,搅拌冷却到25~30℃,过滤,固体再用无水乙醇淋洗两次,真空干燥,得990g含量为99.6%的白色粉状四乙酰基六氮杂异伍兹烷产品,该步加氢的分离收率为87.4%。
实施例6与实施例5区别在于使用的是不同附载量的钯炭催化剂,其中催化剂中金属钯的总重量与实施例5相同,均为1.82g,金属钯与原料TADBIW的重量比均为0.10%。实施例6验证了催化剂中金属钯重量相同时,催化剂中金属钯在催化剂中的不同负载重量比值对反应结果的影响。
表4
a、加氢反应时间是指反应升温至50~55℃,继氢气压力在0.4~0.6Mpa反应至体系基本不耗氢所需要的时间;
b、TAIW的收率为分离产品真空干燥后经过含量折算后的对比中间体TADBIW的摩尔收率。
结果表明:在TADBIW加氢制备TAIW时,因4位和10位两个苄基脱出的活化能较高,低负载的钯炭催化剂反应速率慢,导致该反应的时间延长很多,且有较多的原料及中间体剩余,导致产品分离收率及纯度都偏低。
实施例7对催化剂进行了多次循环利用,验证了TADBIW制备TAIW的7%钯/C加氢催化剂套用对产品收率及产品质量的影响。本实施例加氢操作同实施例5,不同在于催化剂为套用上一次反应液过滤回收,再经去离子水洗涤的回收催化剂,且无需新补加7%(催化剂中钯的重量比)钯/C催化剂:因为每次有第一次加氢反应制备TADBIW过程中带入的3.5%(催化剂中钯的重量比)钯/C催化剂,所以回收催化剂的量随着套用次数增多而变大。
回收方法:经去离子水洗涤后的催化剂,加入2L的氢化反应釜中,再加入1.2Kg去离子水,氮气置换后氢气置换,氢气加压到1.0Mpa,开搅拌(300转/分钟)升温到100℃活化搅拌5小时,搅拌降温到35℃,氮气压滤,在用去离子水洗涤多次,压干水分,取样测量水含量及钯含量。
循环一:
将实施例5中的催化剂进行回收(净重66.0g),完全用于本次制备的步骤S3中。
循环二:
将循环一中的催化剂进行回收(净重106.0g),完全用于本次制备的步骤S3中。
循环三:
将循环二中的催化剂进行回收(净重146.0g),完全用于本次制备的步骤S3中。
循环四:
将循环三中的催化剂进行回收(净重186.g),完全用于本次制备的步骤S3中。
表5
*总反应时间以反应起始至反应基本不耗氢计时结束,前两次催化剂套用计时结束后应液中原料及中间体含量合计小于0.5%,再继续增加催化剂套用次数原料无法反应完全。
1、第一次加氢反应制备TADBIW(四乙酰基二苄基六氮杂异伍兹烷)
1.1、在1000L配料釜中,依次加入140.0Kg经过制备例1重结晶精制过的HBIW,180.0Kg DMF(N,N-二甲基甲酰胺),3.0Kg苄溴,搅拌均匀,大部分HBIW不溶,得到一白色浆状物料;
1.2、向反应釜(1000升碳化硅改性聚四氟乙烯材质内衬加氢反应釜(自吸式四层推进搅拌,转速300转/分钟),搅拌形式为自吸式多层推进搅拌,反应釜配置循环热水加热及冷却盐水)引入溶剂DMF 280.0Kg、乙酸酐115.0Kg、苄溴1.0Kg,以及2.0Kg 3.5%(金属钯在催化剂中的重量比)钯/C催化剂(净重,事先用DMF洗涤置换进行无水处理),反应釜经氮气和氢气置换后,氢气加压到0.5MPa,反应釜开启冷冻水降温,搅拌降温到15℃。
1.3、压料:步骤2.1的配料釜中的白色浆状物料通过隔膜泵打入加氢压力反应釜中,打料速率控制为70~80kg/小时,压料过程中控制氢气压力在0.4~0.6MPa、温度控制在14~17℃,4~5小时原料打入结束后,缓慢升温至17~21℃、继续在0.4~0.6MPa压力下反应6小时,此时反应液取样HPLC分析,原料HBIW剩余<1%,中间体约为75%,TADBIW>20%;缓慢升温至50~53℃,继续在0.4~0.6MPa压力下反应8小时至反应基本不耗氢,此时反应液取样HPLC分析,原料HBIW剩余基本检测不出,中间体剩余<0.5%,TADBIW含量>95%;降温到15~20℃,氮气置换,离心过滤分离,固体产物再用DMF洗涤三次除去杂质,去离子水洗涤两次除去DMF,离心至干,得到108.0Kg水分含15%的灰黑色四乙酰基二苄基六氮杂异伍兹烷产品,无需烘干,可直接用于下一步催化加氢。产品取样HPLC分析,含量为98.7%,扣除水和钯炭催化剂的重量,此步加氢的分离收率为86.9%。
2、第二次加氢反应制备TAIW(四乙酰基六氮杂异伍兹烷)
1000升316L材质氢化反应釜(自吸式四层推进搅拌,转速300转/分钟),用去离子水清洗干净,依次加入80.0Kg去离子水,450.0Kg醋酸,1.30Kg 7.0%(金属钯在催化剂中的重量比,净重)以及上述步骤制备得到的108.0Kg TDABIW湿品,反应釜经氮气和氢气置换后,氢气加压到0.5MPa,搅拌下缓慢升温至50~55℃,继续在此温度下继续在0.4~0.6Mpa反应16小时至基本不耗氢,反应液取样HPLC分析,原料及中间体含量合计为0.2%,氮气置换,趁热过滤分离催化剂,催化剂再用去离子水洗涤两次,合并滤液,转移到1000升316L不锈钢浓缩釜中;滤液减压浓缩至干,加入60.0Kg醋酸,搅拌升温至100~105℃打浆30分钟,加入240.0Kg无水乙醇,继续回流打浆搅拌2小时,搅拌冷却到25~30℃,过滤,固体再用无水乙醇淋洗两次,真空干燥,得51.0Kg含量为99.7%的白色粉状四乙酰基六氮杂异伍兹烷产品,该步加氢的分离收率为88.2%。
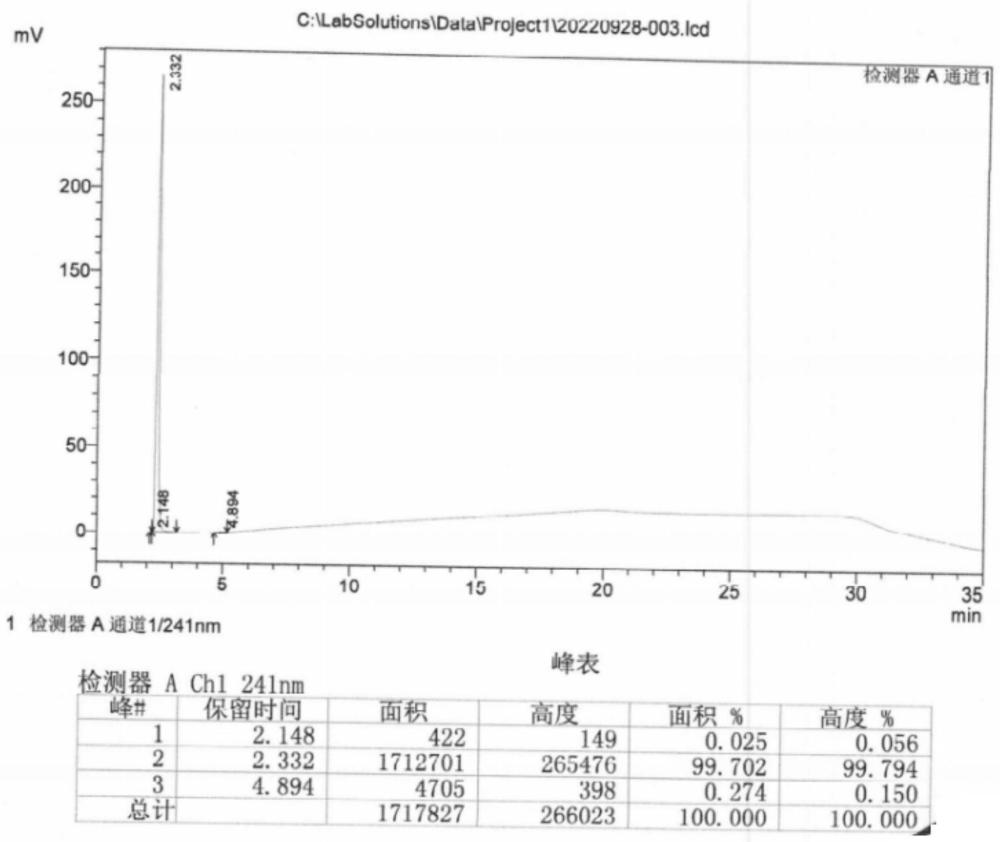
图1所示为本实施例中TAIW液相色谱图。
图2为本实施例中TAIW核磁氢谱图。
以上对本发明的具体实施例进行了描述。需要理解的是,本发明并不局限于上述特定实施方式,本领域技术人员可以在权利要求的范围内做出各种变形或修改,这并不影响本发明的实质内容。
- 一种改性六硝基六氮杂异伍兹烷的制备方法、改性六硝基六氮杂异伍兹烷及应用
- 一种基于电化学路径合成笼状四乙酰基六氮杂异伍兹烷TAIW的方法